

Abstract
Monoclonal antibodies were raised against Daudi B-lymphoblastoid cell line membranes. An mAb (BAT) was selected for its ability to stimulate human and murine lymphocyte proliferation. BAT induced cytotoxicity in human and murine lymphocytes against natural killer cell-sensitive and -resistant tumor cell lines. A single intravenous administration of BAT to mice that had been inoculated with various murine tumors (e.g., B16 melanoma, 3LL carcinoma, and methylcholanthrene fibrosarcoma) resulted in striking antitumor effects as manifested by complete tumor regression and prolonged survival of the treated mice. BAT exhibited a diminished but significant antitumor effect in athymic nude mice, which are deficient in T lymphocytes, and in beige mice, which are deficient in NK cells. Furthermore, selective depletion of T or NK cells in mice reduced the response to the antitumor effect of BAT. These data indicate a dual role for T and NK cells in mediating the antitumor activity of BAT. We report here on the antitumor activity of BAT mAb on human tumor xenografts in mice. BAT demonstrated an antitumor effect in nude mice bearing human colon carcinoma (HT29) xenografts. It failed, however, to inhibit established lung metastases in severe combined immunodeficient (SCID) mice that had been inoculated (i.v.) with SK28 human melanoma. Engraftment of human lymphocytes into SCID mice bearing human melanoma xenografts rendered them responsive to the antitumor effect of BAT. The efficacy of BAT in the regression of human tumors by activation of human lymphocytes indicates its potential clinical use.
Keywords: human melanoma, tumor regression, immune stimulation/nude and beige mice/T or NK cell depletion
Monoclonal antibodies (mAbs) directed against various T cell determinants were previously reported to induce proliferation and differentiation of T cells (1). The most remarkable among them is the mAb against CD3 determinant, which was able to induce clonal proliferation, elicit mitogenic activity, and also trigger the cytolytic process in T lymphocytes (2–4). Additional immunostimulatory mAbs were found to react with CD5 (5), CD69 (6), and CD28. The latter, an antigen on the T cell that interacts with its ligand, B7, present on antigen presenting cells including tumor cells (7–9). In vivo antitumor activity of anti-CD3 and of anti-CD28 was previously reported (10, 11). Activation of T cells, which elicit a variety of effector functions, results from interaction of antigen with the T cell antigen receptor and a costimulation directed to additional surface determinants such as the CD28 (12).
We previously reported a mAb directed against human B-lymphoblastoid cell membranes (BAT) that stimulates human lymphocytes, as manifested by enhanced murine and human lymphocyte proliferation and cytolytic activity against tumor cells in vitro (13). BAT binding protein was identified as a 48- to 50-kDa monomeric protein (13). BAT was found to induce murine tumor regression in C57BL mice that was mediated by its immune stimulatory properties (14).
In the present study, we evaluated the antitumor activity of BAT in nude mice carrying human tumor xenografts and in severe combined immunodeficient (SCID) mice engrafted with human lymphocytes and inoculated with human tumor cells (15, 16).
MATERIALS AND METHODS
Monoclonal Antibodies.
BAT was generated and purified as described (14). In brief, BALB/c mice were immunized with membranes from Daudi cells. Spleen cells were fused with myeloma NS-O cells. BAT was selected by its ability to bind Daudi cells and by its ability to induce proliferation of human peripheral blood mononuclear cells (PBMC). Cells were grown in RPMI 1640 medium supplemented with fetal calf serum (10%), sodium pyruvate, glutamine, and antibiotics and incubated at 37°C in a humidified atmosphere containing 5% CO2. BAT was purified on a protein G Sepharose column according to manufacturer’s instructions (Pharmacia).
Cell Preparations for Engraftment to SCID Mice.
Human PBMC were obtained from blood of healthy donors by Ficoll/Hypaque density centrifugation. Cells were washed and suspended in PBS. Cells (5 × 107) were injected i.p. to each SCID mouse to construct a human immune system in these mice.
