

Abstract
Introduction
Long QT Syndrome (LQTS) is an inherited disorder characterized by prolonged QT intervals and life-threatening polymorphic ventricular tachyarrhythmias. LQT1 caused by KCNQ1 mutations is the most common form of LQTS.
Methods and Results
Patients diagnosed with LQTS were screened for disease-associated mutations in KCNQ1, KCNH2, KCNE1, KCNE2, KCNJ2 and SCN5A. A novel mutation was identified in KCNQ1 caused by a 3 base deletion at the position 824–826, predicting a deletion of phenylalanine at codon 275 in segment 5 of KCNQ1 (ΔF275). Wild-type (WT) and ΔF275-KCNQ1 constructs were generated and transiently transfected together with a KCNE1 construct in CHO-K1 cells to characterize the properties of the slowly activating delayed rectifier current (IKs) using conventional whole-cell patch-clamp techniques. Cells transfected with WT-KCNQ1 and KCNE1 (1:1.3 molar ratio) produced slowly activating outward current with the characteristics of IKs. Tail current density measured at −40mV following a 2 sec step to +60mV was 381.3±62.6 pA/pF (n=11). Cells transfected with ΔF275-KCNQ1 and KCNE1 exhibited essentially no current. (Tail current density: 0.8±2.1 pA/pF, n=11, p=0.00001 vs WT). Co-transfection of WT- and ΔF275- KCNQ1 (50/50) along with KCNE1 produced little to no current (tail current density: 10.3±3.5 pA/pF, n=11, p=0.00001 vs WT alone), suggesting a potent dominant negative effect. Immunohistochemistry showed normal membrane trafficking of ΔF275-KCNQ1.
Conclusion
Our data suggest that a ΔF275 mutation in KCNQ1 is associated with a very potent dominant negative effect leading to an almost complete loss of function of IKs and that this defect underlies a LQT1 form of LQTS.
Keywords: Ion channel, Mutation, Inherited syndrome, Electrophysiology, Torsade de Pointes
INTRODUCTION
Long QT syndrome (LQTS) is characterized by QT prolongation in the surface ECG, syncope, and sudden cardiac death secondary to an atypical polymorphic ventricular tachycardia known as Torsade de Pointes (TdP). Mutations in ten different genes causing inherited LQTS have been identified. Mutations in the KCNQ1 (KvLQT1) gene can cause both the autosomal dominant Romano-Ward syndrome and the autosomal recessive Jervell and Lange-Nielsen syndrome.1 KCNQ1 and KCNE1 (minK) form the slowly activating component of the delayed rectifier K+ current (IKs), which contributes to cardiac repolarization. Functional expression of mutant KCNQ1 channel proteins in heterologous expression system has revealed a loss of channel function in most cases.1 Over one hundred mutations have been identified and shown to be associated with a variety of ion channel dysfunction mechanisms.2 The objective of this study was to evaluate the functional consequences of a novel single amino acid deletion mutant, ΔF275, in a mammalian heterologous expression system.
METHODS
Clinical case presentation
A 14-year-old girl was referred for genetic analysis. Her ECG at annual check-up showed a prolonged QT interval of 520–560 ms. She had syncopal attack at 10 years of age, but no family history of sudden cardiac death and normal hearing. The surface ECG at rest showed broad-based tall T wave typical LQT13,4 and a QTc interval of 515 ms (Figure 1A). Holter recording and treadmill exercise stress test did not reveal a clinically significant arrhythmia. However, her QTc prolonged from a baseline of 475 to 555 ms during a treadmill exercise stress test. Infusion of epinephrine (0.3mg/kg/min) during electrophysiological study prolonged QTc from a baseline of 470 ms to 620ms and induced a polymorphic VT (Figure 1B). Programmed electrical stimulation in the absence of epinephrine did not induce ventricular tachycardia (VT) or fibrillation (VF). Both parents displayed a normal ECG and genetic screening was declined.
Figure 1. Electrocardiograms of the patient.
