

Abstract
Arylamine N-acetyltransferases (NATs) play an important role in the metabolism of arylamine and hydrazine drugs and many arylamine pro-carcinogens. The two human N-acetyltransferases, NAT1 and NAT2, are widely distributed in human tissues and highly polymorphic. While many xenobiotic procarcinogens and drugs are known mammalian NAT substrates, it is unclear what physiological roles these enzymes might play, what endogenous substrates they primarily act upon, or the mechanisms underlying the functional effects of specific NAT gene coding region SNPs. Analyses of mammalian NAT protein structures can greatly help to answer these questions. Homology modeling techniques can be used to approximate mammalian NAT structures using known bacterial NAT crystal structures as templates. In comparison to the bacterial template NATs used for homology modeling, mammalian NATs have a 17 residue insert of unknown structure and function. Homology modeling analyses yielded two different alignments (Modeller 8v1 or 3DCoffee algorithms) that placed this insert in two likely alternative locations. Secondary structure prediction techniques and experimental analyses of a series of human NAT2 mutants with artificial deletions/replacements of the insert region distinguished one of these alternatives as the most likely insert location and provided a better understanding of its structure and function. This study demonstrates both the utility and limitations of computational structural modeling with proteins that differ as much as the mammalian and bacterial NATs.
INTRODUCTION
Arylamine N-acetyltransferases (NATs; EC 2.3.1.5) catalyze the N-acetylation of arylamines and hydrazines and O-acetylation of N-hydroxy-arylamines and heterocyclic amines. These reactions are important for the activation and deactivation of exocyclic amine-containing pro-carcinogens, and for the metabolism of some pharmaceutical drugs (Weber and Hein, 1985). In a ping-pong bi-bi reaction mechanism, the enzyme first acetylates the active site cysteine using acetyl-coenzyme A, and then transfers the acetyl group to the substrate’s exocyclic nitrogen (N-acetylation) or the oxygen of its oxidized exocyclic nitrogen (O-acetylation) (Hein, 1988). While N-acetylation is typically considered a deactivation step, O-acetylation is an activation step, resulting in the formation of reactive arylnitrenium species that can react with DNA to form adducts (Hanna, 1996). Since human NAT genes are highly polymorphic, it is important to understand how different NAT genotypes alter N-acetylation phenotypes, thereby influencing cancer susceptibilities and pharmaceutical drug toxicities (Hein et al., 2000).
Expression of human NAT1 and NAT2 is widely distributed throughout body tissues (Barker et al., 2006; Husain et al., 2007). While many xenobiotic NAT substrates have been discovered, only one potential endogenous substrate, p-aminobenzoylglutamate, has been discovered (Minchin, 1995). Therefore, it is highly probable that other yet unknown endogenous NAT substrates and physiological roles exist. Discovery of potential substrates and useful inhibitors is limited by our knowledge of mammalian NAT enzyme structure.
To-date no publication has described the crystal structure of mammalian NATs. For this reason, investigators have utilized homology modeling techniques to gain clues about the key structural characteristics of mammalian NATs, including the location and potential mechanism(s) of SNPs, and the shape of the active site for computational docking of NAT substrates (Rodrigues-Lima et al., 2001; Rodrigues-Lima and Dupret, 2002; Kawamura et al., 2005; Lau et al., 2006). Quality mammalian NAT structures could be used to rationalize experimental data and screen for potential substrates and inhibitors, potentially pointing to an endogenous physiological role. There are limitations to homology modeling techniques, however, since they sometimes rely on incomplete structural template information.
Bacterial NAT crystal structures were used as templates to create the aforementioned mammalian NAT homology models. Bacterial NAT crystal structures have been solved for Salmonella typhimurium, Pseudomonas aeruginosa, Mycobacterium smegmatis, and Mesorhizobium loti, and can be found with Protein Data Bank accession numbers 1E2T, 1W4T, 1GX3, and 2BSZ, respectively (Sinclair et al., 2000; Sandy et al., 2002; Westwood et al., 2005; Holton et al., 2005). Alignment scores between bacterial and mammalian NAT protein sequences, and among the bacterial NAT sequences, are relatively low (~ 30%). Aligning mammalian and bacterial NAT protein sequences reveals important differences between the bacterial and mammalian NAT protein structures that could limit the utility of the mammalian NAT homology models.
Several studies describe what appears to be an inserted sequence in the second domain of mammalian NATs when aligned with bacterial NATs (Payton et al., 2001; Kawamura et al., 2005; Lau et al., 2006). Although this insert is sometimes referred to as a loop or coil because of its unstructured appearance in homology models, the lack of a template structure for this region is immediately responsible for these designations. Homology modeling can only generate structure for a target protein sequence if a corresponding template structure sequence is provided. Previously reported studies modeled mammalian NATs with this insert adjacent to the active site pocket (Kawamura et al., 2005; Savulescu et al., 2005; Lau et al., 2006), while other studies place it in a different location (Payton et al., 2001) or do not recognize its existence (Rodrigues-Lima et al., 2001).
It is unknown whether this insert has defined secondary structure and is integrated into the second domain beta barrel, or if it takes the form of an unstructured loop or coil. In the latter case, a loop adjacent to the active site pocket could affect the size, shape, and/or accessibility of the active site pocket. In addition, since the location of the loop in a homology model is determined by the placement of the insert in the alignment, as is demonstrated by the differences in previously reported alignments (Payton et al., 2001; Rodrigues-Lima et al., 2001; Savulescu et al., 2005; Lau et al., 2006), it is possible that a slightly different or more optimal alignment could place the insert in a different location.