Splenocytes were obtained from either untreated C57BL mice or from mice 24 h after injection of 100 μg/mouse of anti CD3 (PharMingen) or anti-asialoGM1 (ASGM1) (Wako Chemicals, Dallas). Cells (5 × 107) were injected i.p. to each SCID mouse.
Activation of Lymphocytes by Tumor Cells and BAT.
Human PBL (2 × 106/ml) were incubated on HT29 human colon carcinoma cells monolayers for 1 day. Peripheral blood lymphocyte (PBL) cells were then removed from the tumor cell monolayer, washed twice with medium, and suspended at the initial concentration. Splenocytes obtained from tumor-inoculated mice were suspended at 2 × 106/ml, and BAT at 0.1 μg/ml was added for 3 days in vitro.
Proliferation Assay of Lymphocytes upon Incubation with BAT.
PBLs were separated from PBMC by removing the adherant monocytes after 1-h incubation on plastic Petri dishes. Aliquots of 2 × 106 PBL (200 μl) in culture medium containing 5% human type AB serum were incubated for 3 days in 96-well flat-bottom plates with and without BAT at 0.1 μg/ml. [3H]Thymidine (1 μCi/well; 1 Ci = 37 GBq) was added for 20 h before harvesting. Cultures were harvested into glass filters and radioactivity was counted using a liquid β-scintillation counter.
Mouse Tumor Models.
Five to six female mice, obtained from Harlan Laboratories (Jerusalem), 6–8 weeks old were used in each group for every experiment. SCID mice were maintained in sterile conditions at a controlled temperature.
B16 murine melanoma cells (5 × 104) were injected i.v. into C57BL wild type, nude mice (C57BL nu/nu), beige mice, or SCID mice that had been engrafted with murine splenocytes. BAT was injected i.v. at 10 μg/mouse 14 days later. Twenty-four days after tumor inoculation, mice were killed and lungs metastases were counted and scored by number, size, and lung weight.
HT29 human colon carcinoma cells were injected s.c. at 106/mouse at the axillary region of wild-type or nude mice. BAT (10 μg/mouse) was injected i.p. at days 7, 14, and 21 posttumor inoculation. Beginning at 7 days posttumor inoculation, the tumor size was measured daily until day 30 posttumor inoculation, when the untreated mice died.
SK-mel 28 (SK28), a human melanoma-derived cell line (originally obtained from Sloan–Kettering Institute, New York) was injected i.v. into SCID mice at 5 × 105/mouse. We found that i.v. injection of these cells resulted in tumor lesions in the lungs. SK28 melanoma cells were inoculated 1 day following i.p. administration of anti-ASGM1 (35 μg/mouse) (Wako Chemicals). Anti-ASGM1 is a rabbit polyclonal antibody that binds to murine NK cells and depletes these cells when injected i.p. into mice (17). This antibody was previously used to enhance engraftment of human PBMC into SCID mice (17). Human PBMC (5 × 107/mouse) were engrafted i.p. to SCID mice. Treatment with BAT was done by a single i.v. injection (10 μg/mouse) 14 days posttumor inoculation or at different times as indicated in the tables. Twenty-four days posttumor inoculation, the mice were killed, the lungs removed, melanoma metastases were counted, and lung weight was determined.
RESULTS
We evaluated the antitumor activity of BAT in mice bearing human tumors. Initially we studied the antitumor effect of BAT in athymic nude mice bearing murine B16 melanoma. As demonstrated in Table 1, BAT was found to be effective in this tumor model although to a lesser extent than in the wild-type mice. C57BL mice inoculated with murine B16 melanoma and treated with BAT had zero or only 1 ± 1 melanoma lesion compared with the nude mice that had numerous tumor metastases. The antitumor effect of BAT in nude mice that are deficient in T cells, suggests that non-T cells could mediate its antitumor activity. Similar experiments using beige mice (Table 1) which are deficient in NK cells (18), showed similar results, namely, a reduced number of tumor lesions in the lungs of BAT-treated beige mice although not to the same extent as was observed in the wild-type mice. In the following experiment, C57BL mice were depleted of T or NK cells by in vivo administration of the appropriate antibodies. As seen in Table 2, depletion of either T cells or NK cells increased the tumor resistance to BAT. However prolonged administration of the antibodies as in experiment 1 (Table 2) indicates that NK depletion was effective in rendering mice resistant to the antitumor activity of BAT.
