

Abstract
Human N-acetyltransferase 2 (NAT2) is polymorphic in humans and may associate with cancer risk by modifying individual susceptibility to cancers from carcinogen exposure. Since molecular epidemiological studies investigating these associations usually include determining NAT2 single nucleotide polymorphisms (SNPs), haplotypes, or genotypes, their conclusions can be compromised by the uncertainty of genotype-phenotype relationships. We characterized NAT2 SNPs and haplotypes by cloning and expressing recombinant NAT2 allozymes in mammalian cells. The reference and variant recombinant NAT2 allozymes were characterized for arylamine N-acetylation and O-acetylation of N-hydroxy-arylamines. SNPs and haplotypes that conferred reduced enzymatic activity did so by reducing NAT2 protein without changing NAT2 mRNA levels. Among SNPs that reduced catalytic activity, G191A (R64Q), G590A (R197Q) and G857A (G286E) reduced protein half-life but T341C (I114T), G499A(E167K) and A411T (L137F) did not. G857A (G286E) and the major haplotype possessing this SNP (NAT2*7B) altered the affinity to both substrate and cofactor acetyl coenzyme A, resulting in reduced catalytic activity towards some substrates but not others. Our results suggest that coding region SNPs confer slow acetylator phenotype by multiple mechanisms that also may vary with arylamine exposures.
Introduction
Human arylamine N-acetyltransferase 2 (NAT2) catalyzes the N-acetylation of arylamines and the O-acetylation of N-hydroxylated metabolites of arylamines and heterocyclic amines (HCAs) [1,2]. Arylamines such as 4-aminobiphenyl (ABP) in cigarette smoke, and HCAs such as 2-amino-1-methyl-6-phenylimidazo [4,5-b] pyridine (PhIP) in well-cooked meats, are carcinogenic and DNA adducts following their metabolism have been identified in animals and humans after exposure [3–5]. O-acetylation catalyzed by NAT2 plays an important role in the metabolic bioactivation and the consequent adduct formation of these carcinogens [2,6]. Individuals with slow acetylator NAT2 phenotype have higher frequency of the typical smoking-related DNA adduct pattern in human breast tissue [7] or ABP-hemoglobin levels [8] than those with rapid acetylator phenotype. Individuals with rapid acetylator NAT2 phenotype have higher levels of PhIP-DNA adducts in human breast tissue than slow acetylators [9,10]. Thus, these data suggest that the association of cancer risk with NAT2 phenotype may be exposure specific.
NAT2 capacity varies in humans and is often subdivided into rapid and slow or rapid, intermediate, and slow acetylator phenotypes [11]. NAT2 phenotype variation is thought to be due to single nucleotide polymorphisms (SNPs) in the 870 bp NAT2 open reading frame. The NAT2 acetylation polymorphism modifies toxicity of a number of aromatic and hydrazine drugs in humans [12]. Since NAT2 is also important in the metabolism of carcinogenic arylamines and HCAs, it is plausible that NAT2 polymorphism may modify individual cancer risks following exposures to these carcinogens. Molecular epidemiologic studies have investigated the relationship between NAT2 status and individual risk for various cancers including urinary bladder [11,13, 14], colorectal [15,16], breast [17–20], prostate [21], pancreas [22], lung [23], and non-Hodgkin lymphoma [24]. However, except for the smoking-related urinary bladder cancer, these studies have been unable to establish a consistent association between the acetylator status and human cancers. It needs to be noted, however, that because of the difficulties in determining phenotype, most of these studies used NAT2 phenotypes inferred from genotyping results to group their subjects. Detection of certain SNPs indicates a slow acetylator allele and a person with two such alleles is designated a slow acetylator. Since different NAT2 genotyping methods and genotype-phenotype interpretations are applied in molecular epidemiological studies, the validity of the conclusions derived from these studies may be compromised by the uncertainty of NAT2 genotype-phenotype relationships [25].
In order to investigate these relationships, efforts have been made to assign phenotypes for all the identified genotypes. The in vivo caffeine metabolite test may be influenced by environmental and other genetic factors [26]. Also, the metabolism for caffeine may not be the same as for carcinogens. As an alternative, NAT2 alleles can be cloned and expressed in vitro to determine the corresponding phenotypes. Previous studies have described human NAT2 alleles cloned and expressed in bacteria [27] and yeast [28]. However, discrepancies between these data and some recent studies using a caffeine metabolite test [29] , raise concerns regarding differences between mammals and prokaryotes or single cell eukaryotes, and evoke the importance of investigating haplotypes. Although several alleles were expressed in mammalian cells [30], a comprehensive analysis of NAT2 SNPs and haplotypes in mammalian cells has not been carried out. COS-1 cells, a cell line derived from African green monkey kidney epithelial cells, possess no detectable level of endogenous NAT2 activity. The reference human NAT2 (NAT2 4) recombinantly expressed in COS-1 cells exhibited the same stability, chromatographic behavior, electrophoretic mobility and kinetic characteristics as the native liver NAT2 4 [31], showing that the COS-1 cell line is an ideal expression system to investigate the genotype-phenotype relationship of human NAT2, especially for chemicals that can not be tested in vivo, including certain human carcinogens that are of particular interest.
In the present study, ten SNPs and four common haplotypes of human NAT2 were functionally characterized in COS-1 cells. The effects of each SNP and haplotype on the N- acetylation of SMZ and the O-acetylation of N-hydroxy-ABP and N-hydroxy-PhIP were tested. Possible molecular mechanisms underlying the effects also were investigated.
Materials and methods
Plasmid Construction
Human NAT2 mammalian expression plasmids were constructed as described previously [32]. In brief, the coding region of NAT2*4 was inserted between a cytomegalovirus promoter and a bovine growth hormone polyadenylation sequence of pcDNA5/FRT mammalian expression vector (Invitrogen, Carlsbad, CA). Individual SNPs and haplotypes were introduced into the NAT2*4 plasmid by a PCR-based site-directed mutagenesis method described previously [32] using primers listed in Table I. Correct sequences of the inserts were further confirmed by automated DNA sequencing.
Table I.
Sequences of primers used to introduce SNPs
| Primer name | Sequencea | Amino Acid Change |
|---|---|---|
| 191A-for | 5’-CATTGTAAGAAGAAACCAGGGTGGGTGGTGTCTC-3’ | R64Q |
| 191A-rev | 5’-GAGACACCACCCACCCTGGTTTCTTCTTACAATG-3’ | |
| 282C-for | 5’-GGAGGGTATTTTTATATCCCTCCAGTTAAC-3’ | None |
| 282C-rev | 5’-GTTAACTGGAGGGATATAAAAATACCCTCC-3’ | |
| 341C-for | 5’-CTCCTGCAGGTGACCACTGACGGCAGGAATTAC-3’ | I114T |
| 341C-rev | 5’-GTAATTCCTGCCGTCAGTGGTCACCTGCAGGAG-3’ | |
| 364A-for | 5’-GCAGGAATTACATTGTCAATGCTGGGTCTGGAAGC-3’ | D122N |
| 364A-rev | 5’-GCTTCCAGACCCAGCATTGACAATGTAATTCCTGC-3’ | |
| 411T-for | 5’-GTGGCAGCCTCTAGAATTTATTTCTGGGAAGGATCAG-3’ | L137F |
| 411T-rev | 5’-CTGATCCTTCCCAGAAATAAATTCTAGAGGCTGCCAC-3’ | |
| 481T-for | 5’-GAGAGAGGAATCTGGTACTTGGACCAAATCAGGAGAGAG-3’ | None |
| 481T-rev | 5’-CTCTCTCCTGATTTGGTCCAAGTACCAGATTCCTCTCTC-3’ | |
| 499A-for | 5’-GGACCAAATCAGGAGAAAGCAGTATATTAC-3’ | E167K |
| 499A-rev | 5’-GTAATATACTGCTTTCTCCTGATTTGGTCC-3’ | |
| 590A-for | 5’-TTACGCTTGAACCTCAAACAATTGAAGATTTTGAG-3’ | R197Q |
| 590A-rev | 5’-CTCAAAATCTTCAATTGTTTGAGGTTCAAGCGTAA-3’ | |
| 803G-for | 5’-GAGGTTGAAGAAGTGCTGAGAAATATATTTAAGATTTCCTTGGGG-3 | K268R |
| 803G-rev | 5’-CCCCAAGGAAATCTTAAATATATTTCTCAGCACTTCTTCAACCTC-3’ | |
| 857A-for | 5’-CCAAACCTGGTGATGAATCCCTTACTATTTAG-3’ | G286E |
| 857A-rev | 5’-CTAAATAGTAAGGGATTCATCACCAGGTTTGG-3’ |
The mismatched nucleotide(s) used for mutagenesis is underlined for each primer.
