

Abstract
Polycystic kidney disease describes a heterogeneous collection of disorders that differ significantly with respect to their etiology and clinical presentation. They share, however, abnormal tubular morphology as a common feature, leading to the hypothesis that their respective gene products may function cooperatively in a common pathway to maintain tubular integrity. To study the pathobiology of one major form of human polycystic kidney disease, we generated a mouse line with a floxed allele of Pkhd1, the orthologue of the gene mutated in human autosomal recessive polycystic kidney disease. Cre-mediated excision of exons 3–4 results in a probable hypomorphic allele. Pkhd1del3–4/del3–4 developed a range of phenotypes that recapitulate key features of the human disease. Like in humans, abnormalities of the biliary tract were an invariant finding. Most mice 6 months or older also developed renal cysts. Subsets of animals presented with either perinatal respiratory failure or exhibited growth retardation that was not due to the renal disease. We then tested for genetic interaction between Pkhd1 and Pkd1, the mouse orthologue of the gene most commonly linked to human autosomal dominant polycystic kidney disease. Pkd1+/−;Pkhd1del3–4/del3–4 mice had markedly more severe disease than Pkd1+/+;Pkhd1del3–4/del3–4 littermates. These studies are the first to show genetic interaction between the major loci responsible for human renal cystic disease in a common polycystic kidney disease pathway.
Introduction
Tubules are the fundamental units of structure for many organs in the body. The kidney is perhaps one of the best examples of an organ where tubular structure and function are so closely linked. Under normal conditions, the glomeruli filter 20% of cardiac output to produce approximately 180 liters/day of filtrate. The renal tubules process this bulk flow to reclaim essential electrolytes and nutrients while excreting unwanted metabolic end-products and any excess salt, water or other electrolytes in a volume that may be less than 1% of the original amount. Proper patterning of the tubule is essential for the integrity of this process. Luminal diameter is another key parameter. Too large a lumen could reduce the efficiency of filtrate processing and result in unregulated loss of essential molecules while too narrow a lumen could result in high intra-luminal pressures with a subsequent rise in the hydrostatic pressure in Bowman’s capsule and a decline in glomerular filtration and clearance. Therefore, knowing the factors that regulate the control of luminal diameter and determining the mechanisms by which they do so is fundamental to understanding kidney function.
Polycystic kidney disease (PKD) describes a heterogeneous collection of disorders that share abnormal tubular morphology as their common feature. The study of genetic forms of PKD provides a unique opportunity to define the molecular processes that determine tubular morphology. While there are many different disorders that may present with cystic kidneys, autosomal dominant polycystic kidney disease (ADPKD) and autosomal recessive polycystic kidney disease (ARPKD) are the most representative examples.
ADPKD and ARPKD are genetically distinct entities that are clinically more dissimilar than alike. ADPKD is common (affects approximately 1/1000), its cysts arise from any nephron segment, and it is slowly progressive resulting in renal failure in ~50% by the 6th decade (1). Hepatic cysts are its primary extra-renal manifestation while cardiovascular abnormalities are less common but more life-threatening (2). The disease results from mutation of either of two genes, PKD1 and PKD2 (3, 4). The two forms of ADPKD are nearly indistinguishable with overlapping clinical features, differing only in their severity. PKD1 encodes a large membrane protein that may function as an atypical G-protein coupled receptor (5). PKD2 encodes the founding member of the transient receptor protein polycystin (TRPP) family of calcium channel proteins (4, 6, 7). The two gene products, polycystin-1 (PC1) and polycystin-2 (PC2), are thought to form a receptor channel complex that localizes to the primary cilium (8, 9). PC2 also is present in the endoplasmic reticulum (ER) where it may function as either a calcium release channel or a regulator of the 1,4,5-trisphosphate receptor (IP3R) (10). Molecular genetic studies of human samples suggest that the disease is recessive on a cellular level, likely explaining the focal nature of cyst formation and the variable clinical presentation (11, 12).
ARPKD, on the other hand, has an estimated incidence of ~1:20,000 live births, presents primarily in infancy and childhood, and is typically more severe than ADPKD (13, 14). A significant proportion of affected newborns are born with massively enlarged, cystic kidneys and die in the perinatal period from respiratory failure due to hypoplastic lungs. Unlike in ADPKD, the cystic kidneys retain their reniform shape and the cysts are fusiform dilatations mainly of the collecting ducts that radiate from the renal papilla to the cortex (15). Affected individuals who survive the neonatal period have a variable course. Most develop hypertension and have progressive renal impairment that requires renal replacement by adolescence. Survivors have a spectrum of other problems of variable severity due to ductal plate malformations of the liver, choledochal cysts, ascending cholangitis and growth retardation (16–18). All typical forms of ARPKD, including perinatal and late onset, result from mutations at a single locus, PKHD1 (19, 20). The human gene is large (~500kb) and encodes a complicated set of alternatively spliced RNA transcripts that are most highly expressed in kidney. A 67 exon mRNA transcript of of~13kb encodes the longest predicted open reading frame (4074 amino acids). Mutations have been identified affecting most of the 67 exons. The protein, polyductin-1 (PD1, also known as fibrocystin), is predicted to be a type I membrane protein that contains multiple Ig-like, plexin, transcription factor domains (IPT) and parallel beta-helix repeats (PbH1) in its long, extracellular N-terminus (19, 20). Several studies have localized the protein to the cilia/basal body and possibly other sub-cellular locations (21–23).
There are, however, occasional families with overlapping features of ARPKD and ADPKD. Some individuals with early onset ADPKD present with massive renal enlargement and neonatal respiratory failure while other ADPKD families have congenital hepatic fibrosis (24, 25). Conversely, there also are anecdotal reports of children with ARPKD children with intracranial aneurysms (26) and pancreatic cysts (unpublished observation, LGW). These observations suggest that the two different forms of PKD may share some underlying pathogenic mechanisms. In support of this hypothesis, cystic kidneys of rodent models of both forms of PKD are reported to have similar abnormalities in cAMP activity and similar therapeutic benefit by treatment with V2-receptor antagonists (27, 28). The proteins encoded by each of the genes implicated in these diseases also have been co-localized to the primary cilium (8, 9, 21–23). Finally, two recent studies have reported physical interactions between PC2 and PD1, mediated by kinesin-2 (29, 30). Collectively, these findings suggest that their gene products may function cooperatively to regulate tubular morphology.
In order to better define the pathobiology of ARPKD and directly test this hypothesis, we generated a mouse model of ARPKD by gene targeting of the Pkhd1 locus. We now show that this model fully recapitulates the full spectrum of clinical features of human ARPKD and demonstrate epistasis between the Pkhd1 and Pkd1 loci through the use of genetic crosses.