Table 1.
Antitumor activity of BAT in nude and beige mice bearing B16 melanoma
| BAT treatment | No. of metastases
|
|||
|---|---|---|---|---|
| Exp. 1
|
Exp. 2
|
|||
| − | + | − | + | |
| Effect of BAT in nude mice | ||||
| C57BL | >200 | 0 | >200 | 1 ± 1 |
| Nude | >200 | 48 ± 25 | 95 ± 45 | 10 ± 3 |
| Effect of BAT in beige mice | ||||
| C57Bl | 141 ± 8 | 0 | 49 ± 8 | 0 |
| Beige | >200 | 47 ± 39 | 140 ± 47 | 101 ± 44 |
Mice were injected (i.v.) with B16 melanoma (5 × 104) cells and 14 days later with BAT (10 μg/mouse). Mice were sacrificed 24 days posttumor inoculation and the number of lung metastases were scored. More than 200 represents confluent tumor growth in the lungs. Five mice were used in each group in each experiment, and the results are expressed as mean ± SD.
Table 2.
The effect of T- or NK cell-depletion in mice bearing B16 melanoma on the antitumor activity of BAT
| BAT treatment | Number of metastases
|
|||
|---|---|---|---|---|
| Exp. 1
|
Exp. 2
|
|||
| − | + | − | + | |
| Nondepleted | 113 ± 80 | 0.5 ± 1 | >200 | 0 |
| T cells depleted | >200 | 56 ± 9 | >200 | 37 ± 23 |
| NK cells depleted | >200 | >200 | >200 | 160 ± 60 |
C57BL mice were injected i.p. with either anti-CD3 (100 μg) or anti-asialo GM1 (35 μg) 1 day prior to B16 melanoma injection and on days 7, 14, and 21 days posttumor inoculation. Experiment 1: Mice injected with anti-CD3 or anti-asialoGM1 on day 12 and 22 posttumor inoculation. Experiment 2: Five mice were used in each group. Animals were sacrificed on day 24 posttumor injection and the numbers of metastases were scored. Results are expressed as the means ± SD.
The antitumor activity of BAT in nude mice implanted s.c. with human colon carcinoma (HT29), was also demonstrated (Fig. 1). The growth of the tumor in these mice treated with BAT was delayed up to 24 days posttumor inoculation and was half the size on day 46 compared with the tumor size in untreated mice.
Figure 1.
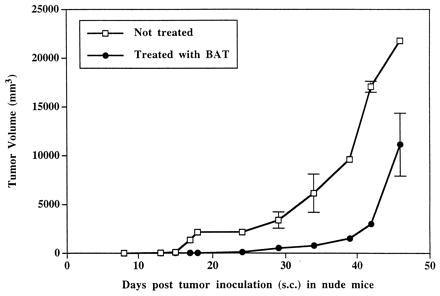
BAT treatment of human colon carcinoma (HT29) growth in nude mice. Nude mice were injected (s.c.) with human HT29 cells (106/mouse) and treated with BAT (10 μg/mouse). BAT was injected i.p. at days 7, 14, and 21 posttumor inoculation.
We have investigated whether BAT is capable of inducing human tumor regression via the stimulation of human lymphocytes. SCID mice injected with murine or human tumors failed to respond to BAT treatment and died within 14 days posttumor inoculation. We then established that engraftment of murine splenocytes enabled BAT to induce regression of murine B16 melanoma in the SCID mice (Table 3). BAT was administered 14 days posttumor inoculation, the time at which BAT was previously found to be most effective in inducing regression of murine tumors in wild-type mice (14). BAT treatment in the engrafted SCID mice reduced the number of lung metastases from 217 ± 65 to an average of only 7 ± 3. Engraftment of splenocytes from C57BL mice, which had been depleted of CD3, rendered the SCID mice recipients resistant to the antitumor effect of BAT. In contrast, engraftment of NK-depleted splenocytes rendered the SCID mice only partially resistant to BAT treatment (Table 3).