Cell Culture, Transfection, and Lysate Preparation
COS-1 cells (CRL-1650, ATCC) were maintained in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% fetal bovine serum and 4 mM L-glutamine. Transfection and lysate preparation were performed as described [32]. Briefly, 1.2 x 106 cells were transfected with 3.6 μg NAT2 plasmid together with 0.4 μg pCMV-sport-βgal plasmid (Invitrogen) using Lipofectamine and Plus reagent (Invitrogen). Cells were harvested 24–48 hrs later and subjected to RNA isolation (described below) or lysis by freeze-thawing.
Measurement of N-acetylation Activity
Sulfamethazine (SMZ) N-acetylation activity was measured with a high performance liquid chromatography method described previously [33]. Cell lysate was incubated with 1 mM acetyl coenzyme A (AcCoA) and 500 μM SMZ at 37°C. Water was substituted for AcCoA as the negative control. SMZ and N-acetyl-SMZ were separated by HPLC and detected by absorbance at 260 nm. Total protein in cell lysate was measured by the Bradford assay using the Bio-Rad protein assay reagent (Bio-Rad, Hercules, CA). β-galactosidase activity was measured using the ortho-nitrophenyl- β-D-galactopyranoside method as described previously [32]. To determine the apparent Km and Vmax for SMZ, reactions were run in the presence of a range of SMZ (10 to 5000 μM) and fixed concentration of AcCoA (1 mM). Similarly, fixed concentration of SMZ (500 μM) and a range of AcCoA (0.1 to 2000 μM) were used to determine the apparent Km and Vmax for AcCoA. Michaelis-Menten kinetic constants were calculated by linear regression of Eadie-Hofstee plots. For heat inactivation assays cell lysates were incubated at either 37°C or 50°C for two hrs. A sample of lysate was removed every 15 min and NAT2 activity was assayed immediately at 37°C. The first order inactivation rate constant was determined by linear regression.
Measurement of O-acetylation Activity
Metabolic activation of N-OH-ABP and N-OH-PhIP via O-acetylation was measured as described previously [33] with minor modifications. Briefly, forty μl of cell lysate was incubated at 37°C with 1 mM N-OH-ABP or 1 mM N-OH-PhIP in the presence of 1 mM AcCoA and 1 mg/ml 2-deoxyguanosine. The reaction was stopped by adding 100 μl of water saturated ethyl acetate. Following centrifugation at 13,000 x g for 8 min at room temperature, 50 μl of the organic phase (upper layer) was carefully transferred to a 1.5 ml centrifuge tube and then put in a speed-vac for 5 min to evaporate the solvent. The resulting lyophilizate was resuspended and dissolved in 100 μl of 10% acetonitrile. The adducts formed in the reactions, dG-C8-ABP or dG-C8-PhIP, were separated by HPLC (Beckman Gold HPLC system) with a Supelco Discovery C18 (Sigma/Aldrich, St. Louis, MO) 25 cm x 4 mm column using a mobile phase of 20 mM sodium percholorate (pH 2.5) (solvent A) and acetonitrile (solvent B). The elution was conducted with a linear gradient from 80% to 50% solvent A over 3 min at a flow rate of 1 ml/min. The dG-C8-ABP and dG-C8-PhIP adducts were detected by a UV detector and quantified by the absorbance at 300 nm and 360 nm, respectively.
Measurement of NAT2 Protein by Western Blot
Cell lysates (20–50 μg of total protein in the supernatant) were subjected to SDS-PAGE on 12% Tris-Glycine gels (Cambrex, Walkersville, MD) and transferred onto nitrocellulose membranes (Amersham, Arlington Heights, IL). The blots were subsequently treated with blocking buffer (20 mM Tris-HCl, 500 mM NaCl, 0.5% Tween 20, 5% non fat dry milk) at room temperature for 1 hr and then probed with anti-human NAT2 primary antibody (1:2000 dilution) raised against a 13-amino acid epitope (residues 175–187; FLNSHLLPKKKHQ) of human NAT2 [28] or with anti-α-tubulin (Sigma). Membranes were then washed and incubated with goat anti-rabbit IgG (1: 50,000 dilution) and visualized with the West Pico chemiluminescent kit (Pierce Biotech. Inc, Rockford, IL).
Measurement of NAT2 mRNA
Total RNA was isolated using the RNeasy Mini Kit (Qiagen, Valencia, CA) from transfected COS-1 cells followed by a treatment with DNA-free DNase Treatment and Removal Reagent (Ambion, Austin, TX). cDNA was synthesized from 1 μg total RNA with oligo (dT18) primer using SuperScript III reverse transcriptase (Invitrogen). Real time quantitative reverse transcriptase-polymerase chain reaction (qRT-PCR) was performed as described previously [32]. Each sample was tested in duplicate with separate tube of African green monkey β-actin as the internal control. Results obtained from three independent experiments were used to calculate the means and standard deviations.
Statistical Analysis
Differences in catalytic activities or mRNA levels among NAT2 allozymes were tested for significance by one way analysis of variance followed by Bonferroni tests for multiple comparisons.
Results
Effect of Individual SNPs and Haplotypes on SMZ N-acetylation Activity
As shown in Figure 1, SMZ N-acetyltransferase activities were detected in COS-1 cells transfected with all variant NAT2 alleles except for the one possessing the G364A(D122N) SNP. Cells transfected with the empty plasmid did not possess measurable levels of SMZ N-acetylation activity (data not shown). The SMZ N-acetylation activity measured in cells transfected with the NAT2*4 allele was designated as the reference level for comparison with activities elicited by all the NAT2 variant alleles. All seven non-synonymous SNPs except A803G (K268R) significantly (p<0.01) reduced NAT2 catalytic activity to various degrees. Among these seven SNPs, G364A (D122N) reduced the enzyme activity below the detection limit (0.3 nmol/min/mg protein). A411T (L137F) exhibited the next largest reduction (>99%) in enzyme activity. Three SNPs, G191A (R64Q), T341C (I114T) and G590A (R197Q), reduced SMZ N-acetyltransferase activity about 85%. G499A (E167K) and G857A (G286E) caused more moderate (40% – 50%) reductions in the SMZ N-acetylation activity. Unlike other non-synonymous SNPs, A803G(K268R) exhibited no significant effect on the SMZ N-acetylation activity (p>0.05). Both allozymes with synonymous SNPs C282T and C481T had SMZ N-acetylation activities comparable to the reference NAT2*4.
Figure 1.