Results
Generation Of Floxed And Mutant Pkhd1 Alleles
Since genotype-phenotype correlations done in humans indicated that bilineal truncating mutations were invariably associated with perinatal lethality (31, 32), we generated a floxed allele of Pkhd1 (Pkhd1flox3–4) by introducing loxP sites into introns 2 and 4 to allow regulated spatio-temporal mutation of the locus (Figure 1A). Three out of ~400 selected ES clones had proper targeting based on initial Southern blot analysis but only one was found to have retained both lox P sites and to be correct using multiple probes. This clone was used to generate highly chimeric mice that produced 129Sv/C57Bl6 offspring carrying the targeted locus with very high frequency (Figure 1B). Pkhd1flox3–4 homozygotes were born at the expected frequency and were healthy without apparent renal or hepatic disease at the oldest time point assayed (1y). We produced the Pkhd1del3–4 allele by crossing Pkhd1flox3–4 mice to a line expressing Cre recombinase under the control of the Meox2 promoter (Figure 1C). RT-PCR analyses of Pkhd1del3–4 tissues confirmed that Pkhd1 transcripts lacked exons 3–4 but identified a number of alternatively spliced products, one of which links exons 2–6 with an intact open reading frame (Figure 1D). The translation product is predicted to include the signal peptide but lack an adjacent 123 amino acid fragment of the extracellular domain (Figure 1E). These data suggest that Pkhd1del3–4 may be a hypomorphic allele rather than a true null. Pkhd1del3–4 heterozygotes also are healthy and lack renal or hepatic disease.
Figure 1. Generation of floxed and mutant alleles of the Pkhd1 locus.

(A) Schematic structures of the targeting construct (TC) and the Pkhd1 locus (Gen) before and after Cre-mediated excision of the floxed sequence. Pkhd1flox3–4 (Pkhd1tm1Ggg) and Pkhd1del3–4 (Pkhd1tm1.1Ggg) identify the floxed and post-Cre mutant alleles, respectively. The positions of primers (“a–b–c”) and probes (“5’ Ext”, “5’ Int” and “3’ Ext”) are as shown. The predicted size of each HindIII genomic fragment and PCR product is as indicated. The Pgk-Neomycin cassette, lox P, frt and HindIII sites are indicated by a blue box, red and yellow triangles, and “H” respectively. Gray ovals identify exons 2–5.
(B) Characterization of the Pkhd1flox3–4 allele by Southern blot. DNA from offspring of a highly chimeric mouse was digested with HindIII, Southern blotted and probed with three markers. Each of the three probes detects the ~17kb HindIII fragment of the wild type allele (M indicates the marker, “+” a mouse that carries the targeted allele and “−“ identifies a wild type littermate). All probes also detect smaller fragments of the predicted sizes for a properly targeted locus.
(C) A PCR strategy was used to identify the wild type, floxed and deleted alleles using the primers “a–b–c” in a single reaction. The ethidium bromide-stained gel shows a representative example of the PCR products amplified from each of the different genotypes. Primer pair “a–b” amplifies185bp and 254bp products from the Pkhd1flox3–4 and Pkhd1del3–4 alleles, respectively, while primer pair “a–c” amplifies a 361bp from the Pkhd1del3–4 allele and nothing from the floxed allele.
(D) An ethidium bromide-stained gel showing a representative example of the RT-PCR products amplified from kidneys of individual mice with each of the different genotypes using primers from exons 2 to 6. Sample “del-3–4/del3–4(cystic)” was isolated from a cystic specimen. The data show that new splice variants are present after deletion of exons 3 and 4. Each of the new bands was excised from the gel, cloned and then sequenced. Three of the bands (red) were found to cause frame shifts either by splicing into the Pgk-Neomycin cassette or into exon 5 (fragments 650, 600 and 196). The sequence of the 87 bp fragment predicts an in-frame splicing event between exons 2–6.
(E) Schematic representation of the predicted polyductin variant that results from splicing between exon 2–6. We have not been able to identify unequivocally either the full-length or mutant protein in mouse kidney using available antibodies.
Pkhd1del3–4 Homozygotes Develop Extra-Renal Disease Of Variable Severity That Recapitulates The Human Phenotype
Initial crosses of Pkhd1del3–4/+ yielded only ~29% of the expected number of liveborn Pkhd1del3–4/del3–4 (18/252; 63 expected) with a subset of neonates subsequently succumbing to respiratory failure (Figure 2A). Interestingly, none of the mice with perinatal respiratory failure had enlarged or cystic kidneys. The prenatal and perinatal lethality disappeared after selectively crossing viable Pkhd1del3–4/del3–4, suggesting an interaction with a major modifier linked to perinatal demise. Mutant homozygotes that survived the neonatal period universally had at least one other disease manifestation. All mutants developed ductal plate malformations with biliary duct proliferation, biliary cysts and mild to severe periportal fibrosis. These changes are characteristic of the ductal plate malformations seen in human ARPKD (33). A subset of mice also developed choledochal cysts and ascending cholangitis (Figure 3A). Pancreatic cysts were also very common, affecting 33.3 % at ≥9 months of age (Figure 3B). The pancreatic cystic disease on occasion was extremely severe, sometimes associated with choledochal cyst and massive dilation of the pancreatic duct, reaching up to 6 cm of diameter in extreme cases. Severity and incidence of these extra-renal phenotypes increased over time, but only the liver phenotype was fully penetrant (Figure 3C). Growth retardation was another common problem, which was sometimes observed in mice with otherwise mild extra-renal disease (Figure 2B). It also preceded the onset of their renal disease (discussed below), suggesting that growth retardation might be a direct result of Pkhd1 mutation.
Figure 2. Pkhd1del3–4/del3–4 mutants have respiratory failure and growth retardation in the absence of significant renal disease.

(A) A subset of Pkhd1del3–4/del3–4 neonates died shortly after birth with dyspnea/apnea, aerophagia, and respiratory failure. Immediate necropsy confirmed aerophagia as stomach and intestines (dashed line and arrows) were filled with air.
(B) A subset of Pkhd1del3–4/del3–4 mutants has severe growth retardation that is out of proportion to any other apparent organ dysfunction. In the top panel on left, gross external appearance of a wild type and Pkhd1del3–4/del3–4 mouse at P4. In right panels, gross anatomy (middle) and histopathology (far right) of the kidney (K), Liver (L) and pancreas (P, not present in middle) of the same mutant reveal little in the way of abnormal pathology. Below are representative examples of a normal and mutant P21 mouse with growth retardation as seen on gross exam (top) and by histopathologic analysis of kidney (K), liver (L) and pancreas (P). Only mild ductal plate malformations were detected (arrow). Scale bars shown correspond to 1mm (yellow) and 100µm (green).
Figure 3. Pkhd1del3–4/del3–4 mice develop abnormalities of the biliary tract and pancreas.