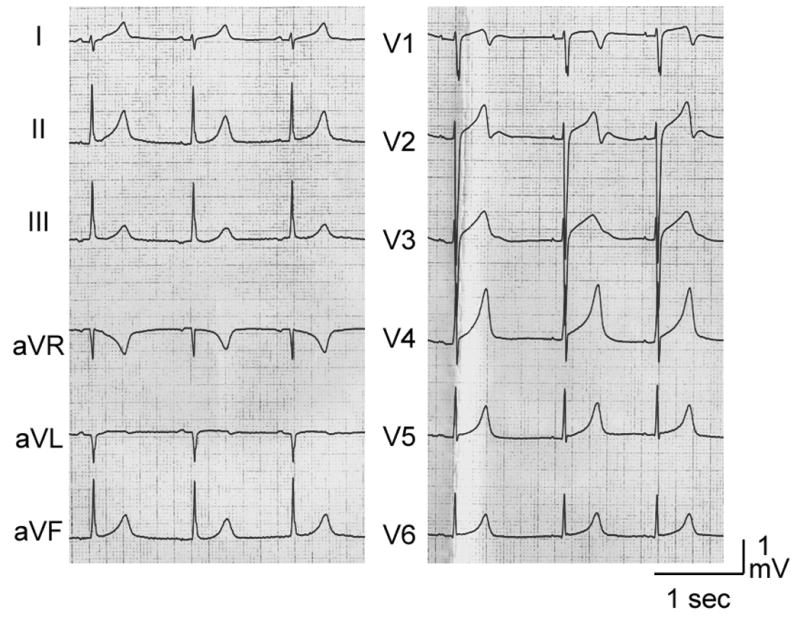
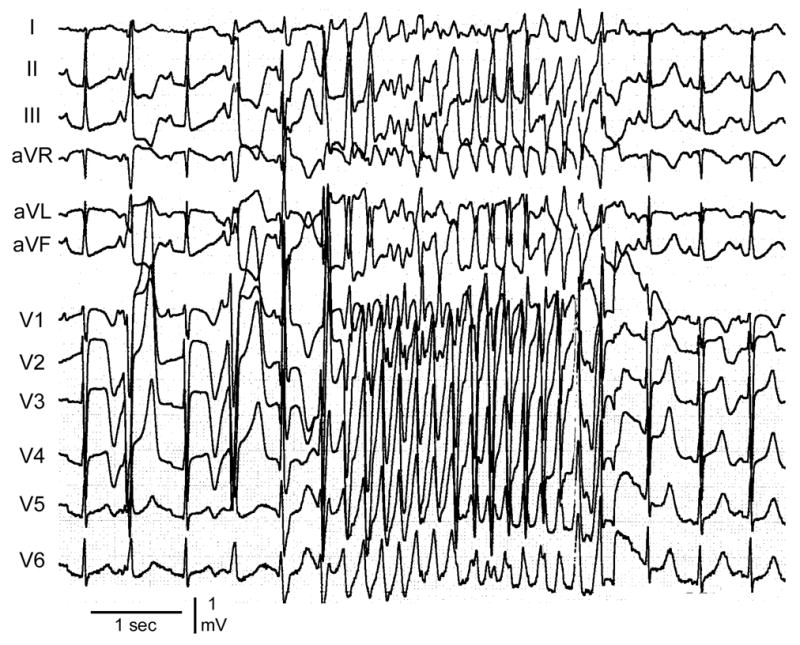
A. 12-Lead ECG of the patient at rest. Broad-based tall T waves are observed. QTc interval is 515 ms at rest. B. Torsade de Pointes arrhythmias induced following administration of epinephrine (0.3mg/kg/min). Her QTc interval prolonged to 620 ms.
Genetic analysis
The protocol of this study was approved by the respective IRBs of the institutions involved in the study. After obtaining written informed consent, genomic DNA was isolated from peripheral blood lymphocytes by conventional methods.5 The genomic DNA was amplified on GeneAmp® PCR System 9700 thermal cycler by standard polymerase chain reaction (PCR) technique using the primers as described previously.6,7 For genetic screening, we used single-strand conformation polymorphism (SSCP) followed by DNA sequencing or PCR-sequencing method for KCNQ1, KCNH2, SCN5A, KCNE1, KCNE2 and KCNJ2 genes. Abnormal conformers were sequenced with ABI377XL genetic analyzer (Applied Biosystems, Foster City, CA). Electropherograms were compared with the KCNQ1 wild-type sequence (GenBank accession number AJ006343) using the DNASIS Ver. 3.7 software (HITACHI, Japan).
Constructs for electrophysiological and confocal studies
The wild-type (WT) KCNQ1 and KCNE1 cDNAs were generated as described previously.8 The deletion of 3 nucleotides, TCT, corresponding to phenylalanine at position 275 was introduced to WT-KCNQ1 cDNA by site-directed mutagenesis with primers of 5′-CGGCTTCCTGGGCCTCATCTCCTCGTACTT-3′ and 5′-AGTACGAGGAGATGAGGCCCAGGAAGCCG-3′, and the ΔF275-KCNQ1 clone was confirmed by sequencing.
Cell culture and transient transfection
CHO-K1 (Chinese hamster ovary) cells were obtained from American Type Cell Collection and cultured in Dulbecco’s modified Eagle’s medium (Invitrogen Corp., Carlsbad, CA) supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin in a humidified 5% CO2 incubator at 37°C. Cultured cells were seeded in 35-mm dishes 1 day before transfection and transiently transfected with various plasmids using FuGENE6 lipid based transfection reagent (Roche Diagnostics Co., Indianapolis, IN). In the electrophysiologic experiments, 0.75, 0.5 or 0.375 μg of WT-KCNQ1 and/or 0.75, 0.375 or 0.25 μg of ΔF275-KCNQ1 together with 0.75 μg of WT-KCNE1 were transfected into CHO-K1 cells. pEGFP-C1 (Clontech Laboratories, Inc., Mountain View, CA) was co-transfected to allow for identification of the transfected cells. Cells displaying green fluorescence 48–72h after transfection were studied electrophysiologically.