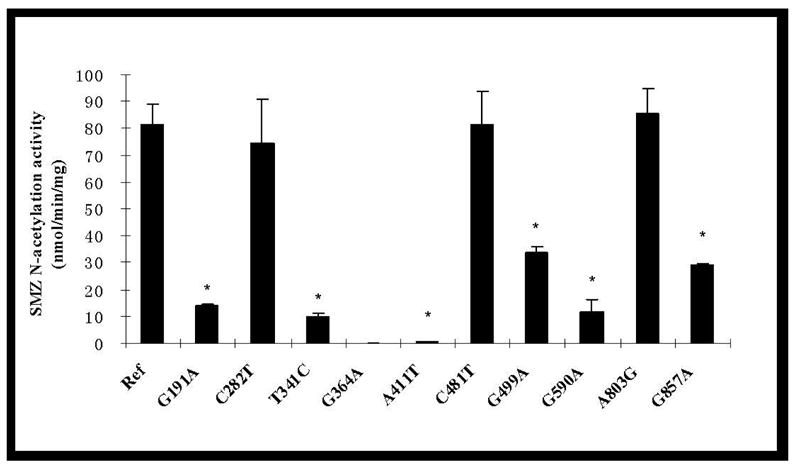
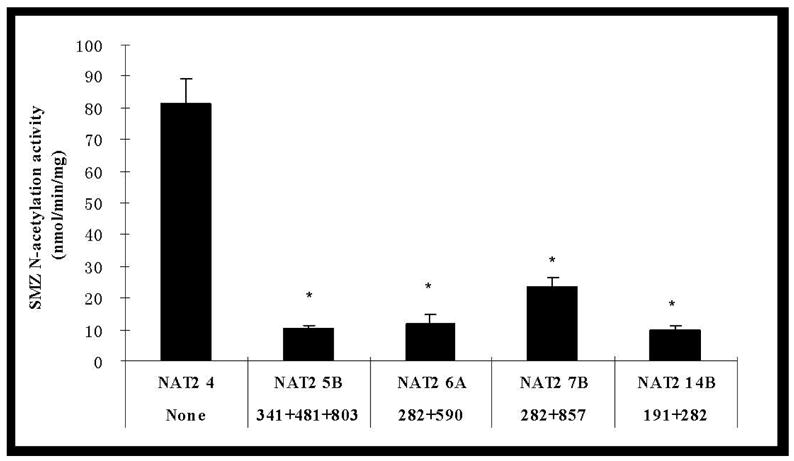
SMZ N-acetylation activities in COS-1 cells expressing NAT2 alleles possessing different SNPs (top) and haplotypes (bottom). Each error bar represents the standard deviation for transfections performed in triplicate on different days. *: Significantly (p<0.01) lower than the reference (Ref) NAT2 4. Activity in COS-1 cells transfected with NAT2 allele possessing G364A was below the detection limit (0.3 nmol/min/mg).
All four NAT2 variant haplotypes were associated with decreased SMZ N-acetylation activity (Figure 1). NAT2*5B (T341C+C282T+ A803G), NAT2*6A (G590A + C282T) and NAT2*14B (G191A+C282T) reduced N-acetylation activity over 85%. More modest (70%) reduction was observed for the cells transfected with NAT2*7B (G857A+C282T).
Effect of Individual SNPs and Haplotypes on O-acetylation Activity
No ABP-deoxyguanosine -adduct was detected in reactions without AcCoA, while a small amount of PhIP- deoxyguanosine -adduct was formed in such reactions. A very low level of basal O-acetylation activity for both N-OH-ABP and N-OH-PhIP was detected in mock-transfected COS-1 cells and was deducted from the activity measured in NAT2-transfected cells. As shown in Figure 2, C282T (synonymous), C481T (synonymous) and A803G (K268R) did not significantly affect the bioactivation of N-OH-ABP or N-OH-PhIP (p>0.05). G191A (R64Q), T341C (I114T), G364A (D122N), A411T (L137F) and G590A (R197Q) each reduced O-acetylation activity towards N-OH-ABP and N-OH-PhIP. G857A (G286E) and G499A (E167K), however, reduced the O-acetylation of N-OH-ABP but not N-OH-PhIP. All four variant haplotypes (NAT2*5B, NAT2*6A, NAT2*7B, and NAT2*14B) reduced N-OH-ABP O-acetylation activity. Similar reductions were observed in N-OH-PhIP O-acetylation activity with the notable exception of NAT2*7B, with which no significant reduction in the O-acetylation of N-OH-PhIP (p>0.05) was observed (Figure 2). N-OH-ABP O-acetylation activities correlated highly (r2=0.90) with SMZ N-acetylation for all NAT2 allozymes. N-OH-PhIP O-acetylation and SMZ N-acetylation activities correlated highly (r2=0.89) across allozymes except those possessing G857A(G286E) or G499A(E167K).
Figure 2.
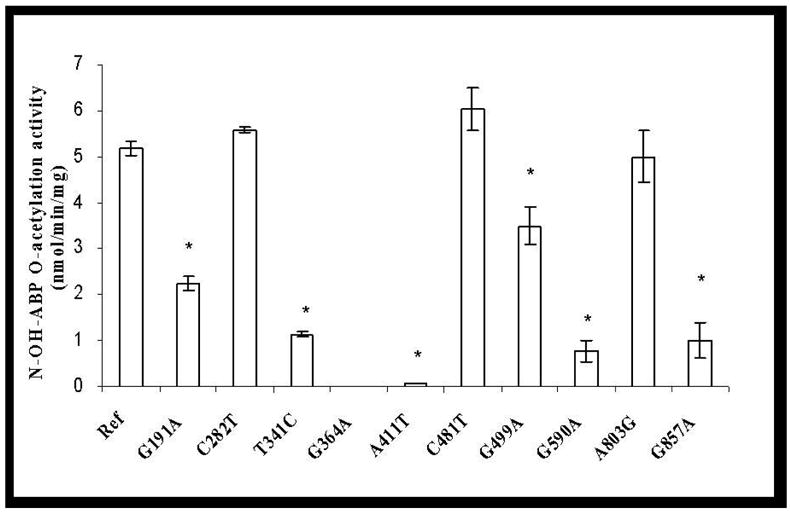
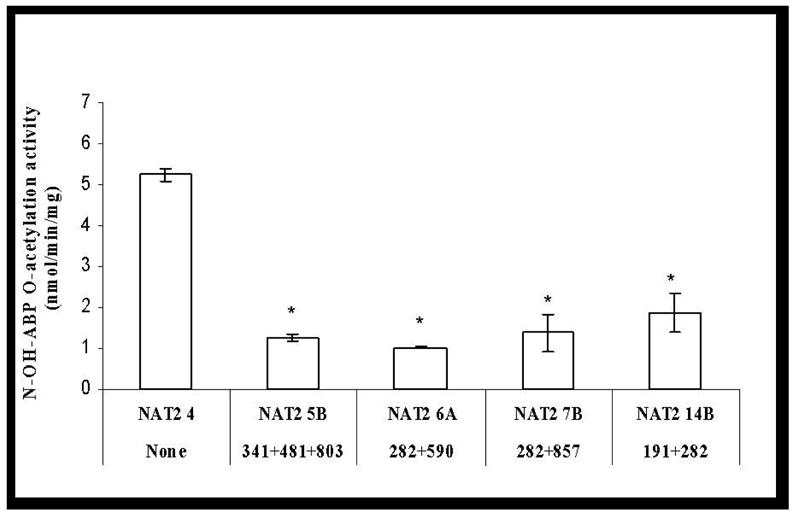
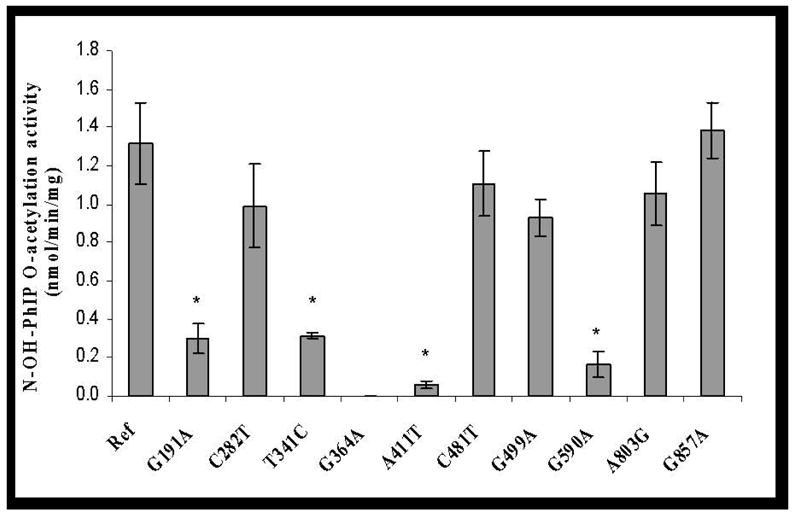
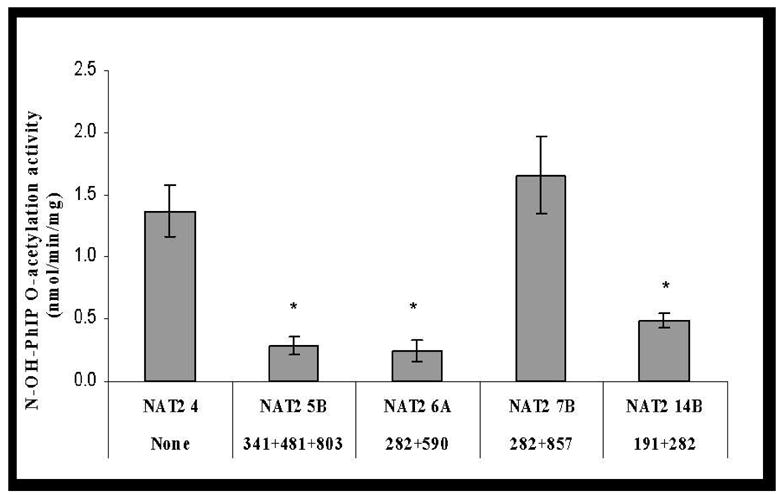
Effects of NAT2 SNPs and haplotypes on the O-acetylation of N-OH-ABP (open bars) and N-OH-PhIP (solid bars). Each error bar represents the standard deviation for transfections performed in triplicate. *: Significantly (p<0.01) lower than the reference (Ref) NAT2 4. Activity in COS-1 cells transfected with NAT2 allele possessing G364A was below the detection limit (0.05 nmol/min/mg).