Our homology modeling analyses yielded two different, and apparently equally valid, alignments that placed this insert in two different locations. We used secondary structure prediction programs and designed a series of human NAT2 deletion mutants to positively define the insert location and better understand its structure and function.
METHODS
Homology Modeling
Human NAT2 and bacterial NAT sequences from Salmonella typhimurium, Pseudomonas aeruginosa, and Mesorhizobium loti were aligned using the Modeller 8v1 auto align function and the 3DCoffee alignment algorithm (Sali and Blundell, 1993; O’Sullivan et al., 2004). The known crystal structures for Salmonella typhimurium, Pseudomonas aeruginosa, and Mesorhizobium loti NATs (PDB accession numbers 1E2T, 1W4T, 2BSZ) were used as template structures for creating homology models of human NAT2. The Modeller and 3DCoffee alignments, with bacterial NAT template structural information, were used to generate homology models by satisfaction of spatial restraints using the program Modeller 8v1 (Sali and Blundell, 1993).
Secondary Structure Prediction
N-acetyltransferase protein sequences from Salmonella typhimurium, Pseudomonas aeruginosa, Mesorhizobium loti, and human NAT2 were evaluated using twelve secondary structure prediction programs: APSSP2, PROF king, PROF sec, PORTER, JPRED, JUFO, SCRATCH, SABLE1, SABLE2, SAM-T99, PSIpred, and YASPIN (McClelland and Rumelhart, 1988; Kneller et al., 1990; Rost, 1996; Karplus et al., 1998; Cuff and Barton, 2000; McGuffin et al., 2000; Ouali and King, 2000; Meiler et al., 2001; Raghava, 2002; Adamczak et al., 2004; Adamczak et al., 2005; Cheng et al., 2005; Lin et al., 2005; Pollastri and McLysaght, 2005). Secondary structure predictions were then aligned based on Modeller or 3DCoffee protein sequence alignments.
Human NAT2 Mutation
A 17 residue segment was deleted from human NAT2, at 157–173 or 167–183 based on Modeller and 3DCoffee alignment results, respectively. Residues immediately surrounding the deletion site were replaced by beta turn-facilitating residues in order to prevent disruption of the beta barrel secondary structure. Eight different deletion mutants were created. For the Modeller-based deletion, turns QEGST, PHAG, ANDH, and SDGSD were inserted on either side of the deletion in four different deletion mutants. For the 3DCoffee-based deletion, turns HHEH, GRSG, GG, and GGSG were inserted on either side of the deletion in four different deletion mutants. In both sets of mutants, the first three turns match turn residues found in the same location in structures 1E2T, 1W4T, 2BSZ, respectively (Figure 1), and the fourth turn was made entirely of “turn promoting” residues (Paul and Rosenbusch, 1985). Codons chosen for the turn residues were based on the codon usage table for Homo sapiens (www.kazusa.or.jp/codon).
Figure 1.
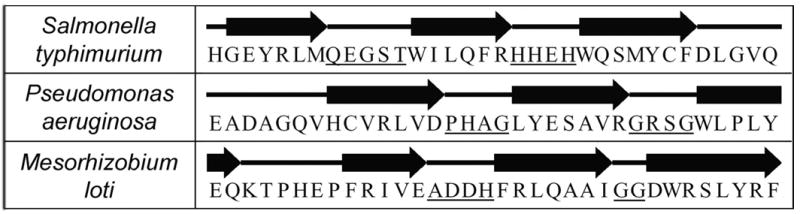
Bacterial template structure beta-barrel turn residues. Residues 146–179 are shown for all three bacterial NATs, with beta sheet residues indicated by arrows and coil/turn residues by straight lines. The first underlined segment on each line corresponds to the turn used for a Modeller-based mutant, and the second for a 3DCoffee-based mutant. This figure was adapted from information on the Protein Data Bank website (www.rcsb.org; accession numbers 1E2T, 1W4T, and 2BSZ).
The human NAT2 nucleotide sequence was mutated by overlap extension (Horton et al., 1989). Human NAT2 coding region sequences on either side of the targeted deletion were amplified independently by polymerase chain reaction (PCR) from a prokaryotic expression vector pKK223-3 containing human NAT2 (Ferguson et al., 1994), using plasmid-specific and human NAT2-specific primers (Table 1). Each set of NAT2-specific primers contained complementary nucleotide sequences coding for the replacement turn residues. For each mutant, the two separate PCR products were combined, denatured at 98ºC, and the complementary sequences allowed to anneal at 30ºC, followed by overlap extension using nested forward and reverse human NAT2-specific PCR primers. The forward and reverse nested primers introduced restriction sites EcoR1 and HindIII, respectively. Independent duplicates of each mutant were cloned and expressed in parallel as cloning and expression controls.
Table 1.