Electrophysiological recordings
To investigate the effects of the ΔF275-KCNQ1 mutation on IKs, we performed whole-cell patch-clamp experiments on CHO-K1 cells transfected with WT- and/or ΔF275-KCNQ1 channel. Briefly, cells were placed in a perfusion chamber (PDMI-2, Medical Systems Corp.) mounted on the stage of an inverted microscope (TE2000, Nikon, USA). Cells were superfused with normal external solution containing (in mmol/L) 132 NaCl, 4.8 KCl, 2 CaCl2, 1.2 MgCl2, 5 Glucose and 10 HEPES-Na (pH=7.4 adjusted with HCL). Patch pipettes were fabricated from borosilicate glass capillaries (1.5 mm O.D., Fisher Scientific, Pittsburgh, PA). Pipettes were pulled using a gravity puller: PP-83 (Narishige, Japan) and filled with pipette solution of the following composition (mmol/L) 110 Aspartic acid, 5 ATP-K2, 11 EGTA, 10 HEPES and 1 MgCl2 (pH=7.35 adjusted with KOH). The pipette resistance ranged from 2–5 MΩ when filled with the internal solution. All recordings were made at room temperature.
After forming a gigaseal, the cell membrane was ruptured by applying negative pressure. Current signals were recorded using an Axopatch 200A amplifier (Axon Instruments Inc., Foster City, CA) and series resistance errors were reduced by 60–70% with electronic compensation. All signals were acquired at 500 Hz to 5 kHz (Digidata 1322, Axon Instruments, Foster City, CA) with a personal computer running Clampex 9 software (Axon Instruments, Foster City, CA) and filtered at 5kHz with a 4 pole Bessel low pass filter. Membrane currents were analyzed with Clampfit 9 software (Axon Instruments, Foster City, CA).
Immunohistochemistry
Immunohistochemistry was performed to assess protein localization of WT- and ΔF275-KCNQ1 channels. Briefly, CHO-K1 cells transfected with WT- and/or ΔF275-KCNQ1 were fixed with an ethanol-acetone mixture and permeabilized with 0.2% Triton X-100. Cells were incubated overnight at 4°C with 1:100/200 dilutions of primary goat polyclonal antibodies against the KCNQ1 channel protein (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) 48 hours after transfection. Cells were incubated with a 1:1,000 dilution of anti-goat alexa-488 conjugated secondary antibody (Molecular Probes, Eugene, OR, USA) for 2 hours at room temperature. Cells were mounted using Pro-Long antifade mounting media (Molecular Probes).
Immunofluorescence stained CHO-K1 cells were visualized by confocal microscopy (Olympus Fluoview FV300). Cells were excited at 488nm using an argon laser and emission was collected via 525 nm band pass filter and photomultiplier tube (40X oil immersion lens).
Statistics
Results are expressed as mean ± S.E.M. Difference between groups were tested by one-way ANOVA followed by Scheffe’s modified F-test for multiple comparisons. Values of p<0.05 were considered statistically significant.
RESULTS
We initially evaluated DNA from the patient for mutations in the KCNQ1 gene using single-strand conformation polymorphism (SSCP) analysis of polymerase chain reaction product. An abnormal pattern of bands was detected in exon 6. Further examination by direct sequencing of the abnormal fragment showed a deletion of TCT at position 824–826 in KCNQ1 (Fig. 2A). This abnormal sequence causes a single amino acid deletion of phenylalanine at position 275 in the S5 segment of KCNQ1 channel. The phenylalanine at position 275 is highly conserved among different species. (Fig. 2B) Predicted topology of WT- and mutant-KCNQ1 channel is shown in Fig. 2C. This mutation was not detected in more than 200 unrelated ethnically-matched healthy individuals. No mutations were identified in any of the other long QT -related genes screened (KCNH2, KCNE1, KCNE2, SCN5A).
Figure 2. Δ275F mutant at KCNQ.