Effect of Individual SNPs and Haplotypes on N-acetylation Kinetics
Michaelis-Menten kinetic constants for different NAT2 allozymes expressed in COS-1 cells are shown in Table II. We could not determine the kinetic parameters for NAT2s encoded by alleles possessing G364A and A411T because of their undetectable or extremely low enzymatic activities. SNPs C282T, C481T and A803G changed neither apparent VmaxSMZ nor VmaxAcCoA. In contrast, all SNPs associated with slow acetylation phenotype reduced both apparent VmaxSMZ and VmaxAcCoA. SMZ N-acetylation activities were highly correlated to VmaxSMZ (r2=0.97) and VmaxAcCoA (r2=0.97) for these allozymes. Allozymes encoded by all four NAT2 variant haplotypes exhibited decreased VmaxSMZ and VmaxAcCoA compared to that of NAT2 4.
Table II.
Apparent Vmax and Km of SMZ and AcCoA for NAT2 allozymes expressed in COS-1 cellsa
| NAT2 allozymes | VmaxSMZ | KmSMZ |
|---|---|---|
| NAT2 4 | 158 ± 34 | 601 ± 141 |
| NAT2-191A | 22.9 ± 10.7 * | 643 ± 127 |
| NAT2-282T | 137 ± 14 | 583 ± 144 |
| NAT2-341C | 30.5 ± 11.7 * | 673 ± 48 |
| NAT2-481T | 126 ± 28 | 708 ± 47 |
| NAT2-499A | 82.2 ± 10.2 * | 765 ± 56 |
| NAT2-590A | 16.4 ± 2.6 * | 529 ± 42 |
| NAT2-803G | 159 ± 31 | 550 ± 115 |
| NAT2-857A | 32.8 ± 3.3 * | 74.2 ± 8.9 * |
| NAT2 5B | 31.9 ± 5.9 * | 640 ± 78 |
| NAT2 6A | 26.9 ± 7.9 * | 480± 49 |
| NAT2 7B | 20.7 ± 4.5 * | 90.7 ± 8.1 * |
| NAT2 14B | 22.0 ± 4.0 * | 778 ± 119 |
| NAT2 allozymes | VmaxAcCoA | KmAcCoA |
|
| ||
| NAT2 4 | 123 ± 25 | 534 ± 37 |
| NAT2-191A | 17.9 ± 2.4 * | 413 ± 47 |
| NAT2-282T | 104 ± 22 | 476± 8 |
| NAT2-341C | 29.9 ± 3.0 * | 553 ± 47 |
| NAT2-481T | 94.0 ± 4.2 | 531 ± 22 |
| NAT2-499A | 55.8 ± 19.4 * | 437 ± 71 |
| NAT2-590A | 12.9 ± 2.3 * | 482± 35 |
| NAT2-803G | 119 ± 9 | 445 ± 52 |
| NAT2-857A | 57.5 ± 10.7 * | 1280 ± 259 * |
| NAT2 5B | 12.4 ± 2.4 * | 430± 125 |
| NAT2 6A | 14.8 ± 2.3 * | 577 ± 60 |
| NAT2 7B | 46.5 ± 9.0 * | 1235 ± 92 * |
| NAT2 14B | 12.5 ± 0.9 * | 309 ± 81 |
COS-1 cells were transfected with NAT2 alleles possessing different SNPs or haplotypes in triplicate. The data represents the mean and standard deviation of three apparent Vmax (nmol/min/mg) or Km (micromolar) calculations determined independently.
: Significantly different from that of NAT2 4, p<0.05.
As shown in Table II, none of the individual SNPs significantly changed the apparent Km except for G857A(G286E). Compared to NAT2 4, the variant containing this SNP displayed an 8-fold lower apparent Km to SMZ but a 3-fold higher apparent Km to AcCoA. The haplotype NAT2*7B (containing G857A and C282T) exhibited similar effects as observed with G857A(G286E) alone.
Effect of Individual SNPs on NAT2 mRNA Level
The levels of NAT2 mRNA in transfected COS-1 cells were measured by qRT-PCR. None of the SNPs had a significant (p>0.05) effect on steady state NAT2 mRNA levels (Figure 3).
Figure 3.
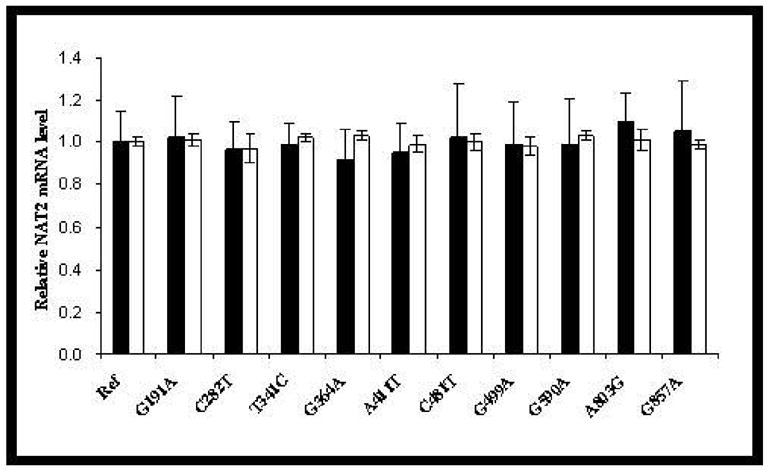
Steady state mRNA levels of NAT2 (solid bars) and β-actin (open bars) in COS-1 cells transfected with NAT2 alleles possessing different SNPs. NAT2 mRNA was first normalized to internal β-actin mRNA and then expressed relative to the NAT2 mRNA level in COS-1 cells transfected with NAT2*4 (defined as 1.0). Each bar shows the mean and the standard deviation of the relative level based on three independent transfections. None of the NAT2 mRNA levels differed significantly (p>0.05) from NAT2*4.
Effect of SNPs on NAT2 Thermostability
Heat inactivation kinetics was studied to compare the intrinsic stability of different NAT2 allozymes expressed in COS-1 cells. The effects of SNPs on the heat inactivation half-life are shown in Figure 4. NAT2 4 exhibited a half-life about six hours at 37°C and one hour at 50°C. Those SNPs which did not affect SMZ N-acetylation activity did not affect NAT2 thermostability at either 37°C or 50°C. Among SNPs that reduced SMZ N-acetylation activity, T341C(I114T), A411T(L137F) and G499A(E167K) also did not affect NAT2 thermostability. In contrast, G191A(R64Q), G590A(R197Q) and G857A(G286E) significantly reduced NAT2 inactivation half-lives by 3-fold, 1.7-fold and 3-fold lower, respectively at 37°C. At 50°C, G590A(R197Q) reduced the heat inactivation half-life 4-fold while more striking decreases in NAT2 heat inactivation half-lives were caused by G191A (21-fold) and G857A (26-fold).