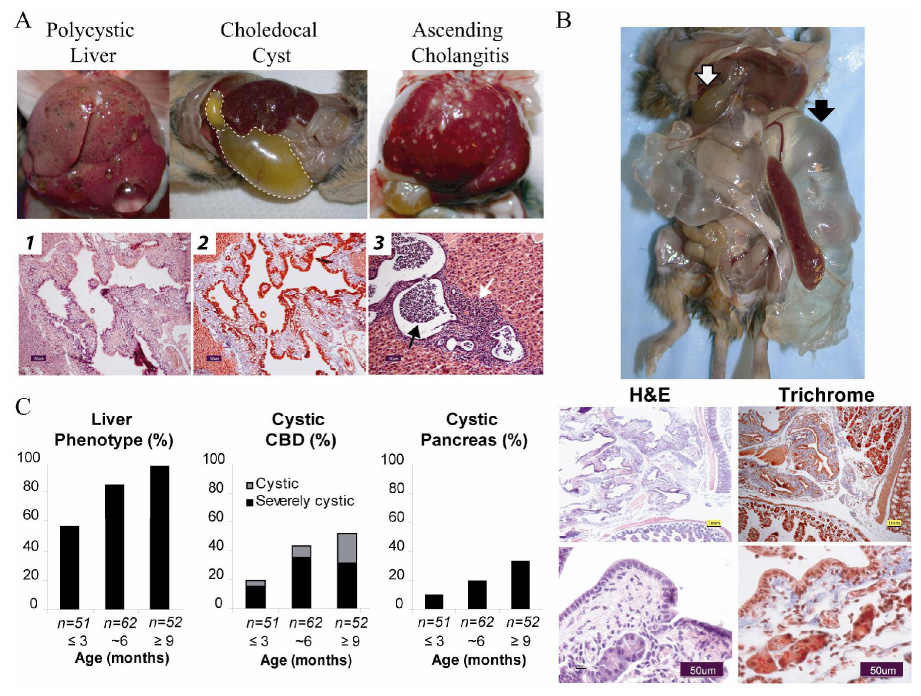
(A) Pkhd1del3–4/del3–4 mutants develop a range of hepatobiliary abnormalities. Gross external appearance of representative examples of macroscopic cystic disease, a massive choledochal cyst (dashed line) and ascending cholangitis. Liver histopathology of a Pkhd1del3–4/del3–4 mouse showing cystic dilation of biliary ducts and dysplastic ducts (b1, Hematoxylin-eosin staining) and periportal fibrosis (b2, Masson’s trichrome). These changes are characteristic of the ductal plate malformations seen in human ARPKD. Acute ascending cholangitis (black arrow, b3) and cholecystitis also are seen (white arrow, b3). Scale bars correspond to 50 µm (black).
(B) Cystic disease of the pancreas was variably present and on occasion extremely severe. Top panel shows a gross external appearance of a 6 month old Pkhd1del3–4/del3–4 mouse with severe pancreatic cystic disease (black arrow) and choledochal cyst (white arrow). Representative histopathology of pancreas stained with hematoxylin/eosin (H&E) and Masson-Trichrome showing massive dilation of the pancreatic duct, peri-ductule fibrosis (blue staining) and remnant acini. Scale bars shown correspond to 1mm (yellow) in the top and 50µm (black) in the lower panel.
(C) Frequency of various phenotypes present on gross exam at different ages from a total of 165 Pkhd1del3–4/del3–4 mutant mice. Liver phenotypes include macroscopic cysts and ascending cholangitis. The common bile duct (CBD) was defined as cystic when the lumen was 3–10x fold normal diameter, and severely cystic when more than 10x.
Pkhd1del3–4/del3–4 Mice Develop Progressive Renal Cystic Disease
In marked contrast to what has been previously reported for another mouse model of ARPKD produced by gene targeting of Pkhd1 (34), the majority of Pkhd1del3–4/del3–4 mice also developed renal cystic disease of variable severity that worsened with age (Figure 4). The kidneys of most mice ≤6 months of age lacked visible cysts on gross exam. However, histopathologic analyses revealed that a substantial fraction of these grossly normal kidneys nonetheless had dilated tubules (Figure 4B). Macroscopic renal cysts, which were present in only 13.7 % of the mice at ≤3 months of age, were found in progressively larger fractions of older mice. The proportion of mice with severe cystic disease also steadily increased from ~10% of young mice to ~55% of older animals (Figure 6B). Cysts radiated from papilla to cortex (Figure 5 and Figure 6) increasing the kidney volume (Figure 6C), in a pattern similar to that seen in human ARPKD. Using nephronsegment specific markers, we confirmed that the cysts were predominantly derived from the collecting duct and Thick Ascending Loop of Henle (TALH, Figure 6D and Supplementary Figure 1) as has been reported for human ARPKD (15).
Figure 4. Pkhd1del3–4/del3–4 mutants develop polycystic kidney disease of variable severity.

(A) The kidney phenotype was classified based on its gross appearance: non-cystic, cystic (< 10 macroscopic cysts) and severely cystic (>10 cysts).
(B) Histopathology confirmed the presence of cysts (lumen >3X that of a normal tubule) in the second and third groups. Two subcategories were defined in the non-cystic group based on their histopathology: kidneys with dilated tubules (lumen 1–3X that of a normal tubule) and kidneys with tubules with normal diameter. Representative examples are shown from 6 month (non-cystic and cystic) and 9 month old mice (Severely cystic).
(C) Gross examination at different ages indicated that the frequency and severity of the kidney phenotype increases with the age in the Pkhd1del3–4/del3–4 mutant mice.
(D) Renal volume correlated with gross phenotype and was significantly higher in the severely cystic group at 6 months of age. ** p<0.005 using the two-tail t-test.
Figure 6. Genetic interaction between Pkd1 and Pkhd1 causes an accentuation of ARPKD-like disease.
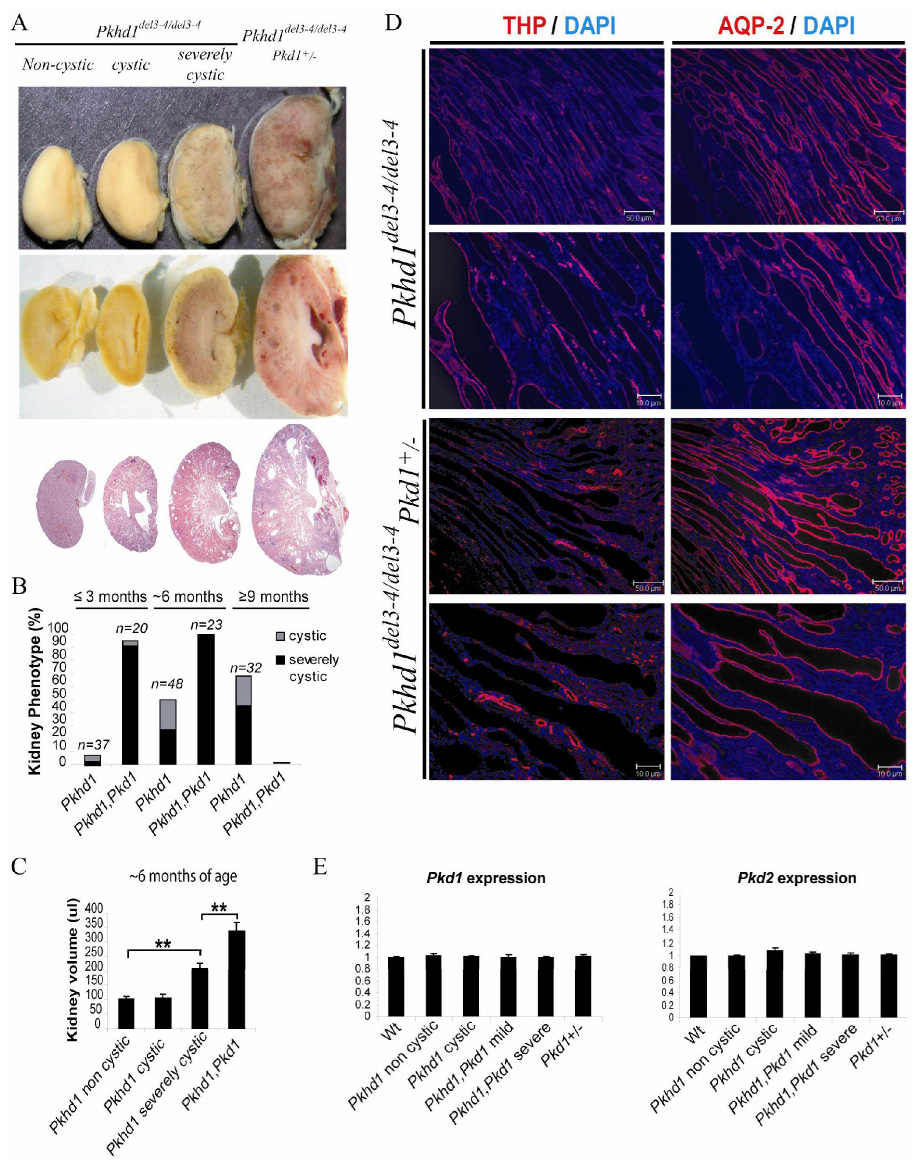
(A) Gross (top) and microscopic examination (bottom) of ~6 month old littermates indicates increased severity of the cystic disease in Pkhd1del3–4/del3–4, Pkd1+/− mice. Cysts radiated from papilla to cortex and were derived from distal tubule and collecting ducts in a pattern similar to that seen in human ARPKD and in severely cystic, late-stage Pkhd1del3–4/del3–4 murine kidneys (Fig 4B).