Table 3.
Regression of murine B16 melanoma in SCID mice engrafted with murine splenocyte subpopulations and treated with BAT
| Splenocyte engraftment | No. of metastases per BAT treatment
|
|
|---|---|---|
| − | + | |
| Nonengrafted | >250 | >250 |
| Nondepleted | 217 ± 65 | 7 ± 3 |
| CD3 depleted | >250 | >250 |
| NK depleted | >250 | 84 ± 38 |
SCID mice were engrafted i.p. with splenocytes (5 × 107/mouse) from C57BL mice or from C57BL mice that were injected with anti-CD3 (100 μg/mouse) or anti-asialoGM1 (100 μg/mouse) 1 day prior to isolation of splenocytes for engraftment. B16 melanoma cells were injected i.v. 5 days later. BAT (10 μg/mouse) was injected i.v. 10 days following tumor administration. Mice were sacrificed 24 days posttumor inoculation and the number of lung metastases was determined. Five SCID mice were used in each group and the results are expressed as the mean ± SD.
We extended these studies to evaluate the antitumor effect of BAT against human tumors. SCID mice were engrafted i.p. with human PBMC along with an inoculation i.v. of human melanoma (SK28) cells. Inoculation i.v. of human SK28 melanoma cells led to development of tumor lesions in the lungs (similar to the previously reported model of established lung metastases using a variety of syngeneic murine tumors) (14). Administration of BAT to these mice, 14 days after tumor inoculation, resulted in a marked regression of tumor lesions in the lungs (Table 4, Fig. 2). Ten out of the 32 mice in five different experiments treated as above were tumor-free. Illustrations of the lungs of BAT-treated mice compared with the lungs of untreated mice are presented in Fig. 2. The distribution of human lymphocyte subpopulations in the lungs of SCID mice engrafted with human PBMC and treated with BAT was investigated. Some 15% of the cells in the lungs were human CD3-positive, whereas 13% were human CD56-positive. Some 7% were CD3- and CD56-positive. The antitumor effect of BAT in the SCID mice was dependent upon the engrafted human lymphoid cells. In these mice, treatment by BAT reduced the number of metastases from an average of 174 ± 53 to 8 ± 9 and lung weight from 702 ± 140 mg to a normal weight of 206 ± 17 mg. It should be noted that the engraftment of the human lymphoid cells by themselves had a slight antitumor effect (Table 4). Treatment with anti-asialoGM1 of nonengrafted mice, which were inoculated with SK28 melanoma, did not change their response to the antitumor effect of BAT. Nonengrafted mice treated or untreated by anti-asialoGM1 died 10–13 days posttumor inoculation.
Table 4.
Number of lung metastases and lung weight in SCID mice engrafted with human lymphocytes, inoculated with human melanoma (SK28), and treated 14 days later with BAT
| BAT treatment | Nonengrafted, − | Engrafted with human PBMC
|
|
|---|---|---|---|
| − | + | ||
| No. lung metastases | >250 | 174 ± 53 | 8 ± 9 |
| (n = 13) | (n = 25) | (n = 32) | |
| Lung weight, mg | 867 ± 82 | 702 ± 140 | 206 ± 17 |
| (n = 12) | (n = 22) | (n = 32) | |
SCID mice were injected with anti-GM1 (25 μg/mouse). On the following day, human PBMC (5 × 107/mouse) were injected i.p. Human melanoma cells (SK28) were inoculated i.v. 3–5 days later at 5–7 × 105/mouse. Mice were treated with BAT (10 μg/mouse) in a single injection i.v. 14 days posttumor inoculation. Mice were sacrificed on day 24 posttumor inoculation, and the extent of lung metastases was evaluated by scoring the number of metastases and lung weights. Average lung weight of untreated mice is 210 ± 10 mg. n, The total number of mice studied in five different experiments.