Figure 4.
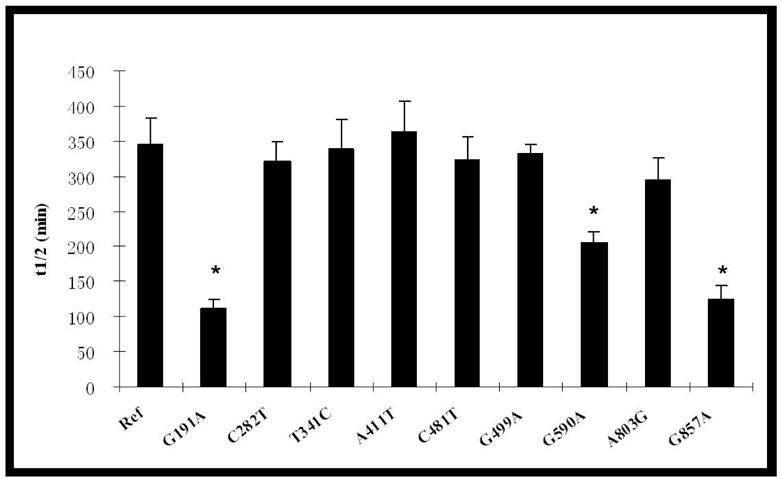
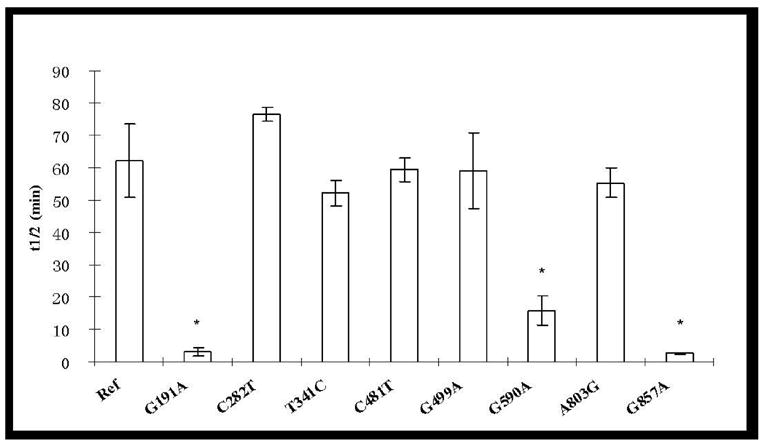
Thermostability of NAT2 allozymes encoded by alleles possessing different SNPs. Heat inactivation assays were performed at 37°C (top) and 50°C (bottom). Due to the extremely low catalytic activity of A411T (Figure 1), thermostability for this variant was not determined at 50°C. Each bar represents the mean and standard deviation of three calculated half-lives using lysates of COS-1 cells transfected separately. *: Significantly (p<0.01) lower than the reference (Ref) NAT2 4.
Effect of NAT2 SNPs and Variant Haplotypes on NAT2 Protein Level
Immunoreactive NAT2 in COS-1 cells transfected with different NAT2 alleles were detected by western blot using an antiserum raised against a 13-amino acid epitope of human NAT2 for which no SNPs have been identified. As shown in Figure 5, C282T(silent), C481T(silent) and A803G(K268R) had no effect on immunoreactive NAT2 protein levels. However, each of the SNPs and variant haplotypes associated with reduced SMZ N-acetyltransferase activity reduced the levels of immunoreactive NAT2 protein.
Figure 5.
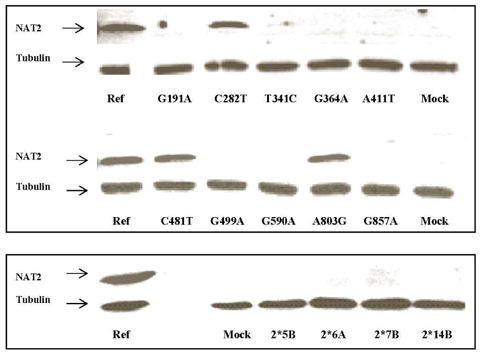
Western blot of NAT2 allozymes expressed in COS-1 cells transfected with plasmids containing NAT2*4 and variant alleles possessing different SNPs (top) and haplotypes (bottom). Immunoblots of NAT2 and α-tubulin are indicated by arrows. Mock: COS-1 cells transfected with the mock pcDNA5/FRT plasmid.
Discussion
All NAT2 SNPs causing reduced SMZ N-acetylation activities were related to reductions in immunoreactive NAT2 protein with unchanged mRNA level. The reductions in activity were not directly proportional to reductions in immunoreactive protein, as measured by Western blot, probably because of the low sensitivity of the NAT2 anti-serum, or differences in substrate affinity between variants. None of these SNPs introduced significant changes in mRNA secondary structure according to the prediction by MFold program [34]. The codon usage frequency for all the changed amino acids are similar between human and African green monkey (www.kazusa.or.jp/codon/), indicating that the decrease in protein level is not due to difference in codon bias between the two species. We previously showed that no protein aggregation is involved in the mechanism for A411T(L137F), the SNP that reduces enzyme activity to the highest extent second only to G364A(D122N) in mammalian cells [35], although a recent paper reported aggregation in several hamster NAT2 variants and proposed it as the mechanism of reduced activity for some human NAT1 variants [36]. The possible mechanisms of each human NAT2 SNP and haplotype are discussed below.
G191A(R64Q) and G590A(R197Q)
Nonsynonymous SNPs, G191A(R64Q) and G590A(R197Q), either alone or in combination with other SNPs as in the NAT2*14 and NAT2*6 haplotype clusters, resulted in slow acetylator phenotype. Neither of these two SNPs changed substrate affinity, but both reduced thermostability and the level of soluble NAT2 protein. Our findings suggest that G590A(R197Q) and G191A(R64Q) share a common mechanism for slow acetylator phenotype. Site-directed mutagenesis and molecular modeling have suggested that Arg64 is structurally important. The interaction between Arg64 and Glu39 helps to maintain the optimal conformation of Cys68 at the catalytic site [37]. Our study confirmed that G191A(R64Q) had a destabilizing effect on NAT2, consistent with previous studies conducted on NAT2 expressed in bacteria [38] and yeast [28]. Similarly, another substitution of Arg64, R64W, also caused reduction in soluble N-acetyltransferase protein in both hamster NAT2 and human NAT1 [36]. However, changed surface property, instead of thermostability, was proposed for the mechanism of aggregation and constitutive ubiquitylation observed in these variant proteins.
T341C(I114T)
This is the signature SNP for alleles in the NAT2*5 cluster. Our study showed that this SNP alone was sufficient to cause slow acetylator phenotype. This conclusion differs from a previous study in which an additional silent base substitution (C481T) was required for reduction in N-acetylation [30], but is consistent with other studies conducted in bacteria [38] and yeast [28] expression systems. The present study also showed that T341C(I114T) and the NAT2*5B haplotype resulted in reduction in O-acetylation of two carcinogens, N-OH-ABP and N-OH-PhIP. Unlike G191A(R64Q) and G590A(R197Q), T341C(I114T) did not reduce NAT2 thermostability. Previous studies have proposed enhanced protein degradation as the mechanism for the reduced NAT2 protein and activity, and showed the importance of hydrophobicity at this position [32].