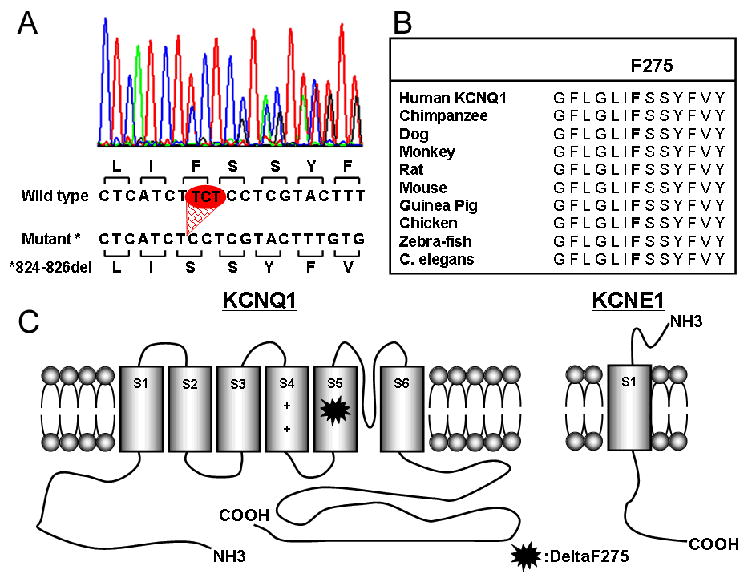
A. Chromatogram showing a heterozygous deletion at the position of 824–826.
B. Amino acid sequence alignment showing conservation of Phenylalanine 275 in multiple species
C. Schematic topology of KCNQ1 and KCNE1 proteins forming IKs. Δ275F mutation was located at the S5 segment of KCNQ1.
Conventional whole-cell patch-clamp experiments were conducted on cells transfected with WT- and/or mutant-KCNQ1. Representative current traces of each transfection protocols are shown in Fig. 3A–E. Cells transfected with 0.75 μg of WT-KCNQ1 together with 0.75 μg of KCNE1 exhibited slowly activating outward current compatible with IKs recorded from native cardiac myocytes (Fig. 3A), whereas cells transfected with same amount of ΔF275-KCNQ1+KCNE1 produced little to no current (Fig. 3E). Cells transfected with 0.375 μg of WT-KCNQ1 (+ KCNE1) expressed slightly less IKs compared to those transfected with 0.75μg of WT-KCNQ1, although the differences were not statistically significant (Fig. 3B). Co-expression of 0.375 μg of WT-KCNQ1 with the same amount of ΔF275-KCNQ1 (+ KCNE1) showed little to no current (Fig. 3D). Increasing the ratio of WT to ΔF275 from 1:1 to 2:1, resulted in a slight recovery of IKs current (Fig. 3C). These observations indicate a potent dominant negative effect of ΔF275-KCNQ1.
Figure 3. Representative current trace of WT- and/or Δ275F-KCNQ1 expressed in CHO-K1 cells.
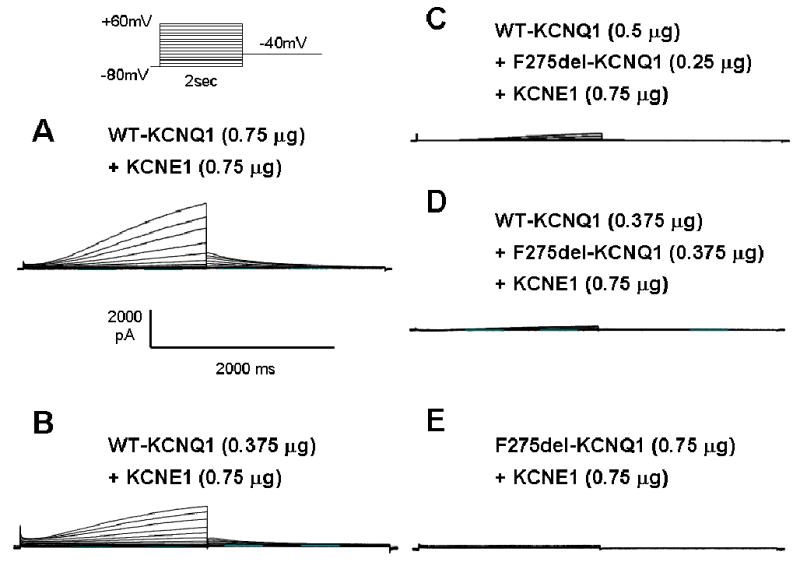
Cells of each panel were transfected as follows:
A. 0.75 μg of WT-KCNQ1 and 0.75 μg of KCNE1.
B. 0.375 μg of WT-KCNQ1 and 0.75 μg of KCNE1.
C. 0.5 μg of WT-KCNQ1, 0.25 μg of Δ275F-KCNQ1 and 0.75μg of KCNE1.
D. 0.375μg of WT-KCNQ1, 0.375 μg of Δ275F-KCNQ1 and 0.75μg of KCNE1.
E. 0.75μg of Δ275F-KCNQ1 and 0.75μg of KCNE1.
Pulse protocol is shown in the inset in the lower right.
Summary data of current-voltage relationships for peak current recorded and tail currents recorded upon repolarization to −40mV for the various transfection protocols are shown graphically in Fig. 4(A, B). Current-voltage relations were not significantly different between 0.75 and 0.375 μg of WT. I-V relation for Δ275F-KCNQ1 channels was nearly flat and significantly different from WT (p=0.00001). Co-transfection of WT- and ΔF275- KCNQ1 (1:1) along with KCNE1 also produced little to no current (p=0.00001 vs WT). Increasing WT to ΔF275-KCNQ1 ratio from 1:1 to 2:1 significantly increased developing current (p=0.00251). Summary data for current intensity recorded during depolarizations to +60 mV and tail currents recorded upon repolarization to −40 mV shown in Fig. 4(C, D). These results once again indicate that the ΔF275-KCNQ1 mutant exerts a very potent dominant-negative effect on channel function or expression.