Figure 2.
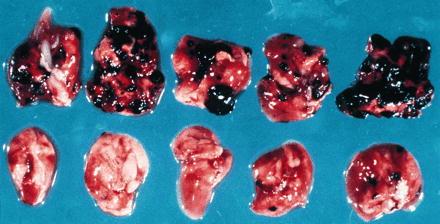
Lungs from SCID mice bearing human melanoma treated with BAT. (Upper) Lungs from human PBMC engrafted SCID mice 24 days postinoculation with SK28 human melanoma. (Lower) Lungs from human PBMC engrafted SCID mice 24 days postinoculation with SK28 human melanoma and 10 days posttreatment with BAT.
We previously demonstrated that the antitumor activity of BAT against murine B16 melanoma was maximally pronounced upon administration of the antibody between days 10 and 14 posttumor inoculation (14). Similar effects of BAT’s antitumor activity, when administered on day 14 posttumor inoculation, in SCID mice engrafted with human PBMC and inoculated with SK28 melanoma, were achieved. Administration of BAT 3 days after tumor inoculation had only a marginal nonsignificant antitumor effect, whereas administration of the antibody after 14 days reduced metastases from confluence to an average of 10 ± 17. BAT administered 5 days and 10 days posttumor inoculation was also effective and reduced metastases to 89 ± 7 and 35 ± 12, respectively (Table 5).
Table 5.
BAT treatment at different times following human melanoma inoculation of SCID mice engrafted with human lymphocytes
| BAT treatment, day | No. metastases | Lung weight, mg |
|---|---|---|
| None | >250 | 750 ± 70 |
| 3 | 206 ± 75 | 665 ± 150 |
| 5 | 89 ± 7 | 315 ± 8 |
| 10 | 35 ± 12 | 219 ± 1 |
| 14 | 10 ± 17 | 217 ± 6 |
BAT was administered (10 μg/mouse) at different times in relation to tumor inoculation at day 0. Human PBMC were engrafted i.p. at 5 × 107/cells per mouse, 1 day postinjection of anti-GM1 (25 μg/mouse). Tumor cells were injected i.v. 3–5 days later at 7 × 105 cells/mouse. Twenty-four-day posttumor inoculation mice were sacrificed and the number of lung metastases and weights were determined. Average lung weight of untreated mice is 210 ± 10 mg.
To study whether BAT elicited its antitumor effect as a result of a stimulatory signal provided by the tumor cells, we conducted experiments in which splenocytes isolated from mice bearing tumors exhibited an enhanced response in vitro to stimulation by BAT (Fig. 3). Splenocytes of C57BL mice injected with B16 melanoma, were co-cultured in the presence of BAT (1 μg/ml) in vitro and the [3H]thymidine uptake was measured. As can be seen, uptake of thymidine in splenocytes from mice bearing tumors for 5 days increased (26633 ± 872) in the presence of BAT, compared with the splenocytes of tumor-free mice (13156 ± 447). To further examine the mechanism of BAT activation, we studied the stimulatory effect of BAT in vitro on human PBLs that were preincubated on HT29 human colon carcinoma cell monolayers. Results of 4 experiments using 4 different human PBLs are shown in Fig. 4. Exposure of lymphocytes to the tumor cells in vitro led to a significant increase in their proliferative response ranging from a 12- to 22-fold increase in [3H]thymidine uptake. Moreover, in lymphocytes that were pre-exposed to tumor cells and then cultured with BAT, thymidine uptake increased 22- to 44-fold. Lymphocytes that were incubated on allogeneic macrophage monolayers did not acquire the enhanced sensitivity to stimulation by BAT (data not shown).
Figure 3.