Primers Used For Mutant Construction and Cloning
| Name | Sequence | Features |
|---|---|---|
| m-h2-1e2tF | 5′-CAGGAGGGCAGCACCCTTAATTCTCATCTCCTGCC-3′b | QEGST peptide |
| m-h2-1e2tR | 5′-GGTGCTGCCCTCCTGTGTCAAGCAGAAAATGCA-3′b | QEGST rc peptide |
| m-h2-1w4tF | 5′-AGACCCCACGCCGGCTTTCTTAATTCTCATCTCCTG-3′b | PHAG peptide |
| m-h2-1w4tR | 5′-AAAGCCGGCGTGGGGTCTCTCTTCTGTCAAGCAGA-3′b | PHAG rc peptide |
| m-h2-2bszF | 5′-GACAGAAGCCAACGACCACCTTAATTCTCATCTCCTGCC-3′b | ANDH peptide |
| m-h2-2bszR | 5′-AAGGTGGTCGTTGGCTTCTGTCAAGCAGAAAATGC-3′b | ANDH rc peptide |
| m-h2-CustF | 5′-ACAAGCGACGCCAGCGACCTTAATTCTCATCTCCTGCC-3′b | SDGSD peptide |
| m-h2-CustR | 5′-GTCGCTGGCGTCGCTTGTCAAGCAGAAAATGCA-3′b | SDGSD rc peptide |
| 3d-h2-1e2tF | 5′-ATCCACCACGAGCACCACCAAAAAATATACTTATTTACGC-3′b | HHEH peptide |
| 3d-h2-1e2tR | 5′-GTGGTGCTCGTGGTGGATTTGGTCCAGGTACCAG-3′b | HHEH rc peptide |
| 3d-h2-1w4tF | 5′-ATCGGCAGAAGCGGCCACCAAAAAATATACTTATTTACGC-3′b | GRSG peptide |
| 3d-h2-1w4tR | 5′-GTGGCCGCTTCTGCCGATTTGGTCCAGGTACCAG-3′b | GRSG rc peptide |
| 3d-h2-2bszF | 5′-ATCAGGGGCGGCAAACACCAAAAAATATACTTATTTACG-3′b | GG peptide |
| 3d-h2-2bszR | 5′-GTGTTTGCCGCCCCTGATTTGGTCCAGGTAC-3′b | GG rc peptide |
| 3d-h2-CustF | 5′-ATCGGCGGCAGCGGCCACCAAAAAATATACTTATTTACGC-3′b | GGSG peptide |
| 3d-h2-CustR | 5′-GCCGCTGCCGCCGATTTGGTCCAGGTACCA-3′b | GGSG rc peptide |
| hNat2EcoRI-F | 5′-ATGCGAGAATTCATGGACATTGAAGCATATTTTGAAAGAATT-3′a | PCR, EcoR1 |
| hNat2HindIII-R | 5′-ATGCGAAAGCTTAAGGGTTTATTTTGTTCCTTATTCTAAAT-3′a | PCR, HindIII |
| pKK-F | 5′-TTATCAGACCGCTTCTGCGT-3′ | PCR, Fwd-Seq |
| h2-Seq1F | 5′-CCGGGGTGGGTGGTGTCT-3′ | Fwd-Seq |
| h2-Seq2F | 5′-CACCTTCTCCTGCAGGTGACCAT-3′ | Fwd-Seq |
| h2-Seq3F | 5′-TTTGGTGGGCTTCATCCTCA-3′ | Fwd-Seq |
| pKK-R | 5′-GCTGTTGACAATTAATCATCGG-3′ | PCR, Rev-Seq |
| h2-Seq1R | 5′-CAACCTCTTCCTCAGTGAGAG-3′ | Rev-Seq |
| h2-Seq2R | 5′-ACATCTGGGAGGAGCTTCCA-3′ | Rev-Seq |
| h2-Seq3R | 5′-TGACCTGGAGACACCACCCA-3′ | Rev-Seq |
Underline indicates restriction site
Underline indicates turn residue codons (rc = reverse complement)
A restriction fragment length analysis using restriction enzymes KpnI and ApoI was used to verify the successful removal of the 17 residue segment in the Modeller-based and 3DCoffee-based deletion mutants, respectively, prior to cloning. Successful deletion of the Modeller-based insert changed the KpnI digestion pattern from one to zero cuts. Successful deletion of the 3DCoffee-based insert changed the ApoI digestion pattern from two to one cut. Digestion products were resolved on a 1% agarose gel.
Cloning, Sequencing, and Expression
Independent duplicates of the reference human NAT2 coding region were cloned and expressed in parallel with the independent duplicates of each mutant. All were directionally cloned into pKK223-3 bacterial expression vector (Pharmacia-LKB Biotechnology, Piscataway, NJ) using EcoRI and HindIII restriction sites as described previously (Doll and Hein, 1995).
The expected nucleotide sequence was confirmed by automated sequencing of sense and antisense strands as previously described (Walraven et al., 2006) using human NAT2 specific sequencing primers (Table 1), prior to transformation and expression in JM105 strain Escherichia coli as previously described (Chung and Miller, 1988; Chung et al., 1989; Doll and Hein, 1995). Bacteria were lysed by sonication in homogenization buffer as previously described (Doll and Hein 1995), and total bacterial lysate protein concentration was determined for each expression (Bradford, 1976).
Enzyme Activity and Stability
As a measure of NAT2 enzyme activity, sulfamethazine (SMZ) N-acetylation assays were performed by incubating bacterial lysate, 1 mM acetyl-coenzyme A, and 300 μM SMZ at 37°C for 10 minutes under linear rate conditions, and the reaction was stopped by adding 1/10 volume of 1 M acetic acid. Control reactions substituted water for acetyl-coenzyme A. Acetylated SMZ product was separated and measured using a Beckman System Gold high-performance liquid chromatography system as described previously (Leff et al, 1999). Sulfamethazine is a NAT2-selective substrate (Grant et al., 1991).
Thermostability was measured by incubating the bacterial lysate (1 mg/ml) at 30°–50°C for 10 minutes, immediately followed by assays for SMZ N-acetylation as described above. Results were normalized to activity of untreated lysate to give the percentage of activity remaining.