(B) The incidence of the severely cystic phenotype was markedly increased in double mutant mice at all time points. All Pkhd1del3–4/del3–4, Pkd1+/− mutants died before 9 months of age. Pkhd1 and Pkhd1, Pkd1 indicate Pkhd1del3–4/del3–4 and Pkhd1del3–4/del3–4, Pkd1+/− genotypes, respectively. All comparisons were based on littermate controls.
(C) Renal volume correlated with gross phenotype and was significantly higher in Pkhd1,Pkd1 double mutants than in severely cystic Pkhd1del3–4/del3–4 mice at 6 months of age. ** p<0.005 using two-tail t-test.
(D) Cysts of Pkhd1del3–4/del3–4 mice universally stained positive for either aquaporin-2 (AQP-2), identifying them as derived from collecting ducts (predominant), or Tamm Horsfall protein/uromodulin (THP), identifying them as derived from the Thick Ascending Limb of Henle. Cysts of Pkhd1del3–4/del3–4, Pkd1+/− double mutants had an identical staining pattern, suggesting an accentuation of the underlying ARPKD-like phenotype. Scale bars correspond to 10µm and 50µm.
(E) Ratio of expression level of Pkd1 and Pkd2 in experimental vs control (wild type) kidneys of 6 month old mice as measured by qPCR. All reactions were done in triplicate using samples from 3 mice for each genotype. Data were normalized to 18s RNA levels and then presented relative to the mean level of Pkd1 and Pkd2 in wild type specimens. All comparisons were based on littermate controls.
Figure 5. Genetic interaction between the two major loci responsible for ADPKD and ARPKD.
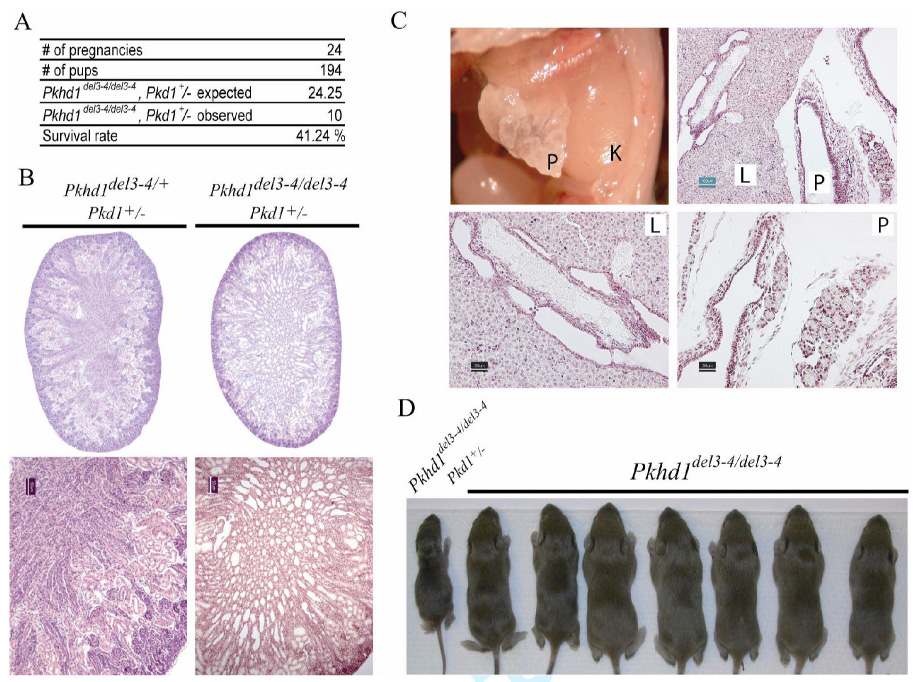
(A) The number of Pkhd1del3–4/del3–4, Pkd1+/− newborn double mutants was <50% of what was expected.
(B) Newborn Pkhd1del3–4/del3–4, Pkd1+/− double mutants frequently had dilated renal tubules and cysts. 1X (top) and 10X (bottom) images of kidneys from Pkhd1del3–4/del3–4, Pkd1+/− and Pkhd1del3–4/+, Pkd1+/− newborn littermates. Scale bars correspond to 50µm (black).
(C) Abnormalities including liver (L) and pancreatic (P) cysts along with fibrosis were often present on the first day of life. Scale bars shown correspond to 100µm (green) in the top and 50µm (black) in the lower panel.
(D) Representative litter at 1 week of age from a cross between Pkhd1del3–4/del3–4, Pkd1+/− and Pkhd1del3–4/del3–4, Pkd1+/+ parents illustrating the gross distortion of the observed genotypes from those that were expected.
Genetic Interaction Between The Major Loci Responsible For ADPKD And ARPKD
To test the hypothesis that the Pkhd1 and Pkd1 gene products may function cooperatively in a common pathway, we set up a series of crosses between Pkhd1del3–4 and Pkd1β-gal null mouse models (35). Double heterozygous mutants were born at the expected frequency and had normal kidneys, livers and pancreas (data not shown). On the other hand, the number of liveborn Pkhd1del3–4/del3–4, Pkd1+/− double mutants was far lower than expected (Figure 5A). Hepatic abnormalities, pancreatic cysts and growth retardation also were present earlier and were more severe in the double mutants (Figure 5C and D), though the pattern was similar to what had been observed in Pkhd1del3–4/del3–4 mice. Mice born with the Pkhd1del3–4/del3–4, Pkd1+/− genotype also had markedly more severe disease of the kidney than littermates with all other genotypes (Figure 5). The collecting ducts were dilated in a substantial fraction of double mutants at the time of birth (Figure 5B), and all had enlarged kidneys with macroscopic cysts that were predominantly derived from the collecting duct and TALH by the third month of life in a pattern identical to that seen in the severely cystic late-stage Pkhd1del3–4/del3–4 murine kidneys (Figure 6A&D and Supplementary Figure 1). All double mutants died before reaching the ninth month of life (Figure 6B). Kidney volumes of the double mutants were more than 50% larger than those of the severely cystic Pkhd1del3–4 homozygotes in the oldest surviving cohort for which comparisons were possible (Figure 6C). In all cases examined (N=11), the more severe phenotype appeared to be an accentuation of the underlying ARPKD phenotype rather than a super-imposition of Pkd1-mutant abnormalities (Figure 6D and Supplementary Figure 1).