Figure 4. Current-voltage relationships of expressed currents.
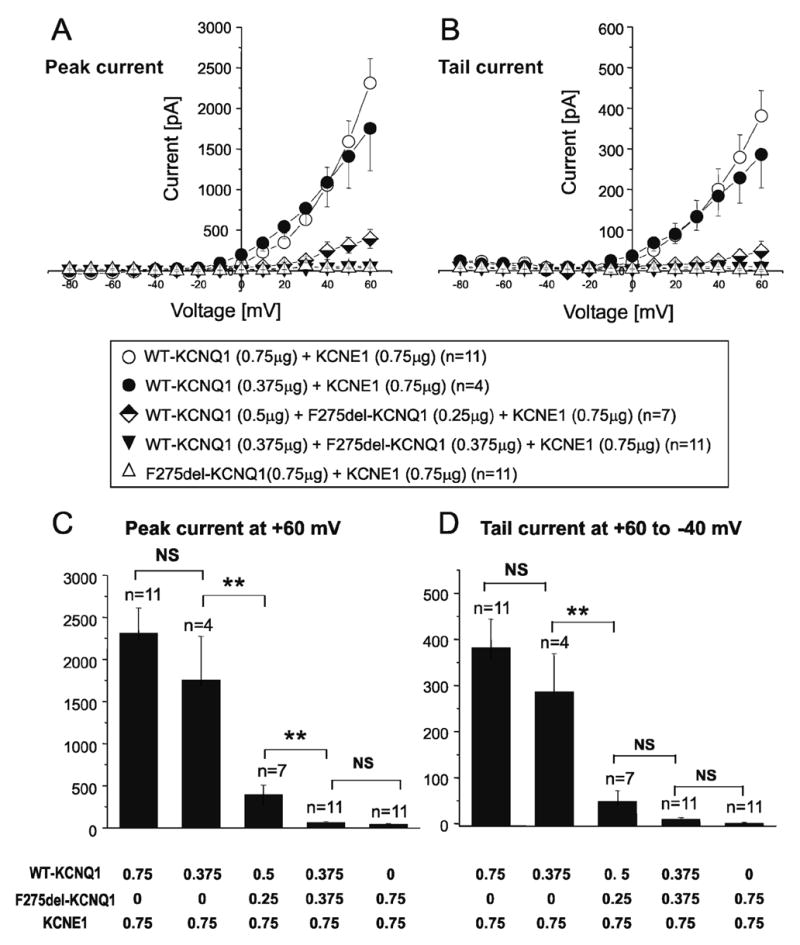
A. Current-Voltage relationship measured at the peak current during the test depolarization pulse.
B. Current-Voltage relationship measured of the tail current upon repolarization to −40mV following test depolarization.
C. Bar graphs showing current densities of developing (peak) recorded current at +60mV.
D. Bar graphs showing current densities of tail current recorded upon repolarization to −40mV from +60mV test depolarization.
To determine whether the changes observed on IKs is due to impaired trafficking of the protein to the plasma membrane, we determined the localization of WT and mutant KCNQ1 channel proteins using immunohistochemical techniques.
CHO-K1 transfected with WT-KCNQ1 and/or ΔF275-KCNQ1 cells were incubated with a goat polyclonal antibody against the KCNQ1 channel protein. Localization of the immunolabeled protein was assessed by confocal imaging, following incubation of the cells with an Alexa-488 conjugated secondary anti-goat antibody. Positive staining was observed in all transfected CHO-K1 cells, but not in non-transfected controls (Fig. 5A). Cells transfected with WT (Fig 5B), ΔF275-KCNQ1 (Fig, 5C) or WT+ΔF275-KCNQ1 (Fig. 5D) all displayed peripheral staining. Similar results were obtained in 15 cells transfected with WT (4 transfections), 14 cells transfected with ΔF275-KCNQ1 (4 transfections) and 5 cells transfected with WT + ΔF275-KCNQ1 (2 transfections).
Figure 5. Protein localization of WT- and/or mutant-KCNQ1 in CHO-K1 cells visualized by immunohistochemistry.