RESULTS
Homology Modeling
Alignments between bacterial template NATs and human NAT2 revealed a 17 residue segment, or insert, in human NAT2 that has no corresponding bacterial template sequence. Modeller and 3DCoffee alignment algorithms placed this 17 residue insert in different locations, 157–173 (GIWYLDQIRREQYITNK) and 167–183 (EQYITNKEFLNSHLLPK), respectively.
The NAT2 homology models were superimposible with the bacterial template structures, with the Cys68-His107-Asp122 catalytic triad positioned as expected in the active site pocket. Due to a lack of structural template information, the 17 residue insert appeared as loops in the second domain of human NAT2. In the models, the Modeller alignment-based loop was located one beta strand away from the 3DCoffee-based loop and away from the active site, whereas the 3DCoffee alignment-based loop was adjacent to the active site pocket (Figure 2).
Figure 2.
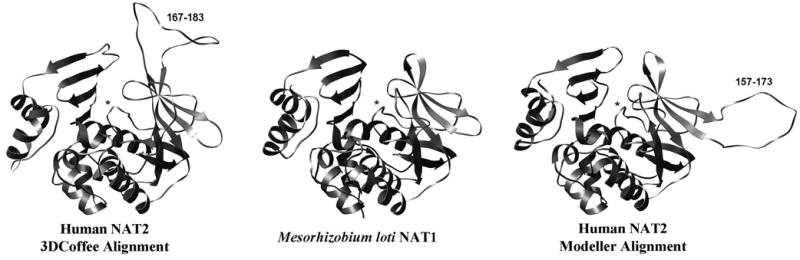
Homology models of human NAT2, and the Mesorhizobium loti NAT1 bacterial template structure (shown for reference). The Modeller alignment produces a second domain loop away from the active site, whereas the 3DCoffee alignment produces a second domain loop adjacent to the active site pocket. Asterisks identify the active site loop, and the residue numbers of each insert are shown.
Since these homology models were not used for detailed evaluation of specific residues or for substrate docking, an analysis of the structural quality was not necessary. Such detailed evaluations would require molecular dynamics simulations for structural optimization. The Modeller program generates models of sufficient quality for the immediate purposes of this study.
Secondary Structure Prediction
Bacterial NAT secondary structure predictions were very similar to their actual secondary structures as defined on the RCSB Protein Data Bank website (www.rcsb.org), thus providing confidence that the human NAT2 predictions also were accurate. The predicted bacterial NAT and human NAT secondary structures were strikingly similar, with nearly all major secondary structures shared (Figure 3). The individual prediction programs gave slightly different variations for each sequence, but the prediction programs were overall very consistent.
Figure 3.
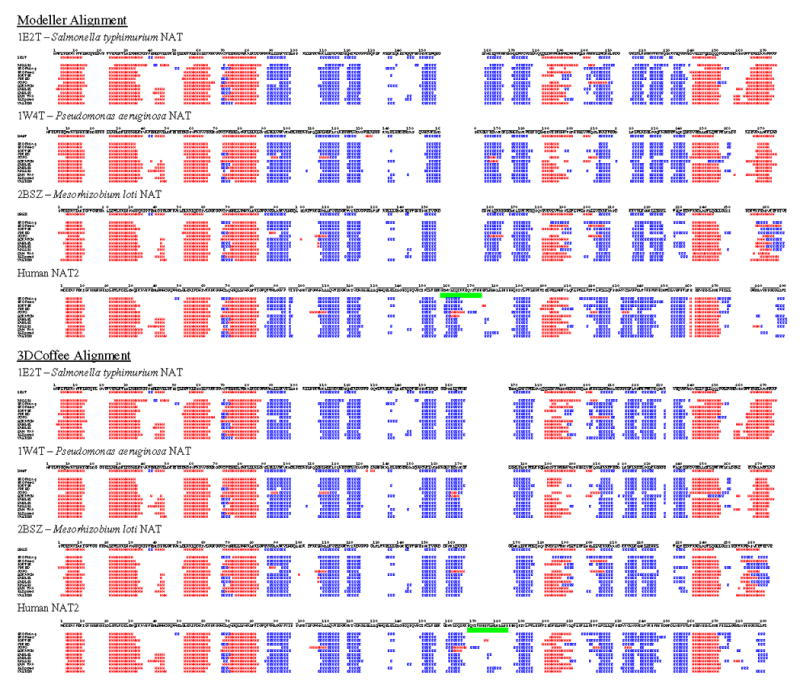
Secondary structure prediction results were aligned based on Modeller and 3DCoffee alignments. This figure is designed to give an overall summary view of how prediction programs assigned secondary structure and how the bacterial NAT and human NAT2 secondary structures align when the Modeller and 3DCoffee alignments are applied. Results from twelve secondary structure prediction programs are listed under each NAT sequence. Actual secondary structure is shown immediately beneath each bacterial NAT sequence. Blue “E” characters represent beta sheets, and red “H” characters represent helices. A green bar spans the insert residues according to each alignment. This figure is also provided as supplementary data in a more legible format.
When the bacterial NAT and human NAT2 predictions were aligned based on the Modeller alignment, the 17 residue human NAT2 insert was located in the same place as a predicted beta strand. In addition, a bacterial NAT beta strand on the c-terminal side of the insert did not match up with a human NAT2 beta strand. This apparent mismatch was corrected when the prediction results were aligned based on the 3DCoffee alignment. In this case, the 17 residue insert did not have any predicted secondary structure and the surrounding predicted secondary structures matched with the bacterial NAT secondary structures.