G857A(G286E)
Unlike other SNPs, G857A(G286E) significantly reduced apparent Km for SMZ and significantly increased apparent Km for AcCoA. It is also of interest that G857A(G286E) reduced the O-acetylation of N-OH-ABP but not N-OH-PhIP. According to a molecular docking based on a human NAT2 structural model, Gly286 is not among the amino acids directly interacting with N-OH-PhIP [39]. Gly286 lies in the C-terminus region, which is highly variable among N-acetyltransferases from different species. A C-terminal undecapeptide truncation mutant of N-acetyltransferase from Salmonella typhimurium is able to hydrolyze AcCoA without the presence of arylamine substrate. This mutant displays a lower Km toward arylamine substrates but higher Km toward AcCoA [40], a finding similar to what we observed for human NAT2 7 variant proteins. Even though the C-terminus secondary structure of prokaryote N-acetyltransferase is different from that of eukaryotes [41], and it is not clear how binding with AcCoA affects binding with the acetyl acceptor or vice versa, we observed that the Kms for both acetyl donor and acceptor substrates were changed. It has been proposed that the C-terminal domain has a shielding effect on bulky arylamine substrates [42]. An amino acid substitution in this region might influence the accessibility of some substrates but not others, leading to substrate-dependent activity changes observed here and in previous studies [43,44]. Kinetic studies have shown that G857A(G268E) reduces apparent Km for some N-acetylation substrates (such as SMZ and dapsone) but not for others (such as 2-aminofluorene and isoniazid) [43]. Based on these findings, the phenotype of NAT2 7 alleles may be both substrate-dependent and concentration-dependent.
Since Gly286 seems to be on the surface of the protein, the overall folding is not likely to be changed by this polymorphism [39]. However, studies on prokaryote N-acetyltransferases suggest that the primary and secondary structures in the C-terminal domain could be important to stability [45], thus accounting for the reduced thermostability and protein related to this SNP. Proteasomal degradation of human NAT1, an enzyme highly homologous to NAT2, is prevented by acetylation of the active site cysteine [46]. Binding with AcCoA is essential for maintaining the structural integrity of NAT1, and very possibly of NAT2 as well.
G499A(E167K)
Like G857A(G286E), this SNP also led to moderate but significant reduction in SMZ N-acetylation activity and decreased the O-acetylation of N-OH-ABP but not N-OH-PhIP. However, G499A(E167K) did not alter the apparent Km towards SMZ or AcCoA, nor did it change the protein thermostability. Based on multiple sequence alignments and computational modelling [39,47], Glu167 lies adjacent to a loop region that is only present in eukaryote N-acetyltransferases. These findings suggest that Glu167 is not functionally essential, but more likely to be important to the conformation of this loop and thus influences interaction with some substrates.
G364A (D122N) and A411T (L137F)
We recently reported that these two SNPs confer slow acetylator phenotype [35], but their effects on O-acetylation of N-hydroxylated-arylamines and -heterocyclic amines have not been reported. In the present study, G364A(D122N) and A411T(L137F) caused the largest reduction in both N-acetylation and O-acetylation activities among all SNPs tested. G364A(D122N) replaces the catalytic aspartate with an asparagine, thus disrupting the catalytic core and eliminating activity. Previous studies suggest that both G364A(D122N) and A411T (L137F) enhance protein degradation [35].
C282T(silent) and C481T(silent)
Synonymous SNP C282T exists either alone as in NAT2*13 allele or in combination with many other SNPs. NAT2*13 is usually defined as a rapid acetylator allele based on recombinant expression in bacterial and yeast cells [27,28,38]. However, caffeine phenotypic tests on Caucasian populations previously suggested that NAT2*13 was related to slow acetylator phenotype [48, 49]. In our study, C282T had no significant effect on mRNA, protein or catalytic activity for N- or O-acetylation. Synonymous SNP C481T was first identified coexisting with T341C(I114T) in the M1/NAT2*5B allele and was suggested to contribute to the corresponding slow acetylator phenotype [30]. C481T is often linked to T341C and in many genotyping methods, restriction enzyme KpnI is applied to detect the existence of C481T as a tag for T341C. However, there are cases when C481T exists either alone (NAT2*11) or in combination with A803G (NAT2*12C). In the present study, C481T had no effect on NAT2 mRNA, protein or catalytic activity.
A803G(K268R)
This SNP results in a conserved amino acid substitution from lysine to arginine. A803G(K268R) is found in combination with other SNPs in NAT2*5, *6, *12 and *14 haplotypes. A803G(K268R) is the sole SNP in the NAT2*12A allele, which has been identified as a rapid acetylator allele following recombinant expression in bacteria and yeast [27,28,38]. This assignment was challenged by two studies on Caucasian populations [48,49] suggesting that A803G(K268R) conferred slow N-acetylation activity in vivo. Both these studies applied a genotyping method in which C481T was detected as a surrogate of T341C. NAT2*5C (T341C + A803G) haplotype could therefore be misclassified as a NAT2*12A haplotype and misclassified as a slow acetylator phenotype. In the present study, we confirmed by mammalian expression that A803G (K268R) did not affect NAT2 mRNA, protein or catalytic activity.
Conclusions
This is the first comprehensive functional study of NAT2 genetic polymorphisms carried out in mammalian cells. Seven SNPs (G191A, T341C, G364A, A411T, G499A, G590A, and G857A) reduced SMZ-acetylation activity to various extents. Although the mechanisms may differ, each of these SNPs reduced NAT2 protein, consistent with slow acetylator phenotype secondary to reduction of NAT2 protein. This supports previous findings in human liver samples [50]. The functional effects of the individual SNPs did not change in haplotype combinations. Our results provided further evidence that the A803G is associated with rapid acetylator phenotype. A particularly interesting observation was that the functional effects of G857A were substrate-dependent. Since numerous past, ongoing, and future molecular epidemiological investigations assess associations between NAT2 SNPs and cancer risk, understanding of the functional consequences of these SNPs is critical to the interpretations of these studies.
Whereas HCAs such as PhIP do not undergo significant N-acetylation, O-acetylation has an important role in the bioactivation of N-hydroxylated metabolites of both aromatic and heterocyclic amine carcinogens. For the purpose of evaluating individualized cancer susceptibility upon carcinogen exposure, deduction of O-acetylation capacity of study subjects from their genotype/N-acetylation phenotype is needed. For most alleles, SMZ N-acetylation activity correlated very well with N-OH-ABP and N-OH-PhIP O-acetylation activity, suggesting that NAT2 genotype could be used to deduce O-acetylation phenotype. However, a notable exception was observed for the O-acetylation of N-OH-PhIP by NAT2s encoded by haplotypes possessing G857A(K268R), either alone or in combination with C282T. Thus, while this SNP reduces N-acetylation reflective of slow acetylator phenotype for arylamines such as SMZ, nevertheless its effect on the O-acetylation differed between N-OH-ABP, where the phenotype was also reduced, and N-OH-PhIP, in which the phenotype was not reduced. Due to the inconsistent effect and the changes in apparent Km with different substrates, the phenotype of individuals with NAT2*7 haplotypes need to be interpreted more carefully. Our data suggest that both substrate and exposure concentration may modify phenotype of individuals with this haplotype.
Acknowledgments
This work was partially supported by USPHS grant CA-34627, a dissertation research award from the Susan G. Komen Breast Cancer Foundation (DISS0403147); and a grant from the Kentucky Lung Cancer Research Program. Portions of this work constituted partial fulfillment for the award of the PhD in pharmacology and toxicology to Yu Zang at the University of Louisville.