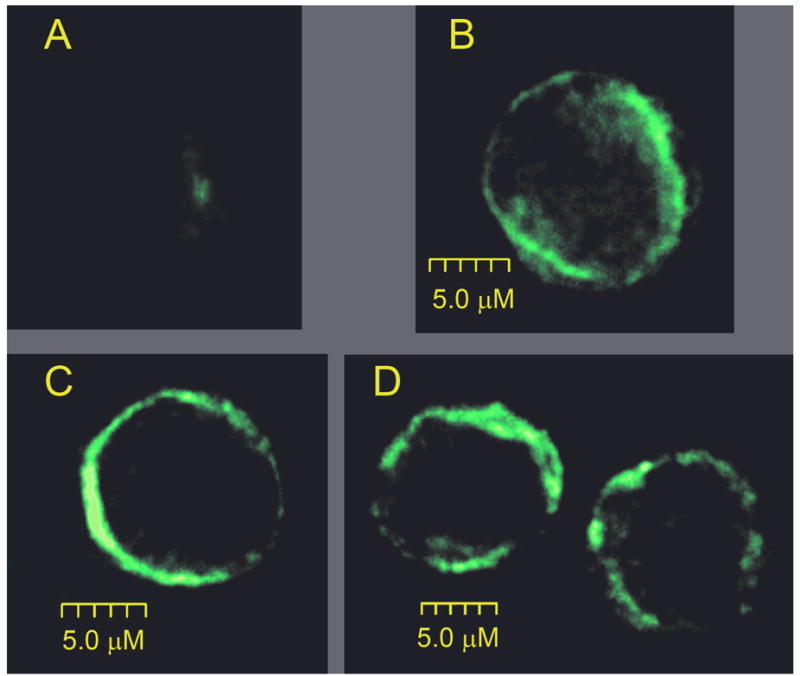
Figure shows 0.15μm optical sections from the center of CHO-K1 cells transfected with WT- KCNQ1 (B), ΔF275-KCNQ1 (C) or ΔF275-KCNQ1 + WT (D). A brighter fluorescence intensity near the plasma membrane reveals positive immunostaining of the KCNQ1 channel. Panel A shows the lack of any fluorescence signal from non transfected cells.
These findings suggest that the loss of function observed with the ΔF275 mutant is not due to a defect in trafficking of mature KCNQ1 channels from the ER/Golgi complex to the cell membrane.
DISCUSSION
LQT1 is caused by gene mutations in KCNQ1 and it is the most common form of congenital LQTS, responsible for approximately 43% of LQTS-linked mutations. KCNQ1 encodes the pore-forming α-subunit of the IKs potassium channel. KCNE1 encodes the β-subunit. Over one hundred mutations in KCNQ1 have been reported to cause a loss of function and thus to predispose to the development of LQTS. Reduced levels of IKs lead to prolongation of action potential duration (APD) and thus to prolongation of the QT interval and the development of TdP. The present study identifies a novel KCNQ1 mutation (F275del or ΔF275) caused by a 3 nucleotide deletion, resulting in deletion of a phenylalanine in the S5 segment of KCNQ1. Functional analysis of the mutated channel protein revealed a major loss of function consistent with the LQT1 phenotype of the patient.
The ΔF275-KCNQ1 channel failed to produce any current when co-expressed with KCNE1, indicating that the mutant was unable to form functional homo-multimeric channels, or could not be expressed on the surface membrane due to a trafficking defect. Subcellular localization of the channel proteins revealed normal trafficking of the mutant protein, thus pointing to a functional dysfunction of the channel as the cause for the loss of current. Co-expression of WT- and ΔF275-KCNQ1 channel produced little to no IKs, suggesting that the ΔF275-KCNQ1 suppresses the function of the WT channel in a potent dominant-negative manner. Thus, the heteromultimeric channel containing mutant subunits is largely non-functional. The strong dominant negative suppression is consistent with the clinical phenotype observed in our patient with QTc intervals at rest as long as 560 msec, which prolonged by 150 ms in response to epinephrine infusion, leading to induction of TdP. The ΔQTc induced by epinephrine is longer than that observed in LQT1 patients with the Romano-Ward syndrome.9,10 These characteristics approach those described for patients with Jervell and Lange-Nielsen syndrome, the homozygous recessive form of LQT1.11 Although several dominant-negative KCNQ1 mutants have been reported,8,12–15 the ΔF275-KCNQ1 mutation herein described appears to result in the most prominent dominant-negative suppression of IKs.
A number of mutations in KCNQ1 have been shown to cause trafficking defects, leading to haploinsufficiency (T587M and ΔS276) or a dominant-negative effect (A178fs/105). These variations are often missense mutations in the N-terminus, S5, pore region or C-terminus.8,16,17 Recent studies indicate that specific regions of KCNQ1 (N-terminal juxtamembranous domain or C-terminus amino acids 610–620) are critical for channel surface expression.18,19 However, mutations outside these domains can lead to trafficking problems of varying degrees.20 In the case of the KCNH2 channel, the C-terminus region, including the cyclic nucleotide binding domain, is thought to be essential for channel trafficking, because it contains crucial sequence linked to endoplasmic reticulum retention and the interaction site with GM130 required for channel protein trafficking.21 In the present study, the mutation affected the mid-portion of the S5 segment and no trafficking defect was detected.