Another important observation was made regarding the C-terminal tail. The bacterial NAT C-terminal helix found in all four known bacterial NAT crystal structures was not predicted for the human NAT2 sequence by any of the prediction programs.
Human NAT2 Deletion Mutants
Based on restriction fragment length analyses and complete sequencing results, the eight different human NAT2 deletion mutants were successfully constructed and cloned into bacterial expression vector pKK223-3. Duplicate clones of the reference and deletion mutant human NAT2 provided highly reproducible results. This validates the cloning process and expression system used in this study as highly consistent, as demonstrated previously (Walraven et al., 2006).
Enzyme activity results (Figure 4) clearly demonstrated that deletion of the Modeller alignment insert resulted in complete loss of enzyme activity, regardless of the turn used to replace the residues surrounding the deletion. On the other hand, all of the 3DCoffee deletion mutants had measurable N-acetylation activity. The activities for these mutants were highly variable, with one turn variant exhibiting SMZ NAT activity nearly equivalent to the reference activity.
Figure 4.
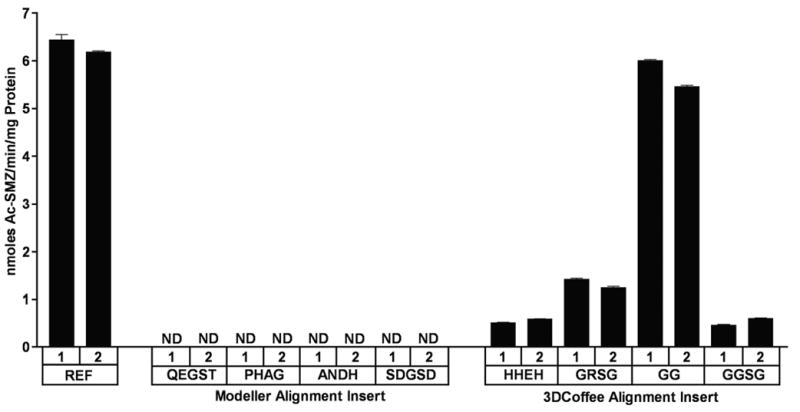
SMZ N-acetyltransferase activities of reference and mutant human NAT2. Mutants are labeled according to the turn sequence used. Duplicate clones are indicated with “1” and “2” above the sequence for each turn. Activity is nanomoles of product per minute reaction time per milligram of total protein. Activity was not detected for substrates marked ND (limit of detection = 0.06 nmoles/min/mg). Error bars indicate standard error (n = 3).
Thermostability measurements for the reference human NAT2 and the highest NAT2-activity 3DCoffee deletion mutant revealed that the deletion mutant was much less stable than reference human NAT2 (Figure 5). The reference NAT2 enzyme had only slightly reduced activity at the higher temperatures, whereas the deletion mutant began losing activity at lower temperatures and completely lost activity at the higher temperatures. One of the deletion mutants with lower activity was also tested for thermostability, but no SMZ NAT2 activity was detectable after incubation at 32°C (data not shown).
Figure 5.
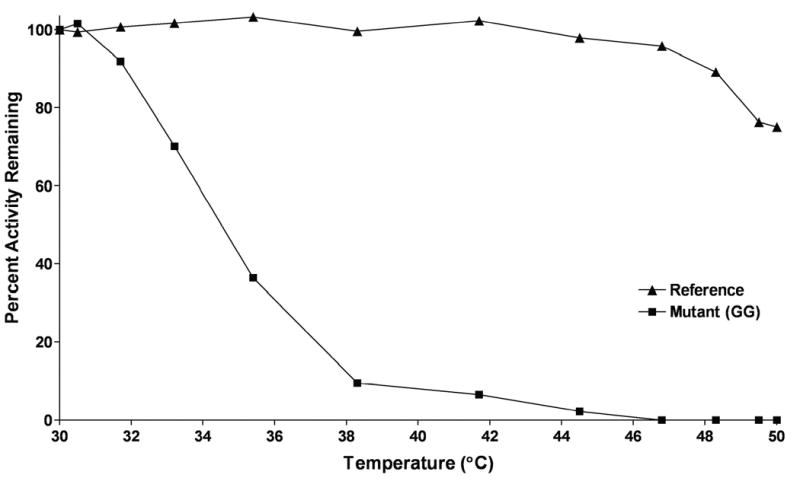
Thermostability of reference and mutant human NAT2. Percentage of SMZ activity remaining plotted on the ordinate versus 10 minute incubation temperature on the abscissa. Error bars indicate standard error (n = 3).
Deleting the 17 residue insert resulted in decreased activity due at least in part to reduced stability. The choice of turn residues was apparently also critical to the mutant enzyme’s stability. The mutant with the highest activity was also the mutant with the shortest turn substituted in for the segment, which was composed entirely of glycines (GG).
DISCUSSION
Previous studies have used bacterial template structures to create mammalian NAT homology models (Rodrigues-Lima et al., 2001; Rodrigues-Lima and Dupret, 2002; Kawamura et al., 2005; Lau et al., 2006). However, since the generation of a homology model is not the primary focus of this study, many of the structural details discussed in those studies will not be revisited here. This study focused on the undefined and not well understood mammalian NAT insert. Since homology models will conform to the template structure(s) provided, the regions that lack a template require more attention. This is especially true when the regions in question could influence the active site pocket’s shape and substrate accessibility.