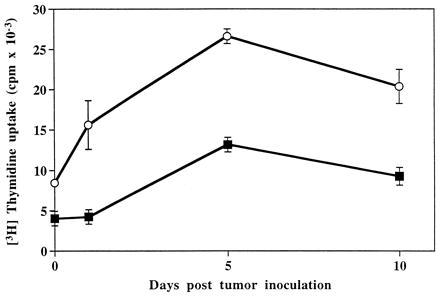
Proliferation of splenocytes from B16 melanoma bearing mice in the presence and absence of BAT. Splenocytes obtained from C57BL mice at various days postinjection with murine B16 melanoma cells were cultured in the presence of BAT (1 μg/ml) for 3 days (○) and without BAT for 3 days (▪). [3H]Thymidine uptake was determined as described.
Figure 4.
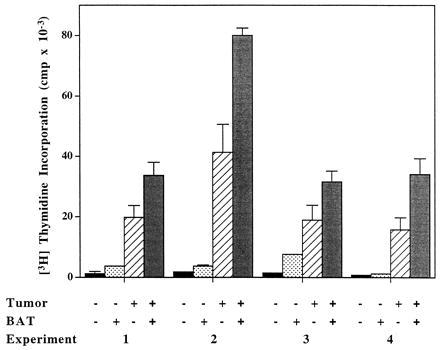
Thymidine uptake in human PBLs incubated on human HT29 colon carcinoma cells in the presence and absence of BAT. Four experiments using four different human PBLs are shown. PBLs were cocultured on HT29 human colon cells. The lymphocytes were removed after 1 day, washed, and resuspended to 2 × 106/ml. Lymphocytes were cultured in the absence and presence of BAT at 0.1 μg/ml for 3 more days and [3H]thymidine uptake was determined as described.
The cell surface markers (CD3 for T cells and CD56 for NK cells) of the lymphocytes from the different experimental groups described in Fig. 4 were analyzed by fluorescence cell analyzer. The percent of CD3/CD56-positive double-labeled cells increased to 25 ± 2 following preincubation on tumor monolayers and incubation with BAT. The percent of CD3/CD56 cells of the lymphocytes that were incubated on tumor cells alone was 17 ± 1%, whereas it was 9 ± 3% of the lymphocytes treated with BAT alone. In control untreated group 6 ± 1% of CD3/CD56-positive cells were detected.
DISCUSSION
The BAT mAb generated against human B-lymphoblastoid cell line was found to bind to and stimulate human T cells. The stimulation was manifested by induction of cell proliferation and cytolytic activity to NK-resistant and NK-sensitive tumor target cells (13). This antibody also stimulated murine splenocytes in vitro and was found to induce in vivo regression of a variety of murine tumors (14). Tumor regression was related to the immune-stimulatory properties of the antibody. This conclusion is strongly supported by the observation that the antitumor activity could be transferred with splenocytes from mice treated with the antibody. Furthermore, mice bearing tumors that had been cured by treatment with BAT were refractory to a tumor rechallenge (14).
To evaluate the potential clinical use of this antibody in human cancer, experiments were initiated in mice bearing human tumors. First, we investigated whether BAT would elicit an antitumor effect in nude mice, which are commonly employed for studies involving human tumor xenografts (19). Nude mice are deficient in T cells, and the question was raised whether BAT would induce antitumor activity in these immune-compromised animals. As observed in Table 1, BAT exhibited antitumor activity in nude mice bearing the B16 melanoma. This effect, however, was incomplete as compared with the curing effect that was attained in wild-type (C57BL) animals bearing the same tumor. A similar incomplete antitumor effect of BAT was also observed in nude mice implanted subcutaneously with human colon carcinoma (Fig. 1).