The IKs channel is comprised of 4 KCNQ1 subunits. In the case of a heterozygous mutation in KCNQ1, as in the present case, mutant and WT subunits may combine to form a heteromultimeric channel. When the mutant KCNQ1 subunits interfere with the function of the wild-type subunits, IKs will be reduced by greater than 50%. This greater than expected loss of function is referred to as a dominant-negative effect. Mutations in the pore region, as in the case of ΔF275-KCNQ1, are more likely to produce a dominant negative effect.12,22 In contrast to ΔF275-KCNQ1, the molecular phenotype of ΔS276-KCNQ1 has a very weak dominant negative effect leading to a recessive form of LQTS.17
In summary, we have identified a novel single amino acid deletion in domain 5 of KCNQ1 in a patient with LQTS. Functional analysis revealed normal trafficking nonfunctioning channel with very strong dominant-negative suppression of IKs, consistent with a pronounced LQTS phenotype and sensitivity to epinephrine.
Acknowledgments
We thank Tabitha Carrier for expert technical support and Drs. Yoshifusa Aizawa, Serge Sicouri and Guido Pollevick for helpful discussions.
Supported by a grant from Japan Heart Foundation/Bayer Yakuhin Research Grant Abroad (YA), grant # HL 47678 from the National Institutes of Health (CA), and the Masons of New York State and Florida.
Reference List
- 1.Kass RS, Moss AJ. Long QT syndrome: novel insights into the mechanisms of cardiac arrhythmias. J Clin Invest. 2003;112:810–815. doi: 10.1172/JCI19844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roden DM. Long QT syndrome: reduced repolarization reserve and the genetic link. J Intern Med. 2006;259:59–69. doi: 10.1111/j.1365-2796.2005.01589.x. [DOI] [PubMed] [Google Scholar]
- 3.Moss AJ, Zareba W, Benhorin J, Locati EH, Hall WJ, Robinson JL, Schwartz PJ, Towbin JA, Vincent GM, Lehmann MH, Keating MT, MacCluer JW, Timothy KW. ECG T-wave patterns in genetically distinct forms of the hereditary long QT syndrome. Circulation. 1995;92:2929–2934. doi: 10.1161/01.cir.92.10.2929. [DOI] [PubMed] [Google Scholar]
- 4.Zhang L, Timothy KW, Vincent GM, Lehmann MH, Fox J, Giuli LC, Shen J, Splawski I, Priori SG, Compton SJ, Yanowitz F, Benhorin J, Moss AJ, Schwartz PJ, Robinson JL, Wang Q, Zareba W, Keating MT, Towbin JA, Napolitano C, Medina A. Spectrum of ST-T-wave patterns and repolarization parameters in congenital long-QT syndrome: ECG findings identify genotypes. Circulation. 2000;102:2849–2855. doi: 10.1161/01.cir.102.23.2849. [DOI] [PubMed] [Google Scholar]
- 5.Schulze-Bahr E, Haverkamp W, Wiebusch H, Schulte H, Hordt M, Borggrefe M, Breithardt G, Assmann G, Funke H. Molecular analysis at the Harvey Ras-1 gene in patients with long QT syndrome. J Mol Med. 1995;73:565–569. doi: 10.1007/BF00195141. [DOI] [PubMed] [Google Scholar]
- 6.Splawski I, Shen J, Timothy KW, Vincent GM, Lehmann MH, Keating MT. Genomic structure of three long QT syndrome genes: KVLQT1, HERG, and KCNE1. Genomics. 1998;51:86–97. doi: 10.1006/geno.1998.5361. [DOI] [PubMed] [Google Scholar]
- 7.Neyroud N, Richard P, Vignier N, Donger C, Denjoy I, Demay L, Shkolnikova M, Pesce R, Chevalier P, Hainque B, Coumel P, Schwartz K, Guicheney P. Genomic organization of the KCNQ1 K+ channel gene and identification of C-terminal mutations in the long-QT syndrome. Circ Res. 1999;84:290–297. doi: 10.1161/01.res.84.3.290. [DOI] [PubMed] [Google Scholar]
- 8.Aizawa Y, Ueda K, Wu LM, Inagaki N, Hayashi T, Takahashi M, Ohta M, Kawano S, Hirano Y, Yasunami M, Aizawa Y, Kimura A, Hiraoka M. Truncated KCNQ1 mutant, A178fs/105, forms hetero-multimer channel with wild-type causing a dominant-negative suppression due to trafficking defect. FEBS Lett. 2004;574:145–150. doi: 10.1016/j.febslet.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 9.Shimizu W, Noda T, Takaki H, Nagaya N, Satomi K, Kurita T, Suyama K, Aihara N, Sunagawa K, Echigo S, Miyamoto Y, Yoshimasa Y, Nakamura K, Ohe T, Towbin J, Priori SG, Kamakura S. Diagnostic value of epinephrine test for genotyping LQT1, LQT2 an LQT3 forms of congenital long QT syndrome. Heart Rhythm. 2004;1:276–283. doi: 10.1016/j.hrthm.2004.04.021. [DOI] [PubMed] [Google Scholar]