Previous studies have generated alignments that placed the insert in different locations. Lau et al. (2006) placed the insert residues at either 172–188 or 170–186 (in the alignment, QP is repeated on both sides of the insert) which is modeled as a loop in the second domain, adjacent to the active site pocket. The alignment published by Savulescu et al. (2005) places the insert in the same location, and Kawamura et al. (2005) reported models with a loop also in the second domain, adjacent to the active site. These three studies placed the loop close to the 3DCoffee-based insert (167–183) described in our study. Payton et al. (2001) published an alignment with the insert located at 156–172, which is nearly identical to the Modeller-based insert (157–173) described in our study.
The results of the present study clearly identify the correct insert location and confirm that the insert is a loop. The loop is not integrated into the secondary structure of the protein and has no defined secondary structure. Since bacterial NATs function well without an insert, it is conceivable that deleting the loop in a way that facilitated the folding of the beta barrel at the deletion site could result in active protein, probably with slightly altered characteristics. On the other hand, it is much less likely that deleting the wrong insert location would yield an active protein, since it would remove a significant portion of the surrounding beta barrel secondary structure in the second domain. If the insert were not a loop, but were integrated into the secondary structure of the protein, both deletions would significantly alter the fold of the protein or prevent it from folding altogether.
When the 3DCoffee-assigned insert (167–183) was deleted, the enzyme remained active, though with altered characteristics. On the other hand, deletion of the Modeller-assigned insert (157–173) resulted in undetectable activity. These results support the conclusion that residues 167–183 are not only the correct insert location, but they form a loop that does not play a critical role in the integrity of the beta barrel secondary structure. Deleting residues 157–173 probably removed a portion of the second domain beta barrel and altered the folding and/or function of the protein. For the purposes of this study, it is not necessary to know whether the loss of activity was due to misfolding of the protein, altered protein function, or a combination of both.
Secondary structure predictions supported and confirmed these conclusions. According to the alignments in Figure 3, deletion of the Modeller-assigned insert resulted also in the deletion of a beta strand in the second domain beta barrel. This is consistent with the loss of activity seen for the Modeller alignment-based mutants. In addition, no secondary structure was predicted for the 3DCoffee-assigned insert, which is consistent with the conclusion that the insert must be a loop or coil with no defined secondary structure, and not integrated into the surrounding beta barrel. These results are also consistent with our hypothesis that the mammalian NAT proteins can function without the loop.
The secondary structure prediction that the bacterial NAT C-terminal helix is not shared by human NAT2 suggests that the C-terminal tail is free to interact with the active site. The C-terminal helix is likewise not predicted for human NAT1 or any of the rat Nats (unpublished data). This would be consistent with previous reports that the 10 C-terminal residues are critical for control of acetyl-coenzyme A hydrolysis (Mushtaq et al., 2002). Since acetyl-coenzyme A hydrolysis takes place in the active site, it is reasonable to assume that the C-terminal tail must interact with the active site pocket.
Although the loop is not required for NAT catalytic activity, the reduced stability of loop-less human NAT2 suggests that this loop may serve a role in stabilizing the protein’s structure. How the loop might accomplish this task is beyond the scope of homology modeling. Our models suggest that the loop could also play a role in active site access since it is adjacent to the active site and flexible enough to extend over the bacterial NAT-like active site cavity. Combined with the possibility that the C-terminal tail interacts with the active site, the active site pocket could look much different than its representation in current homology models.
After our experiments were completed, the crystal structure for a mutant human NAT1 protein was deposited into the RCSB Protein Data Bank under the code 2IJA. Although the publication describing this structure has not yet been released, it substantiates the findings of our human NAT2 study. The overall structure of our NAT2 model compares favorably to 2IJA, with a root mean square deviation of 1.4 Å between the two structures. In addition, the NAT2 loop location as determined by our study (167–183) is nearly identical to the 21JA loop residues (165–185). Two structural differences between 2IJA and our model are the association of the loop with a beta strand in domain three, and the insertion of the C-terminal tail adjacent to the active site pocket. These interesting structural features have a dramatic impact on the size and shape of the active site pocket and rationalize the influence of the C-terminal tail on acetyl-coenzyme A hydrolysis. These two major differences also support our study’s finding that the loop may have a stabilizing effect on the protein’s structure, and make sense of the predicted lack of secondary structure in the C-terminal NAT2 sequence. More information is needed about the flexibility of these important structural features in solution.
This study demonstrates the strength of combining computational and experimental approaches for determining protein structure and function. Based on the initial success of mammalian crystallization attempts (2IJA), it might be expected that the results of this study will be confirmed by the eventual crystallization of human NAT2. This study identified the location and potential function of the mammalian NAT loop and contributes to our understanding of arylamine N-acetyltransferase structure and function.
Acknowledgments
We thank Dr. David Barker for his technical assistance and helpful advice.
This work was partially supported by U.S. Public Health Service grant CA34627 from the National Cancer Institute and training grant ES011564 from the National Institute of Environmental Health Sciences. This work constitutes partial fulfillment by Jason Walraven for the Ph.D. in Pharmacology and Toxicology at the University of Louisville School of Medicine.