The lymphocyte cell type that mediates the antitumor effect in nude mice may involve NK cells that are present in these mice. This possibility was further supported by the experiment depicted in Table 2, which indicated that continuous depletion of NK cells diminishes the antitumor effect of BAT. However the possibility that T cells could mediate the antitumor effect of BAT is indicated from experiments (Table 1) demonstrating that BAT exhibited antitumor response in the beige mouse that is deficient in NK cells (18). Supporting this view are experiments in which SCID mice bearing murine B16 melanoma engrafted with splenocytes depleted of CD3 lymphocytes failed to respond to BAT (Table 3). In these experiments engraftment of splenocytes which were NK-depleted rendered the SCID mice partially responsive to BAT. Recent studies have shown that CD3/CD56-positive lymphocytes exhibited a potent antitumor activity (20–23). We have found that CD3/CD56-positive cells are generated in vitro upon stimulation of human lymphocytes by BAT and human tumor cells. Thus it is possible that this cell type plays a role in the antitumor effect induced by BAT. Taken together, it is possible that either NK or T cells can mediate the antitumor effect of BAT. Furthermore cross talk between these sub-populations (24) may also play an important role in mediating the effect of BAT.
A question of utmost clinical importance was whether BAT could induce human tumor regression mediated by activation of human lymphocytes. To address this question, we evaluated the effect of BAT in SCID mice engrafted with human lymphocytes and inoculated with human SK28 melanoma cells. BAT alone failed to induce tumor regression in SCID mice. Engraftment of PBL alone into SCID mice had a slight antitumor effect, which most probably is related to the genetic disparity between the tumor and the engrafted PBLs. The most pronounced antitumor effect was achieved when BAT was administered into the SCID mice that had been engrafted with human PBLs. In contrast to the curative effect that was attained in the wild strain of mice, the antitumor effect in the SCID mouse engrafted with PBL, although most pronounced, was incomplete in a few cases. This may result from the incomplete reconstitution of the immune system in the SCID mice (25) and was also demonstrated in the experiment in SCID mice that had been inoculated with B16 melanoma and engrafted with syngeneic splenocyctes (Table 3).
Our previous results in normal C57BL mice bearing murine tumors indicated that maximal antitumor effect was obtained upon administration of BAT late after tumor inoculation. A similar effect was also observed when BAT was administered to human PBMC-engrafted SCID mice at different times after inoculation of SK28 human melanoma. It is possible that lymphocytes sensitization by the tumor cells led to their enhanced response to BAT. Support for this notion was obtained from experiments in which the stimulatory response to BAT was assessed in vitro in splenocytes derived from tumor bearing mice and was also demonstrated in our in vitro experiments which showed that human lymphocytes sensitized by human tumor cells increased proliferation in the presence BAT (Fig. 4).
The nature of the two signals elicited by the tumor cells and by BAT is not known. It is possible that the first signal elicited by the tumor involves the presentation of a tumor antigen to the T cell receptor. The second signal provided by BAT may represent an accessory signal. The finding that BAT induced regression of human tumors mediated by activation of human lymphocytes points to its therapeutic potential in cancer patients.
Acknowledgments
We thank Dr. Yehuda Shoenfeld for helpful discussions and his critical review of the manuscript. We thank Mrs. Sara Domintz for the preparation of the manuscript.