References
- 1.Hanna PE. Metabolic activation and detoxification of arylamines. Curr Med Chem. 1996;3:195–210. [Google Scholar]
- 2.Hein DW. Molecular genetics and function of NAT1 and NAT2: role in aromatic amine metabolism and carcinogenesis. Mutat Res. 2002;506–507:65–77. doi: 10.1016/s0027-5107(02)00153-7. [DOI] [PubMed] [Google Scholar]
- 3.Chen T, Mittelstaedt RA, Beland FA, Heflich RH, Moore MM, Parsons BL. 4-Aminobiphenyl induces liver DNA adducts in both neonatal and adult mice but induces liver mutations only in neonatal mice. Int J Cancer. 2005;117:182–7. doi: 10.1002/ijc.21173. [DOI] [PubMed] [Google Scholar]
- 4.Snyderwine EG, Venugopal M, Yu M. Mammary gland carcinogenesis by food-derived heterocyclic amines and studies on the mechanisms of carcinogenesis of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) Mutat Res. 2002;506–507:145–52. doi: 10.1016/s0027-5107(02)00161-6. [DOI] [PubMed] [Google Scholar]
- 5.Underwood PM, Zhou Q, Jaeger M, Reilman R, Pinney S, Warshawsky D, Talaska G. Chronic, topical administration of 4-aminobiphenyl induces tissue-specific DNA adducts in mice. Toxicol Appl Pharmacol. 1997;144:325–31. doi: 10.1006/taap.1997.8158. [DOI] [PubMed] [Google Scholar]
- 6.Snyderwine EG. Mammary gland carcinogenesis by 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine in rats: possible mechanisms. Cancer Lett. 1999;143:211–5. doi: 10.1016/s0304-3835(99)00127-5. [DOI] [PubMed] [Google Scholar]
- 7.Firozi PF, Bondy ML, Sahin AA, Chang P, Lukmanji F, Singletary ES, Hassan MM, Li D. Aromatic DNA adducts and polymorphisms of CYP1A1, NAT2, and GSTM1 in breast cancer. Carcinogenesis. 2002;23:301–6. doi: 10.1093/carcin/23.2.301. [DOI] [PubMed] [Google Scholar]
- 8.Probst-Hensch NM, Bell DA, Watson MA, Skipper PL, Tannenbaum SR, Chan KK, Ross RK, Yu MC. N-acetyltransferase 2 phenotype but not NAT1*10 genotype affects aminobiphenyl-hemoglobin adduct levels. Cancer Epidemiol Biomarkers Prev. 2000;9:619–23. [PubMed] [Google Scholar]
- 9.Williams JA, Stone EM, Fakis G, Johnson N, Cordell JA, Meinl W, Glatt H, Sim E, Phillips DH. N-Acetyltransferases, sulfotransferases and heterocyclic amine activation in the breast. Pharmacogenetics. 2001;11:373–88. doi: 10.1097/00008571-200107000-00002. [DOI] [PubMed] [Google Scholar]
- 10.Zhu J, Chang P, Bondy ML, Sahin AA, Singletary SE, Takahashi S, Shirai T, Li D. Detection of 2-amino-1-methyl-6-phenylimidazo[4,5-b]-pyridine-DNA adducts in normal breast tissues and risk of breast cancer. Cancer Epidemiol Biomarkers Prev. 2003;12:830–837. [PubMed] [Google Scholar]
- 11.Hein DW. N-acetyltransferase 2 genetic polymorphism: effects of carcinogen and haplotype on urinary bladder cancer risk. Oncogene. 2006;25:1649–58. doi: 10.1038/sj.onc.1209374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boukouvala S, Fakis G. Arylamine N-acetyltransferases: what we learn from genes and genomes. Drug Metab Rev. 2005;37:511–64. doi: 10.1080/03602530500251204. [DOI] [PubMed] [Google Scholar]
- 13.Gago-Dominguez M, Bell DA, Watson MA, Yuan JM, Castelao JE, Hein DW, Chan KK, Coetzee GA, Ross RK, Yu MC. Permanent hair dyes and bladder cancer: risk modification by cytochrome P4501A2 and N-acetyltransferases 1 and 2. Carcinogenesis. 2003;24:483–9. doi: 10.1093/carcin/24.3.483. [DOI] [PubMed] [Google Scholar]
- 14.Garcia-Closas M, Malats N, Silverman D, Dosemeci M, Kogevinas M, Hein DW, Tardon A, Serra C, Carrato A, Garcia-Closas R, Lloreta J, Castano-Vinyals G, Yeager M, Welch R, Chanock S, Chatterjee N, Wacholder S, Samanic C, Tora M, Fernandez F, Real FX, Rothman N. NAT2 slow acetylation, GSTM1 null genotype, and risk of bladder cancer: results from the Spanish Bladder Cancer Study and meta-analyses. Lancet. 2005;366:649–59. doi: 10.1016/S0140-6736(05)67137-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moslehi R, Chatterjee N, Church TR, Chen J, Yeager M, Weissfeld J, Hein DW, Hayes RB. Cigarette smoking, N-acetyltransferase genes and the risk of advanced colorectal adenoma. Pharmacogenomics. 2006;7:819–29. doi: 10.2217/14622416.7.6.819. [DOI] [PubMed] [Google Scholar]
- 16.Ognjanovic S, Yamamoto J, Maskarinec G, Le Marchand L. NAT2, meat consumption and colorectal cancer incidence: an ecological study among 27 countries. Cancer Causes Control. 2006;17:1175–82. doi: 10.1007/s10552-006-0061-3. [DOI] [PubMed] [Google Scholar]
- 17.Ambrosone CB, Freudenheim JL, Graham S, Marshall JR, Vena JE, Brasure JR, Michalek AM, Laughlin R, Nemoto T, Gillenwater KA, Shields PG. Cigarette smoking, N-acetyltransferase 2 genetic polymorphisms, and breast cancer risk. JAMA. 1996;276:1494–501. [PubMed] [Google Scholar]
- 18.Deitz AC, Zheng W, Leff MA, Gross M, Wen WQ, Doll MA, Xiao GH, Folsom AR, Hein DW. N-Acetyltransferase-2 genetic polymorphism, well-done meat intake, and breast cancer risk among postmenopausal women. Cancer Epidemiol Biomarkers Prev. 2000;9:905–10. [PubMed] [Google Scholar]
- 19.van der Hel OL, Peeters PHM, Hein DW, Doll MA, Grobbee DE, Kromhout D, de Mesquita HBB. NAT2 slow acetylation and GSTM1 null genotypes may increase postmenopausal breast cancer risk in long-term smoking women. Pharmacogenetics. 2003;13:399–407. doi: 10.1097/00008571-200307000-00005. [DOI] [PubMed] [Google Scholar]
- 20.Sillanpaa P, Hirvonen A, Kataja V, Eskelinen M, Kosma VM, Uusitupa M, Vainio H, Mitrunen K. NAT2 slow acetylator genotype as an important modifier of breast cancer risk. Int J Cancer. 2005;114:579–84. doi: 10.1002/ijc.20677. [DOI] [PubMed] [Google Scholar]
- 21.Rovito PM, Jr, Morse PD, Spinek K, Newman N, Jones RF, Wang CY, Haas GP. Heterocyclic amines and genotype of N-acetyltransferases as risk factors for prostate cancer. Prostate Cancer Prostatic Dis. 2005;8:69–74. doi: 10.1038/sj.pcan.4500780. [DOI] [PubMed] [Google Scholar]
- 22.Li D, Jiao L, Li Y, Doll MA, Hein DW, Bondy ML, Evans DB, Wolff RA, Lenzi R, Pisters PW, Abbruzzese JL, Hassan MM. Polymorphisms of cytochrome P4501A2 and N-acetyltransferase genes, smoking, and risk of pancreatic cancer. Carcinogenesis. 2006;27:103–11. doi: 10.1093/carcin/bgi171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gemignani F, Landi S, Szeszenia-Dabrowska N, Zaridze D, Lissowska J, Rudnai P, Fabianova E, Mates D, Foretova L, Janout V, Bencko V, Gaborieau V, Gioia-Patricola L, Bellini I, Barale R, Canzian F, Hall J, Boffetta P, Hung RJ, Brennan P. Development of lung cancer before the age of 50: the role of xenobiotic metabolism genes. Carcinogenesis. 2007;28:1287–93. doi: 10.1093/carcin/bgm021. [DOI] [PubMed] [Google Scholar]
- 24.Morton LM, Schenk M, Hein DW, Davis S, Zahm SH, Cozen W, Cerhan JR, Hartge P, Welch R, Chanock SJ, Rothman N, Wang SS. Screening and characterizing human NAT2 variants. Methods Enzymol. 2005;400:192–215. doi: 10.1016/S0076-6879(05)00011-X. [DOI] [PubMed] [Google Scholar]