Non-standard Abbreviations Used in this Paper
- NAT
Arylamine N-acetyltransferase
- SNP
single nucleotide polymorphism
- SMZ
sulfamethazine
- PCR
polymerase chain reaction
Footnotes
Send reprint requests to: David W. Hein, Ph.D., Department of Pharmacology and Toxicology, University of Louisville School of Medicine, Louisville, KY 40292. E-mail: d.hein@louisville.edu
References
- Adamczak R, Porollo A, Meller J. Combining Prediction of Secondary Structure and Solvent Accessibility in Proteins. Proteins. 2005;59:467–75. doi: 10.1002/prot.20441. [DOI] [PubMed] [Google Scholar]
- Barker DF, Husain A, Neale JR, Martini BD, Zhang X, Doll MA, States JC, Hein DW. Functional properties of an alternative, tissue-specific promoter for human arylamine N-acetyltransferase 1. Pharmacogenet Genomics. 2006;16:515–525. doi: 10.1097/01.fpc.0000215066.29342.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Cheng J, Randall A, Sweredoski M, Baldi P. SCRATCH: a Protein Structure and Structural Feature Prediction Server. Nucleic Acids Research. 2005;33:72–76. doi: 10.1093/nar/gki396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung CT, Miller RH. A rapid and convenient method for the preparation and storage of competent bacterial cells. Nucleic Acids Res. 1988;16:3580. doi: 10.1093/nar/16.8.3580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung CT, Niemela SL, Miller RH. One-step preparation of competent Escherichia coli: transformation and storage of bacterial cells in the same solution. Proc Natl Sci USA. 1989;86:2172–2175. doi: 10.1073/pnas.86.7.2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuff JA, Barton GJ. Application of Multiple Sequence Alignment Profiles to Improve Protein Secondary Structure Prediction. Proteins. 2000;40:502–511. doi: 10.1002/1097-0134(20000815)40:3<502::aid-prot170>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- Doll MA, Hein DW. Cloning, sequencing and expression of NAT1 and NAT2 encoding genes from rapid and slow acetylator inbred rats. Pharmacogenetics. 1995;5:247–251. doi: 10.1097/00008571-199508000-00009. [DOI] [PubMed] [Google Scholar]
- Ferguson RJ, Doll MA, Rustan TD, Gray K, Hein DW. Cloning, expression, and functional characterization of two mutant (NAT2(191) and NAT2(341/803)) and wild-type human polymorphic N-acetyltransferase (NAT2) alleles. Drug Metab Dispos. 1994;22(3):371–376. [PubMed] [Google Scholar]
- Grant DM, Blum M, Beer M, Meyer UA. Monomorphic and polymorphic human arylamine N-acetyltransferases: a comparison of liver isozymes and expressed products of two cloned genes. Mol Pharmacol. 1991;39(2):184–191. [PubMed] [Google Scholar]
- Hanna PE. Metabolic activation and detoxification of arylamines. Curr Med Chem. 1996;3:195–210. [Google Scholar]
- Hein DW. Acetylator genotype and arylamine-induced carcinogenesis. Biochim Biophys Acta. 1988;948:37–66. doi: 10.1016/0304-419x(88)90004-2. [DOI] [PubMed] [Google Scholar]
- Hein DW, Doll MA, Fretland AJ, Leff MA, Webb SJ, Xiao GH, Devanaboyina US, Nangju NA, Feng Y. Molecular genetics and epidemiology of the NAT1 and NAT2 acetylation polymorphisms. Cancer Epidemiol Biomarkers Prev. 2000;9(1):29–42. [PubMed] [Google Scholar]
- Holton SJ, Dairou J, Sandy J, Rodrigues-Lima F, Dupret J-M, Noble ME, Sim E. Structure of Mesorhizobium Loti Arylamine N-Acetyltransferase 1. Acta Crystallograph Sect F Struct Biol Cryst Commun. 2005;61:14–16. doi: 10.1107/S1744309104030659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton RM, Hunt HD, Ho SN, Pullen JK, Pease LR. Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene. 1989;77(1):61–68. doi: 10.1016/0378-1119(89)90359-4. [DOI] [PubMed] [Google Scholar]
- Husain A, Zhang X, Doll MA, States JC, Barker DF, Hein DW. Identification of N-acetyltransferase 2 (NAT2) transcription start sites and quantitation of NAT2-specific mRNA in human tissues. Drug Metab Dispos. 2007;35:721–727. doi: 10.1124/dmd.106.014621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karplus K, Barret C, Hughey R. Hidden (Markov) models for detecting remote protein homologies. Bioinformatics. 1998;14(10):846–856. doi: 10.1093/bioinformatics/14.10.846. [DOI] [PubMed] [Google Scholar]
- Kawamura A, Graham J, Mushtaq A, Tsiftsoglou SA, Vath GM, Hanna PE, Wagner CR, Sim E. Eukaryotic arylamine N-acetyltransferase investigation of substrate specificity by high-throughput screening. Biochem Pharmacol. 2005;69:347–359. doi: 10.1016/j.bcp.2004.09.014. [DOI] [PubMed] [Google Scholar]
- Kneller DG, Cohen FE, Langridge R. Improvements in Protein Secondary Structure Prediction by an Enhanced Neural Network. J Mol Biol. 1990;214:171–182. doi: 10.1016/0022-2836(90)90154-E. [DOI] [PubMed] [Google Scholar]
- Lau EY, Felton JS, Lightstone FC. Insights into the O-acetylation reaction of hydroxylated heterocyclic amines by human arylamine N-acetyltransferases: a computational study. Chem Res Toxicol. 2006;19:1182–1190. doi: 10.1021/tx0600999. [DOI] [PubMed] [Google Scholar]