- 10.Vyas H, Hejlik J, Ackerman MJ. Epinephrine QT stress testing in the evaluation of congenital long-QT syndrome: diagnostic accuracy of the paradoxical QT response. Circulation. 2006;113:1385–1392. doi: 10.1161/CIRCULATIONAHA.105.600445. [DOI] [PubMed] [Google Scholar]
- 11.Goldenberg I, Moss AJ, Zareba W, McNitt S, Robinson JL, Qi M, Towbin JA, Ackerman MJ, Murphy L. Clinical course and risk stratification of patients affected with the Jervell and Lange-Nielsen syndrome. J Cardiovasc Electrophysiol. 2006;17:1161–1168. doi: 10.1111/j.1540-8167.2006.00587.x. [DOI] [PubMed] [Google Scholar]
- 12.Shalaby FY, Levesque PC, Yang WP, Little WA, Conder ML, Jenkins-West T, Blanar MA. Dominant-negative KvLQT1 mutations underlie the LQT1 form of long QT syndrome. Circulation. 1997;96:1733–1736. doi: 10.1161/01.cir.96.6.1733. [DOI] [PubMed] [Google Scholar]
- 13.Wang Z, Tristani-Firouzi M, Xu Q, Lin M, Keating MT, Sanguinetti MC. Functional effects of mutations in KvLQT1 that cause long QT syndrome. J Cardiovasc Electrophysiol. 1999;10:817–826. doi: 10.1111/j.1540-8167.1999.tb00262.x. [DOI] [PubMed] [Google Scholar]
- 14.Thomas D, Wimmer AB, Karle CA, Licka M, Alter M, Khalil M, Ulmer HE, Kathofer S, Kiehn J, Katus HA, Schoels W, Koenen M, Zehelein J. Dominant-negative I(Ks) suppression by KCNQ1-DeltaF339 potassium channels linked to Romano-Ward syndrome. Cardiovasc Res. 2005;37:487–497. doi: 10.1016/j.cardiores.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 15.Boulet IR, Raes AL, Ottschytsch N, Snyders DJ. Functional effects of a KCNQ1 mutation associated with the long QT syndrome. Cardiovasc Res. 2006;70:466–474. doi: 10.1016/j.cardiores.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 16.Yamashita F, Horie M, Kubota T, Yoshida H, Yumoto Y, Kobori A, Ninomiya T, Kono Y, Haruna T, Tsuji K, Washizuka T, Takano M, Otani H, Sasayama S, Aizawa Y. Characterization and subcellular localization of KCNQ1 with a heterozygous mutation in the C terminus. J Mol Cell Cardiol. 2001;33:197–207. doi: 10.1006/jmcc.2000.1300. [DOI] [PubMed] [Google Scholar]
- 17.Gouas L, Bellocq C, Berthet M, Potet F, Demolombe S, Forhan A, Lescasse R, Simon F, Balkau B, Denjoy I, Hainque B, Baro II, Guicheney P. New KCNQ1 mutations leading to haploinsufficiency in a general population; Defective trafficking of a KvLQT1 mutant. Cardiovasc Res. 2004;63:60–68. doi: 10.1016/j.cardiores.2004.02.011. [DOI] [PubMed] [Google Scholar]
- 18.Kanki H, Kupershmidt S, Yang T, Wells S, Roden DM. A structural requirement for processing the cardiac K+ channel KCNQ1. J Biol Chem. 2004:1–37. doi: 10.1074/jbc.M404539200. [DOI] [PubMed] [Google Scholar]
- 19.Dahimene S, Alcolea S, Naud P, Jourdon P, Escande D, Brasseur R, Thomas A, Baro I, Merot J. The N-terminal juxtamembranous domain of KCNQ1 is critical for channel surface expression: implications in the Romano-Ward LQT1 syndrome. Circ Res. 2006;99:1076–1083. doi: 10.1161/01.RES.0000250262.12219.95. [DOI] [PubMed] [Google Scholar]
- 20.Wilson AJ, Quinn KV, Graves FM, Bitner-Glindzicz M, Tinker A. Abnormal KCNQ1 trafficking influences disease pathogenesis in hereditary long QT syndromes (LQT1) Cardiovasc Res. 2005;67:476–486. doi: 10.1016/j.cardiores.2005.04.036. [DOI] [PubMed] [Google Scholar]
- 21.Roti EC, Myers CD, Ayers RA, Boatman DE, Delfosse SA, Chan EK, Ackerman MJ, January CT, Robertson GA. Interaction with GM130 during HERG ion channel trafficking. Disruption by type 2 congenital long QT syndrome mutations. Human Ether-a-go-go-Related Gene. J Biol Chem. 2002;277:47779–47785. doi: 10.1074/jbc.M206638200. [DOI] [PubMed] [Google Scholar]
- 22.Wollnik B, Schroeder BC, Kubisch C, Esperer HD, Wieacker P, Jentsch TJ. Pathophysiological mechanisms of dominant and recessive KVLQT1 K+ channel mutations found in inherited cardiac arrhythmias. Hum Mol Genet. 1997;6:1943–1949. doi: 10.1093/hmg/6.11.1943. [DOI] [PubMed] [Google Scholar]