- 25.Deitz AC, Rothman N, Rebbeck TR, Hayes RB, Chow WH, Zheng W, Hein DW, Garcia-Closas M. Impact of misclassification in genotype-exposure interaction studies: example of N-acetyltransferase 2 (NAT2), smoking, and bladder cancer. Cancer Epidemiol Biomarkers Prev. 2004;13:1543–6. [PubMed] [Google Scholar]
- 26.Le Marchand L, Sivaraman L, Franke AA, Custer LJ, Wilkens LR, Lau AF, Cooney RV. Predictors of N-acetyltransferase activity: should caffeine phenotyping and NAT2 genotyping be used interchangeably in epidemiological studies? Cancer Epidemiol Biomarkers Prev. 1996;5:449–55. [PubMed] [Google Scholar]
- 27.Hein DW, Doll MA, Rustan TD, Ferguson RJ. Metabolic activation of N-hydroxyarylamines and N-hydroxyarylamides by 16 recombinant human NAT2 allozymes: effects of 7 specific NAT2 nucleic acid substitutions. Cancer Res. 1995;55:3531–6. [PubMed] [Google Scholar]
- 28.Fretland AJ, Leff MA, Doll MA, Hein DW. Functional characterization of human N-acetyltransferase 2 (NAT2) single nucleotide polymorphisms. Pharmacogenetics. 2001;11:207–15. doi: 10.1097/00008571-200104000-00004. [DOI] [PubMed] [Google Scholar]
- 29.Bolt HM, Selinski S, Dannappel D, Blaszkewicz M, Golka K. Re-investigation of the concordance of human NAT2 phenotypes and genotypes. Arch Toxicol. 2005;79:196–200. doi: 10.1007/s00204-004-0622-8. [DOI] [PubMed] [Google Scholar]
- 30.Blum M, Demierre A, Grant DM, Heim M, Meyer UA. Molecular mechanism of slow acetylation of drugs and carcinogens in humans. Proc Natl Acad Sci U S A. 1991;88:5237–41. doi: 10.1073/pnas.88.12.5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grant DM, Blum M, Beer M, Meyer UA. Monomorphic and polymorphic human arylamine N-acetyltransferases: a comparison of liver isozymes and expressed products of two cloned genes. Mol Pharmacol. 1991;39:184–91. [PubMed] [Google Scholar]
- 32.Zang Y, Zhao S, Doll MA, States JC, Hein DW. The T341C (Ile114Thr) polymorphism of N-acetyltransferase 2 yields slow acetylator phenotype by enhanced protein degradation. Pharmacogenetics. 2004;14:717–23. doi: 10.1097/00008571-200411000-00002. [DOI] [PubMed] [Google Scholar]
- 33.Leff MA, Fretland AJ, Doll MA, Hein DW. Novel human N-acetyltransferase 2 alleles that differ in mechanism for slow acetylator phenotype. J Biol Chem. 1999;274:34519–22. doi: 10.1074/jbc.274.49.34519. [DOI] [PubMed] [Google Scholar]
- 34.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–15. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zang Y, Zhao S, Doll MA, States JC, Hein DW. Functional characterization of the A411T (L137F) and G364A (D122N) genetic polymorphisms in human N-acetyltransferase 2. Pharmacogenet Genomics. 2007;17:37–45. doi: 10.1097/01.fpc.0000236325.73186.2c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu F, Zhang N, Zhou X, Hanna PE, Wagner CR, Koepp DM, Walters KJ. Arylamine N-acetyltransferase aggregation and constitutive ubiquitylation. J Mol Biol. 2006;361:482–492. doi: 10.1016/j.jmb.2006.06.029. [DOI] [PubMed] [Google Scholar]
- 37.Rodrigues-Lima F, Dupret JM. 3D model of human arylamine N-acetyltransferase 2: structural basis of the slow acetylator phenotype of the R64Q variant and analysis of the active-site loop. Biochem Biophys Res Commun. 2002;291:116–23. doi: 10.1006/bbrc.2002.6414. [DOI] [PubMed] [Google Scholar]
- 38.Hein DW, Ferguson RJ, Doll MA, Rustan TD, Gray K. Molecular genetics of human polymorphic N-acetyltransferase: enzymatic analysis of 15 recombinant wild-type, mutant, and chimeric NAT2 allozymes. Hum Mol Genet. 1994;3:729–34. doi: 10.1093/hmg/3.5.729. [DOI] [PubMed] [Google Scholar]
- 39.Lau EY, Felton JS, Lightstone FC. Insights into the O-acetylation reaction of hydroxylated heterocyclic amines by human arylamine N-acetyltransferases: a computational study. Chem Res Toxicol. 2006;19:1182–90. doi: 10.1021/tx0600999. [DOI] [PubMed] [Google Scholar]
- 40.Mushtaq A, Payton M, Sim E. The COOH terminus of arylamine N-acetyltransferase from Salmonella typhimurium controls enzymic activity. J Biol Chem. 2002;277:12175–81. doi: 10.1074/jbc.M104365200. [DOI] [PubMed] [Google Scholar]
- 41.Zhang N, Liu L, Liu F, Wagner CR, Hanna PE, Walters KJ. NMR-based model reveals the structural determinants of mammalian arylamine N-acetyltransferase substrate specificity. J Mol Biol. 2006;363:188–200. doi: 10.1016/j.jmb.2006.08.026. [DOI] [PubMed] [Google Scholar]
- 42.Sandy J, Mushtaq A, Holton SJ, Schartau P, Noble ME, Sim E. Investigation of the catalytic triad of arylamine N-acetyltransferases: essential residues required for acetyl transfer to arylamines. Biochem J. 2005;390:115–23. doi: 10.1042/BJ20050277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hickman D, Palamanda JR, Unadkat JD, Sim E. Enzyme kinetic properties of human recombinant arylamine N-acetyltransferase 2 allotypic variants expressed in Escherichia coli. Biochem Pharmacol. 1995;50:697–703. doi: 10.1016/0006-2952(95)00182-y. [DOI] [PubMed] [Google Scholar]
- 44.Hein DW, Fretland AJ, Doll MA. Effects of single nucleotide polymorphisms in human N-acetyltransferase 2 on metabolic activation (O-acetylation) of heterocyclic amine carcinogens. Int J Cancer. 2006;119:1208–1211. doi: 10.1002/ijc.21957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dairou J, Flatters D, Chaffotte AF, Pluvinage B, Sim E, Dupret JM, Rodrigues-Lima F. Insight into the structure of Mesorhizobium loti arylamine N-acetyltransferase 2 (MLNAT2): A biochemical and computational study. FEBS Lett. 2006;580:1780–1788. doi: 10.1016/j.febslet.2006.02.033. [DOI] [PubMed] [Google Scholar]
- 46.Butcher NJ, Arulpragasam A, Minchin RF. Proteasomal Degradation of N-acetyltransferase 1 is prevented by acetylation of the active site cysteine: a mechanism for the slow acetylator phenotype and substrate-dependent down-regulation. J Biol Chem. 2004;279:22131–22137. doi: 10.1074/jbc.M312858200. [DOI] [PubMed] [Google Scholar]
- 47.Savulescu MR, Mushtaq A, Josephy PD. Screening and Characterizing Human NAT2 Variants. Methods in Enzymology. In: Helmut Sies aLP., editor. Phase II Conjugation Enzymes and Transport Systems. Academic Press; 2005. pp. 192–215. [DOI] [PubMed] [Google Scholar]
- 48.Gross M, Kruisselbrink T, Anderson K, Lang N, McGovern P, Delongchamp R, Kadlubar F. Distribution and concordance of N-acetyltransferase genotype and phenotype in an American population. Cancer Epidemiol Biomarkers Prev. 1999;8:683–92. [PubMed] [Google Scholar]
- 49.Cascorbi I, Drakoulis N, Brockmoller J, Maurer A, Sperling K, Roots I. Arylamine N-acetyltransferase (NAT2) mutations and their allelic linkage in unrelated Caucasian individuals: correlation with phenotypic activity. Am J Hum Genet. 1995;57:581–92. [PMC free article] [PubMed] [Google Scholar]
- 50.Grant DM, Morike K, Eichelbaum M, Meyer UA. Acetylation pharmacogenetics. The slow acetylator phenotype is caused by decreased or absent arylamine N-acetyltransferase in human liver. J Clin Invest. 1990;85:968–72. doi: 10.1172/JCI114527. [DOI] [PMC free article] [PubMed] [Google Scholar]