- Leff MA, Epstein PN, Doll MA, Fretland AJ, Devanaboyina US, Rustan TD, Hein DW. Prostate-specific human N-acetyltransferase 2 (NAT2) expression in the mouse. J Pharmacol Exp Ther. 1999;290:182–187. [PubMed] [Google Scholar]
- Lin K, Simossis VA, Taylor WR, Heringa J. A Simple and Fast Secondary Structure Prediction Algorithm using Hidden Neural Networks. Bioinformatics. 2005;21(2):152–9. doi: 10.1093/bioinformatics/bth487. [DOI] [PubMed] [Google Scholar]
- McClelland JL, Rumelhart DE. Explorations in Parallel Distributed Processing. Vol. 3. MIT Press; Cambridge MA: 1988. pp. 318–362. [Google Scholar]
- McGuffin LJ, Bryson K, Jones DT. The PSIPRED protein structure prediction server. Bioinformatics. 2000;16:404–405. doi: 10.1093/bioinformatics/16.4.404. [DOI] [PubMed] [Google Scholar]
- Meiler J, Muller M, Zeidler A, Schmaschke F. Generation and evaluation of dimension-reduced amino acid parameter representations by artificial neural networks. J Mol Model. 2001;7(9):360–369. [Google Scholar]
- Minchin RF. Acetylation of para-aminobenzoylglutamate, a folate catabolite, by recombinant human NAT and U937 cells. Biochem J. 1995;307:1–3. doi: 10.1042/bj3070001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mushtaq A, Payton M, Sim E. The COOH terminus of arylamine N-acetyltransferase from Salmonella typhimurium controls enzymatic activity. J Biol Chem. 2002;277:12175–12181. doi: 10.1074/jbc.M104365200. [DOI] [PubMed] [Google Scholar]
- O’Sullivan O, Suhre K, Abergel C, Higgins DG, Notredame C. 3DCoffee: Combining protein sequences and structures within multiple sequence alignments. J Mol Biol. 2004;340:385–395. doi: 10.1016/j.jmb.2004.04.058. [DOI] [PubMed] [Google Scholar]
- Ouali M, King RD. Cascaded multiple classifiers for secondary structure prediction. Prot Sci. 2000;9:1162–1176. doi: 10.1110/ps.9.6.1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul C, Rosenbusch JP. Folding patterns of porin and bacteriorhodopsin. EMBO J. 1985;4(6):1593–1597. doi: 10.1002/j.1460-2075.1985.tb03822.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payton M, Mushtaq A, Yu T-W, Wu L-J, Sinclair J, Sim E. Eubacterial arylamine N-acetyltransferase identification and comparison of 18 members of the protein family with conserved active site cysteine, histidine, and aspartate residues. Microbiology. 2001;147:1137–1147. doi: 10.1099/00221287-147-5-1137. [DOI] [PubMed] [Google Scholar]
- Pollastri G, McLysaght A. Porter: a new, accurate server for protein secondary structure prediction. Bioinformatics. 2005;21(8):1719–20. doi: 10.1093/bioinformatics/bti203. [DOI] [PubMed] [Google Scholar]
- Raghava GPS. CASP5. 2002. APSSP2: A combination method for protein secondary structure prediction based on neural network and example based learning; p. A132. [Google Scholar]
- Rodrigues-Lima F, Delomenie C, Goodfellow GH, Grant DM, Dupret J-M. Homology modelling and structural analysis of human arylamine N-acetyltransferase NAT1: Evidence of the conservation of a cysteine protease catalytic domain and an active site loop. Biochem J. 2001;356:327–334. doi: 10.1042/0264-6021:3560327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues-Lima F, Dupret J-M. 3D model of human arylamine N-acetyltransferase 2: Structural basis of the slow acetylator phenotype of the R64Q variant and analysis of the active-site loop. Biochem Biophys Res Commun. 2002;291:116–123. doi: 10.1006/bbrc.2002.6414. [DOI] [PubMed] [Google Scholar]
- Rost B. PHD: predicting one-dimensional protein structure by profile based neural networks. Meth Enzymol. 1996;266:525–539. doi: 10.1016/s0076-6879(96)66033-9. [DOI] [PubMed] [Google Scholar]
- Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- Sandy J, Mushtaq A, Kawamura A, Sinclair J, Sim E, Noble M. The structure of arylamine N-acetyltransferase from Mycobacterium smegmatis – an enzyme which inactivates the anti-tubercular drug, isoniazid. J Mol Biol. 2002;318:1071–1083. doi: 10.1016/S0022-2836(02)00141-9. [DOI] [PubMed] [Google Scholar]
- Savulescu MR, Mushtaq A, Josephy PD. Screening and characterizing human NAT2 variants. Methods Enzymol. 2005;400:192–215. doi: 10.1016/S0076-6879(05)00011-X. [DOI] [PubMed] [Google Scholar]
- Sinclair JC, Sandy J, Delgoda R, Sim E, Noble ME. Structure of arylamine N-acetyltransferase reveals a catalytic triad. Nat Struct Biol. 2000;7:560–564. doi: 10.1038/76783. [DOI] [PubMed] [Google Scholar]
- Walraven JM, Doll MA, Hein DW. Identification and characterization of functional rat arylamine N-acetyltransferase 3: comparisons with rat arylamine N-acetyltransferases 1 and 2. J Pharmacol Exp Ther. 2006;319:369–375. doi: 10.1124/jpet.106.108399. [DOI] [PubMed] [Google Scholar]
- Weber WW, Hein DW. N-acetylation pharmacogenetics. Pharmacol Rev. 1985;37:25–27. [PubMed] [Google Scholar]
- Westwood IM, Holton SJ, Rodrigues-Lima F, Dupret J-M, Bhakta S, Noble ME, Sim E. Expression, purification, characterization and structure of Pseudomonas aeruginosa arylamine N-Acetyltransferase. Biochem J. 2005;385:605–612. doi: 10.1042/BJ20041330. [DOI] [PMC free article] [PubMed] [Google Scholar]