ABBREVIATIONS
- SCID mice
severe combined immunodeficient mice
- PBMC
peripheral blood mononuclear cells
- PBL
peripheral blood lymphocyte
References
- 1.Clark E A, Ledbetter J A. Immunol Today. 1986;7:267–270. doi: 10.1016/0167-5699(86)90008-3. [DOI] [PubMed] [Google Scholar]
- 2.Meurer S C, Hussey R E, Cantrel D A, Hodgdon J C, Schlossman S F, Smith K A, Reinherz E L. Proc Natl Acad Sci USA. 1984;81:1509–1513. doi: 10.1073/pnas.81.5.1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Van Wauwe J P, DeMey J P, Goossens J G. J Immunol. 1980;124:2708–2713. [Google Scholar]
- 4.Jung G, Martin D E, Muller-Eberhard J H. J Immunol. 1987;139:639–644. [PubMed] [Google Scholar]
- 5.Ledbetter J A, Martin P J, Spooner C E, Wofsy D, Tsu T T, Beatty P G, Gladstone P. J Immunol. 1985;135:2331–2336. [PubMed] [Google Scholar]
- 6.Moretta A, Poggi A, Pende D, Tripodi G, Orengo A M, Pella N, Augugliaro R, Bottino C, Ciccone E, Moretta L. J Exp Med. 1991;174:1393–1398. doi: 10.1084/jem.174.6.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Razi-Wolf Z, Freeman G J, Galvin F, Benacerraf B, Nadler L, Reiser J. Proc Natl Acad Sci USA. 1992;89:4210–4214. doi: 10.1073/pnas.89.9.4210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Norton S D, Zuckerman L, Urdahl K B, Shefner R, Miller J, Jenkins M K. J Immunol. 1992;149:1556–1561. [PubMed] [Google Scholar]
- 9.Li Y, McGowan P, Hellstrom I, Hellstrom K E, Chen L. J Immunol. 1994;153:421–428. [PubMed] [Google Scholar]
- 10.Ellenhorn J D, Hirsch R, Schreiber H, Bluestone J A. Science. 1988;242:569–571. doi: 10.1126/science.2902689. [DOI] [PubMed] [Google Scholar]
- 11.Townsend S E, Allison J P. Science. 1993;259:368–380. doi: 10.1126/science.7678351. [DOI] [PubMed] [Google Scholar]
- 12.Jenkins M K, Taylor P S, Norton S D, Urdahl K B. J Immunol. 1991;147:2461–2466. [PubMed] [Google Scholar]
- 13.Hardy B, Galli M, Rivlin E, Goren L, Novogrodsky A. Cancer Immunol Immunother. 1995;40:376–382. doi: 10.1007/BF01525388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hardy B, Yampolski I, Kovjazin R, Galli M, Novogrodsky A. Cancer Res. 1994;54:5793–5796. [PubMed] [Google Scholar]
- 15.Mueller B M, Reisfeld R A. Cancer Metastasis Rev. 1991;10:1930–200. doi: 10.1007/BF00050791. [DOI] [PubMed] [Google Scholar]
- 16.Sandhu J, Shpitz B, Gallinger S, Hozumi N. J Immunol. 1994;152:3806–3813. [PubMed] [Google Scholar]
- 17.Shpitz B, Chambers C A, Singhal A B, Hozumi N, Fernandes B J, Roifman C M, Weiner L M, Roder J C, Gallinger S. J Immunol Methods. 1994;169:1–15. doi: 10.1016/0022-1759(94)90119-8. [DOI] [PubMed] [Google Scholar]
- 18.Stutman O, Cuttito M J. Nature (London) 1981;290:254–257. doi: 10.1038/290254a0. [DOI] [PubMed] [Google Scholar]
- 19.Garofalo A, Chirivi R G S, Scanziani E, Mayo J G, Vecchi A, Giavazzi R. Invasion Metastasis. 1993;13:82–91. [PubMed] [Google Scholar]
- 20.Lu P-H, Negrin R S. J Immunol. 1994;153:1687–1696. [PubMed] [Google Scholar]
- 21.Schmidt-Wolf I G H, Lefterova P, Johnston V, Huhn D, Blum K G, Negrin R S. Br J Haematol. 1994;87:453–458. doi: 10.1111/j.1365-2141.1994.tb08297.x. [DOI] [PubMed] [Google Scholar]
- 22.Mehta B A, Schmidt-Wolf G H, Weissman I L, Negrin R S. Blood. 1995;86:3493–3499. [PubMed] [Google Scholar]
- 23.Takeda K, Dennert G. Transplantation. 1994;58:496–504. doi: 10.1097/00007890-199408270-00017. [DOI] [PubMed] [Google Scholar]
- 24.Kurosawa S, Harada M, Matsuzaki G, Shinomiya Y, Terao H, Kobayashi N, Nomoto K. Immunology. 1995;85:338–346. [PMC free article] [PubMed] [Google Scholar]
- 25.Hendrickson E A. Am J Pathol. 1993;143:1511–1522. [PMC free article] [PubMed] [Google Scholar]