The apparent genetic interaction between the Pkd1 and Pkhd1 loci could have multiple possible explanations. While background effects due to the use of mixed strains might theoretically confound the analysis, this is a very unlikely explanation for our findings since the double mutant phenotype was significantly different from that of littermates who were derived in a similar manner. It has been previously reported that there may be a PKD transcriptional network (36). If such exists, Pkd1 could be downstream of Pkhd1 and mutations of the latter might result in further down-regulation of Pkd1 expression. Two lines of evidence argue against this possibility. First, one might expect that the double mutants would have more Pkd1-mutant features rather than the exacerbation of the Pkhd1-mutant phenotype that was observed. Secondly, Pkd1 expression levels in tissues of double mutants are no different than those in tissues of other genotypes as assayed by quantitative PCR (qPCR, Figure 6E). An alternative possibility is that their gene products directly interact to form a molecular complex. We tested for this possibility by co-expressing recombinant, epitope-tagged versions of polycystin-1 and polyductin in human embryonic kidney cells (Hek 293) and testing for co-precipitation. Neither molecule was capable of bringing down the other using C-termini tags (data not shown). Our findings are more consistent with an indirect relationship between the two gene products, either by functioning cooperatively in a linear pathway or independently in pathways that converge to regulate tubular morphology.
Discussion
We have developed the first mouse model of human ARPKD that fully recapitulates all of the latter’s key features, including polycystic kidney disease. These results are in striking contrast to what has been previously reported for another Pkhd1 mutant mouse line with targeted deletion of exon 40 (34). Homozygotes for the latter developed pathology limited to the hepatobiliary system. Our model develops a wide spectrum of clinical features and provides the first direct evidence that mutation of Pkhd1 is sufficient to cause renal cystic disease in mice. The results of the two studies suggest that various organs have different requirements for specific exons, consistent with our previous studies that suggested alternative splicing is likely to be a functionally important aspect of Pkhd1 biology (37).
Respiratory insufficiency is a major cause of neonatal death of ARPKD infants. The prevailing wisdom is that this problem is an indirect consequence of the renal cystic disease, in part because the greatly enlarged kidneys impair lung expansion and in part because of the associated oliguria. Fetal urine production is thought to be a major source of amniotic fluid and the latter is required for proper lung development. The observation that our mutant mice develop respiratory insufficiency in the absence of significant renal disease suggests that the Pkhd1 gene product may have a primary function in the pulmonary system. Because Pkhd1del3–4 mutant mice do not develop severe renal disease until later in life, this model can for the first time be used to distinguish primary versus secondary effects resulting from Pkhd1 mutation. The floxed allele is a particularly useful feature that can be exploited to allow regulated mutation of the locus.
A substantial proportion of the Pkhd1del3–4 mutant mice also were significantly growth retarded. This finding is commonly associated with human ARPKD but is generally attributed to the well-known adverse effects of renal disease on body growth (18, 38). We now show that this problem can occur in mice prior to the onset of significant renal disease, excluding the latter as the underlying explanation. Growth retardation in these animals is also unlikely to be secondary to disease of the biliary tract or pancreas since a substantial fraction of the growth retarded mice had only minimal or mild disease of these organs. We note that our findings are consistent with two previously published clinical reports that described ARPKD children with growth abnormalities discordant with their renal phenotype (14, 39). Collectively, these data indicate that Pkhd1 may play a more direct role in the control of body growth, and the Pkhd1del3–4 line of mice will be a useful genetic tool for studying the pathobiology of this process.
Another feature common to our model and human ARPKD is the clinical variability with which it presents. In humans, this has been in part attributed to the pattern of mutation (31, 32). For example, we have previously shown that bilineal truncating mutations are invariably associated with severe disease. In the Pkhd1del3–4 mouse model, all affected mice have an identical mutation, excluding this factor as an explanation. Because the mutant allele gives rise to a set of alternatively spliced transcripts, it is theoretically possible that differences in the splicing pattern could account for the observed phenotypic variability. However, we have compared the pattern of Pkhd1 splicing in tissues of severely cystic versus non-cystic Pkhd1del3–4 mutants and found that they were no different. Therefore, we think genetic modifiers likely account for much of the variability. There is strong evidence that multiple loci can influence disease expression in other rodent models and they have been postulated as likely relevant for human disease as well (40). Congenic lines bearing the Pkhd1d3–4 mutation will be useful for identifying key factors that modulate polyductin’s function in utero, in the developing respiratory system, and in the regulation of tubular structure and body growth.
One benefit of having the exon 3–4 deletion result in a hypomorphic allele rather than a true null is that the former often results in a sensitized pathway that is highly susceptible to modest perturbation of other pathway factors. Hypomorphic alleles have been used to great effect in simple model organisms like Drosophila to identify potential pathway partners by testing for genetic epistasis with candidate loci in either a directed fashion or by a non-specific mutagenesis screen. We have used the former approach to show that Pkd1 can be a major modifier of Pkhd1 disease expression in mice. Our findings suggest that variants at the PKD1 locus could potentially be important modifiers of ARPKD disease severity in humans. Our data also prompt speculation as to whether variants of PKHD1 might explain the occasional individual with ADPKD and congenital hepatic fibrosis or a severe, neonatal presentation.
In conclusion, these studies are the first to show genetic interaction between the major loci responsible for human ADPKD and ARPKD. Our data strongly support the hypothesis that their respective gene products function cooperatively in a common pathway to maintain tubular integrity.
Materials & Methods
Generation of the Pkhd1 exon2–4 targeting construct
We assembled the targeting construct from four fragments. The ~5.0 kb 3’ end of the construct was generated by PCR using a forward primer based in intron 4 and a reverse primer in intron 5. The forward primer included the sequence of a naturally occurring SacII restriction site. The amplified fragment was cloned into the Topo II vector (Invitrogen). A second genomic product of 1.2kb containing exons 2–3 was amplified using a forward primer in intron 2 that contained sequence for a synthetic XbaI site at its 5’ end and a reverse primer based in intron 3 that included the sequence of the same SacII site described above. This fragment was then added to the 5’ end of the construct by cloning into the SacII site. A Pgk-Neo selection cassette flanked by FRT sites and a single lox P site (gift of Dr. Gail Martin (41)) was then cloned into the single SacII site. A final 3.0 kb genomic fragment was amplified using a forward primer in intron 1 and a reverse primer in intron 2 that included sequence for a synthetic XbaI site at its 5’ end. This fragment was then added to the rest of the targeting construct. In the final step, the construct was linearized at the XbaI site, blunt ended, and then a blunt-ended fragment containing a single lox P site was ligated to the targeting construct. The entire insert was sequenced and verified to be correct with the lox P sites in the correct orientation. Primer sequences available upon request.
Electroporation of ES cells and selection of homologous recombinant clones
The linearized construct was electroporated into 129Svev ES cells and 400 clones were isolated. Two clones were identified by Southern blotting as having correct integration without rearrangement or duplication, and only one of the two had retained both lox P sites. The ES clone was injected into C57Bl6 blastocysts to produce highly chimeric male and female mice. The clone was expanded and used to generate highly chimeric animals that transmitted the targeted allele (Pkhd1flox3–4) with high frequency in test crosses to C57Bl6 mice.
Mouse Lines
Highly chimeric male mice were crossed to 129Svev females to produce the Pkhd1Tm1Ggg (Pkhd1flox3–4) mouse line. The Pkhd1Tm1.1Ggg line (Pkhd1del3–4) was generated by crossing Pkhd1flox3–4/flox3–4 to mixed strain mice (Black Swiss, 129SvEv) that were positive for Meox2Cre. This line (Meox-Cre B6.129S4-Meox2 tm1[cre]Sor), originally obtained from Jackson Laboratory (42), has the Cre recombinase gene inserted into the Meox2 gene (mesenchyme homeobox 2 gene) locus. Cre expression is regulated by the Meox2 native promoter and induces deletion with high efficiency in somatic tissues. Pkhd1del3–4 heterozygotes were crossed to produce Pkhd1del3–4 homozygotes. The Pkd1β-gal null line of mice has been previously described (35). Briefly, the 3’ end of exon 2 and all of exon 3 of the mouse Pkd1 gene was replaced by the β-galactoside which was knocked in-frame into the 5’ end of exon 2. The 5’ end of the targeted Pkd1 allele is expressed normally but transcriptional termination sequences in the 3’UTR of the β-galactoside gene prevent expression of more 3’ Pkd1 exons (Supplementary Figure 2). All studies were performed using protocols approved by the University Animal Care and Use Committee, and mice were housed and cared in pathogen-free facilities accredited by the American Association for the Accreditation of Laboratory Animal Care and meet federal (NIH) guidelines for the humane and appropriate care of laboratory animal.
Genotyping
Genomic DNA was isolated and genotyped using the REDExtract-N-Amp tissue PCR Kit (SIGMA) according to the manufacturer’s protocols, using a 3-primer PCR strategy (Figure 1A) with the following primers: (primer-a) M/lox P-F: GGG AAG CAG AAA TTC AGG; (primer-b) M/lox P C2R: AGA TGA AGC ACG GAT CAG TGG G; (primer-c) PgkNEO-R1: GCT CAT TCC TCC CAC TCA T. Pkhd1 wild-type, Pkhd1flox3–4 and Pkhd1del3–4 alleles were identified in 2% 3:1 NuSieve agarose gels as 185-, 254- and 361-bp products, respectively (Figure 1C). Details regarding PCR conditions are available upon request. Genomic Southern blots of tail and ES cell DNA (10µg/sample) were prepared using standard techniques and probed with P32-labeled probes generated using gel-purified fragments of cloned DNA.
Reverse Transcription-PCR
Total RNA was isolated from kidney tissues using the Qiagen RNA extraction kit. We determined RNA concentration by spectrophotometry (NanoDrop) and then used 5µg of total RNA for first strand cDNA using Superscript II (Invitrogen). We used 1/10 of the final product as template for PCR amplification of Pkhd1 transcripts (Platinum HiFi, Invitrogen) using the following primers: (primer-d) MG/Exon2F: GTC TCT CTG CTG AGT ATG G, (primer-e) MG/Exon6R: CCA CTT GCT GGG TAA ACT.
Histopathology
Kidney, liver, and pancreas specimens from neonate and adult animals were collected and immediately fixed in 4% Paraformaldehyde (PFA) at 4°C overnight. To remove the excess of Paraformaldehyde (PFA), samples were washed in water for 2 h, dehydrated in 50% ethanol for 2 hours and 70% ethanol for 2 h and then stored at room temperature in 70% ethanol. Following rehydration in water for 2h, all samples were dehydrated in a graded alcohol series and embedded in paraffin for histological analysis. Gross pictures of the specimens were taken with the Nikon Coolpix 995 digital camera. We established a tissue phenotype classification according to the organ’s gross appearances. Kidney phenotype was classified as non-cystic, cystic (<10 cysts) and severely cystic (>10 cysts). The non-cystic group was sub-classified microscopically as non-dilated and dilated (when the lumen was between 3 and 5 times the diameter of a normal tubule). The common bile was classified as cystic (when the bile duct lumen was between 3–10 fold larger than the normal diameter) and severely cystic (more than 10 times the normal diameter). Liver phenotype included macroscopic cysts and ascending cholangitis. Hematoxylin/eosin (H&E) and Masson-trichrome stains were applied using standard protocols. Kidney volume measurements were developed using the Sherle method (43)
Immunofluorescent Staining
For immunolabeling, tissues were fixed, sectioned and stained as previously described (35). Antibodies to Aquaporin-2 (rabbit α-AQP2, 1:500, a gift from M. Knepper) and Tamm-Horsfall protein (rabbit α-THP, 1:500, a gift from J.R. Hoyer) were incubated overnight at 4°C. Secondary antibodies were conjugated to cyanine 3 and were used at 1:300 (Jackson ImmunoResearch Laboratories Inc., West Grove, Pennsylvania, USA). For each antibody, specificity was confirmed using negative control slides omitting primary antibody. Indirect fluorescence microscopy was performed using a Nikon Eclipse E600, and images were captured using a SPOT-RT monochromic camera (SPOT Diagnostic Instruments).
Real-time quantitative PCR (qPCR) analysis
We isolated the kidney RNA and generated cDNA as previously described. We developed the qPCR analysis on an AB Prism 7900HT sequence detection system according to the manufacturer methods using primers designed by Applied Biosystems (Pkd1 Mm00465434_m1; Pkd2 Mm00435829_m1; 18S Hs99999901_s1). We used 50 ng of cDNA template for each reaction and performed each in triplicate for each of three different samples/genotype. Data were normalized to 18s RNA levels and then presented relative to the mean level of Pkd1 and Pkd2 in wild type specimens. We intentionally used primers located in the boundary of exon 1 and 5’ region of exon 2 to detect possible transcriptional modifications by the Pkhd1del3–4 allele (see Supplementary Figure 2A).
Supplementary Material
Acknowledgments
This work was funded by the National Institutes of Health (DK48006, DK51259, DK57325), and the investigators are members of the Johns Hopkins NIDDK PKD Center of Excellence. We thank Dr. Sandra Guggino and the Mouse Physiology Core of the Hopkins Digestive Disease Basic Research Development Center grant (DK064388) for technical support. We thank members of the Qian, Watnick and Germino laboratory for helpful discussions.
Nonstandard abbreviations
- ADPKD
autosomal dominant polycystic kidney disease
- ARPKD
autosomal recessive polycystic kidney disease
- AQP2
aquaporin-2
- ER
endoplasmic reticulum
- H&E
Hematoxylin/eosin
- Hek 293
human embryonic kidney cell line
- IP3R
1,4,5-trisphosphate receptor
- IPT domains
Ig-like, plexin, transcription factor
- Meox2
mesenchyme homeobox-2 gene
- qPCR
quantitative polymerase chain reaction
- PbH1 repeats
parallel beta-helix
- PC1
polycystin-1
- PC2
polycystin-2
- PD1
Polyductin/fibrocystin-1
- PFA
Paraformaldehyde
- PKD
polycystic kidney disease
- Pkhd1
mouse Polycystic Kidney and Hepatic Disease-1 gene
- Pkd1
mouse Polycystic Kidney Disease-1 gene
- PKD1
human Polycystic Kidney Disease Gene-1
- PKD2
human Polycystic Kidney Disease Gene-2
- TALH
Thick Ascending Loop of Henle
- THP
Tamm-Horsfall protein
- TRPP
polycystin sub-class of the transient receptor protein family
References
- 1.Gabow PA. Autosomal Dominant Polycystic Kidney Disease. 1993;329:332–342. doi: 10.1056/NEJM199307293290508. [DOI] [PubMed] [Google Scholar]
- 2.Fick GM, Johnson AM, Hammond WS, Gabow PA. Causes of death in autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 1995;5:2048–2056. doi: 10.1681/ASN.V5122048. [DOI] [PubMed] [Google Scholar]
- 3.The European Polycystic Kidney Disease Consortium. The polycystic kidney disease 1 gene encodes a 14 kb transcript and lies within a duplicated region on chromosome 16. Cell. 1994;77:881–894. doi: 10.1016/0092-8674(94)90137-6. [DOI] [PubMed] [Google Scholar]
- 4.Mochizuki T, Wu G, Hayashi T, Xenophontos SL, Veldhuisen B, Saris JJ, Reynolds DM, Cai Y, Gabow PA, Pierides A, et al. PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science. 1996;272:1339–1342. doi: 10.1126/science.272.5266.1339. [DOI] [PubMed] [Google Scholar]
- 5.Sandford R, Mulroy S, Foggensteiner L. The polycystins: a novel class of membrane-associated proteins involved in renal cystic disease. Cell. Mol. Life Sci. 1999;56:567–579. doi: 10.1007/s000180050454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsiokas L, Arnould T, Zhu C, Kim E, Walz G, Sukhatme VP. Specific association of the gene product of PKD2 with the TRPC1 channel. Proc. Natl. Acad. Sci. U.S.A. 1999;96:3934–3939. doi: 10.1073/pnas.96.7.3934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Montell C. Physiology, phylogeny, and functions of the TRP superfamily of cation channels. Sci. S.T.K.E. 2001;2001:RE1. doi: 10.1126/stke.2001.90.re1. [DOI] [PubMed] [Google Scholar]
- 8.Nauli SM, Alenghat FJ, Luo Y, Williams E, Vassilev P, Li X, Elia AE, Lu W, Brown EM, Quinn SJ, et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat. Genet. 2003;33:129–137. doi: 10.1038/ng1076. [DOI] [PubMed] [Google Scholar]
- 9.Watnick T, Germino G. From cilia to cyst. Nat. Genet. 2003;34:355–356. doi: 10.1038/ng0803-355. [DOI] [PubMed] [Google Scholar]
- 10.Li Y, Wright JM, Qian F, Germino GG, Guggino WB. Polycystin 2 interacts with type I inositol 1,4,5-trisphosphate receptor to modulate intracellular Ca2+ signaling. J. Biol. Chem. 2005;280:41298–41306. doi: 10.1074/jbc.M510082200. [DOI] [PubMed] [Google Scholar]
- 11.Qian F, Watnick TJ, Onuchic LF, Germino GG. The molecular basis of focal cyst formation in human autosomal dominant polycystic kidney disease type I. Cell. 1996;87:979–987. doi: 10.1016/s0092-8674(00)81793-6. [DOI] [PubMed] [Google Scholar]
- 12.Pei Y, Watnick T, He N, Wang K, Liang Y, Parfrey P, Germino G, St George-Hyslop P. Somatic PKD2 mutations in individual kidney and liver cysts support a "two-hit" model of cystogenesis in type 2 autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 1999;10:1524–1529. doi: 10.1681/ASN.V1071524. [DOI] [PubMed] [Google Scholar]
- 13.Zerres K, Rudnik-Schoneborn S, Steinkamm C, Becker J, Mucher G. Autosomal recessive polycystic kidney disease. J. Mol. Med. 1998;76:303–309. doi: 10.1007/s001090050221. [DOI] [PubMed] [Google Scholar]
- 14.Guay-Woodford LM, Desmond RA. Autosomal recessive polycystic kidney disease: the clinical experience in North America. Pediatrics. 2003;111:1072–1080. doi: 10.1542/peds.111.5.1072. [DOI] [PubMed] [Google Scholar]
- 15.Osathanondh V, Potter EL. Pathogenesis of Polycystic Kidneys. Type 1 Due to Hyperplasia of Interstitial Portions of Collecting Tubules. Arch. Path. 1964;77:466–473. [PubMed] [Google Scholar]
- 16.Reuss A, Wladimiroff JW, Stewart PA, Niermeijer MF. Prenatal diagnosis by ultrasound in pregnancies at risk for autosomal recessive polycystic kidney disease. Ultrasound in medicine & biology. 1990;16:355–359. doi: 10.1016/0301-5629(90)90065-k. [DOI] [PubMed] [Google Scholar]
- 17.Guay-Woodford L. Autosomal recessive disease: clinical and genetic profiles. In: Torres V, Watson M, editors. Polycystic kidney disease. 1996. pp. 237–267. [Google Scholar]
- 18.Zerres K, Rudnik-Schoneborn S, Deget F, Holtkamp U, Brodehl J, Geisert J, Scharer K. Autosomal recessive polycystic kidney disease in 115 children: clinical presentation, course and influence of gender. Arbeitsgemeinschaft fur Padiatrische, Nephrologie. Acta Paediatr. 1996;85:437–445. doi: 10.1111/j.1651-2227.1996.tb14056.x. [DOI] [PubMed] [Google Scholar]
- 19.Onuchic LF, Furu L, Nagasawa Y, Hou X, Eggermann T, Ren Z, Bergmann C, Senderek J, Esquivel E, Zeltner R, et al. PKHD1, the polycystic kidney and hepatic disease 1 gene, encodes a novel large protein containing multiple immunoglobulin-like plexin-transcription-factor domains and parallel beta-helix 1 repeats. Am. J. Hum. Genet. 2002;70:1305–1317. doi: 10.1086/340448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ward CJ, Hogan MC, Rossetti S, Walker D, Sneddon T, Wang X, Kubly V, Cunningham JM, Bacallao R, Ishibashi M, et al. The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor-like protein. Nat. Genet. 2002;30:259–269. doi: 10.1038/ng833. [DOI] [PubMed] [Google Scholar]
- 21.Menezes LF, Cai Y, Nagasawa Y, Silva AM, Watkins ML, Da Silva AM, Somlo S, Guay-Woodford LM, Germino GG, Onuchic LF. Polyductin, the PKHD1 gene product, comprises isoforms expressed in plasma membrane, primary cilium, and cytoplasm. Kidney Int. 2004;66:1345–1355. doi: 10.1111/j.1523-1755.2004.00844.x. [DOI] [PubMed] [Google Scholar]
- 22.Kaimori J, Nagasawa Y, Menezes LF, Garcia-Gonzalez M, Deng J, Imai E, Onuchic LF, Guay-Woodford L, Germino GG. Polyductin undergoes Notch-Like processing and regulated release from primary cilia. Hum. Mol. Genet. 2007 doi: 10.1093/hmg/ddm039. [In Press] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ward CJ, Yuan D, Masyuk TV, Wang X, Punyashthiti R, Whelan S, Bacallao R, Torra R, LaRusso NF, Torres VE, et al. Cellular and subcellular localization of the ARPKD protein; fibrocystin is expressed on primary cilia. Hum. Mol. Genet. 2003;12:2703–2710. doi: 10.1093/hmg/ddg274. [DOI] [PubMed] [Google Scholar]
- 24.Chevalier RL, Garland TA, Buschi AJ. The neonate with adult-type autosomal dominant polycystic kidney disease. The International journal of pediatric nephrology. 1981;2:73–77. [PubMed] [Google Scholar]
- 25.Martinez JR, Grantham JJ. Polycystic kidney disease: etiology, pathogenesis, and treatment. Dis. Mon. 1995;41:693–765. doi: 10.1016/s0011-5029(05)80007-0. [DOI] [PubMed] [Google Scholar]
- 26.Neumann HP, Krumme B, van Velthoven V, Orszagh M, Zerres K. Multiple intracranial aneurysms in a patient with autosomal recessive polycystic kidney disease. Nephrol. Dial. Transplant. 1999;14:936–939. doi: 10.1093/ndt/14.4.936. [DOI] [PubMed] [Google Scholar]
- 27.Gattone VH, 2nd, Wang X, Harris PC, Torres VE. Inhibition of renal cystic disease development and progression by a vasopressin V2 receptor antagonist. Nat. Med. 2003;9:1323–1326. doi: 10.1038/nm935. [DOI] [PubMed] [Google Scholar]
- 28.Torres VE, Wang X, Qian Q, Somlo S, Harris PC, Gattone VH., 2nd Effective treatment of an orthologous model of autosomal dominant polycystic kidney disease. Nat. Med. 2004;10:363–364. doi: 10.1038/nm1004. [DOI] [PubMed] [Google Scholar]
- 29.Wu Y, Dai XQ, Li Q, Chen CX, Mai W, Hussain Z, Long W, Montalbetti N, Li G, Glynne R, et al. Kinesin-2 mediates physical and functional interactions between polycystin-2 and fibrocystin. Hum. Mol. Genet. 2006;15:3280–3292. doi: 10.1093/hmg/ddl404. [DOI] [PubMed] [Google Scholar]
- 30.Wang S, Zhang J, Nauli SM, Li X, Starremans PG, Luo Y, Roberts KA, Zhou J. Fibrocystin/Polyductin, Found in the Same Protein Complex with Polycystin-2, Regulates Calcium Responses in Kidney Epithelia. Mol. Cell. Biol. 2007;27:3241–3252. doi: 10.1128/MCB.00072-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Furu L, Onuchic LF, Gharavi A, Hou X, Esquivel EL, Nagasawa Y, Bergmann C, Senderek J, Avner E, Zerres K, et al. Milder presentation of recessive polycystic kidney disease requires presence of amino acid substitution mutations. J. Am. Soc. Nephrol. 2003;14:2004–2014. doi: 10.1097/01.asn.0000078805.87038.05. [DOI] [PubMed] [Google Scholar]
- 32.Bergmann C, Senderek J, Sedlacek B, Pegiazoglou I, Puglia P, Eggermann T, Rudnik-Schoneborn S, Furu L, Onuchic LF, De Baca M, et al. Spectrum of mutations in the gene for autosomal recessive polycystic kidney disease (ARPKD/PKHD1) J. Am. Soc. Nephrol. 2003;14:76–89. doi: 10.1097/01.asn.0000039578.55705.6e. [DOI] [PubMed] [Google Scholar]
- 33.Desmet VJ. What is congenital hepatic fibrosis? Histopathology. 1992;20:465–477. doi: 10.1111/j.1365-2559.1992.tb01031.x. [DOI] [PubMed] [Google Scholar]
- 34.Moser M, Matthiesen S, Kirfel J, Schorle H, Bergmann C, Senderek J, Rudnik-Schoneborn S, Zerres K, Buettner R. A mouse model for cystic biliary dysgenesis in autosomal recessive polycystic kidney disease (ARPKD) Hepatology. 2005;41:1113–1121. doi: 10.1002/hep.20655. [DOI] [PubMed] [Google Scholar]
- 35.Piontek KB, Huso DL, Grinberg A, Liu L, Bedja D, Zhao H, Gabrielson K, Qian F, Mei C, Westphal H, et al. A functional floxed allele of Pkd1 that can be conditionally inactivated in vivo. J. Am. Soc. Nephrol. 2004;15:3035–3043. doi: 10.1097/01.ASN.0000144204.01352.86. [DOI] [PubMed] [Google Scholar]
- 36.Gresh L, Fischer E, Reimann A, Tanguy M, Garbay S, Shao X, Hiesberger T, Fiette L, Igarashi P, Yaniv M, et al. A transcriptional network in polycystic kidney disease. Embo J. 2004;23:1657–1668. doi: 10.1038/sj.emboj.7600160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nagasawa Y, Matthiesen S, Onuchic LF, Hou X, Bergmann C, Esquivel E, Senderek J, Ren Z, Zeltner R, Furu L, et al. Identification and characterization of Pkhd1, the mouse orthologue of the human ARPKD gene. J. Am. Soc. Nephrol. 2002;13:2246–2258. doi: 10.1097/01.asn.0000030392.19694.9d. [DOI] [PubMed] [Google Scholar]
- 38.Konrad M, Zerres K, Wuhl E, Rudnik-Schoneborn S, Holtkamp U, Scharer K. Body growth in children with polycystic kidney disease. Arbeitsgemeinschaft fur Padiatrische Nephrologie. Acta Paediatr. 1995;84:1227–1232. doi: 10.1111/j.1651-2227.1995.tb13538.x. [DOI] [PubMed] [Google Scholar]
- 39.Lilova M, Kaplan BS, Meyers KE. Recombinant human growth hormone therapy in autosomal recessive polycystic kidney disease. Pediatr. Nephrol. 2003;18:57–61. doi: 10.1007/s00467-002-0986-z. [DOI] [PubMed] [Google Scholar]
- 40.Guay-Woodford LM. Murine models of polycystic kidney disease: molecular and therapeutic insights. Am. J. Physiol. Renal Physiol. 2003;285:F1034–F1049. doi: 10.1152/ajprenal.00195.2003. [DOI] [PubMed] [Google Scholar]
- 41.Meyers EN, Lewandoski M, Martin GR. An Fgf8 mutant allelic series generated by Cre- and Flp-mediated recombination. Nat. Genet. 1998;18:136–141. doi: 10.1038/ng0298-136. [DOI] [PubMed] [Google Scholar]
- 42.Tallquist MD, Soriano P. Epiblast-restricted Cre expression in MORE mice: a tool to distinguish embryonic vs. extra-embryonic gene function. Genesis. 2000;26:113–115. doi: 10.1002/(sici)1526-968x(200002)26:2<113::aid-gene3>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 43.Scherle W. A simple method for volumetry of organs in quantitative stereology. Mikroskopie. 1970;26:57–60. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.