

Abstract
The independent influences of invariant chain (Ii) and HLA-DM molecules on the array of naturally processed peptides displayed by HLA-DR molecules were studied using transfected cell lines. The absence of Ii led to an altered set of HLA-DR-bound peptides as judged by the discriminating responses of alloreactive T cell clones. While most T cell clones raised against DR+Ii+DM+ peripheral blood mononuclear cells (PBMC) failed to respond to DR+Ii−DM− cells, T cell clones raised against DR+Ii−DM− transfectants were not stimulated by DR+Ii+DM+ cells. Furthermore, coexpression of HLA-DM with HLA-DR1 in the absence of Ii augmented responses of anti-PBMC T cell clones but inhibited allorecognition by T cell clones raised against DR+Ii−DM− transfectants. The conformational integrity of the class II molecules, as judged by serology, suggests that the patterns of reactivity of the T cell clones reflect specificity for different alloantigen-bound peptides. Hence, discordant regulation of expression of major histocompatibility complex class II, Ii, and HLA-DM molecules in vivo may lead to the display of novel self-peptides and possible interruption of self-tolerance.
Major histocompatibility complex (MHC) class II molecules present peptide antigens to MHC class II-restricted CD4+ T cells. The peptides presented are usually derived from internalized exogenous or membrane-bound proteins (1) which are unfolded, denatured, and degraded within the progressively acidic endosomal pathway. Class II molecules are assembled in the endoplasmic reticulum (ER), where they associate noncovalently with the invariant chain (Ii), a type 2 transmembrane protein (reviewed in ref. 2). The CLIP region of Ii (amino acids 81–104) binds in the groove of nascent MHC class II molecules, thereby inhibiting the binding of peptides in the ER (3–5). An endosomal targeting motif within the amino terminus of the p33 form of Ii directs the Ii–class II complexes to specialized endosomal compartments, MIIC (6, 7). Here, Ii is sequentially degraded by proteases, and the catalytic activity of HLA-DM promotes exchange of CLIP for peptides derived from endocytosed proteins (8–11).
In the absence of Ii, as shown in Ii knockout mice (Ii0/0), few class II molecules reach the cell surface, as most are retained in the ER and degraded (12, 13). The studies of antigen presentation by Ii-negative (Ii−) cells reported to date have largely used T cell clones or hybridomas raised against conventional antigen-presenting cells (APC) as responders. The results of such studies have shown that a degree of overlap exists between the peptides displayed by MHC class II molecules expressed in the presence and the absence of Ii (for example, see ref. 14). However, this approach does not inform whether the complete or relative absence of Ii leads to the display of novel peptide epitopes by MHC class II molecules, an important issue if circumstances can arise in vivo in which Ii is limiting in MHC class II+ cells.
In this study we used alloreactive T cell clones as probes of peptide occupancy of class II molecules expressed in the presence or absence of Ii and HLA-DM on the basis that anti-MHC allorecognition generally involves the corecognition of the allogeneic MHC molecule and bound peptide (for review see ref. 15). The results suggest that in the absence of Ii, MHC class II molecules display a distinct array of peptides. Furthermore, HLA-DM influenced allorecognition of DR molecules in the absence of Ii, suggesting that HLA-DM can influence MHC class II peptide occupancy independently of Ii. Taken together, these findings support the prediction that discordant regulation of MHC class II, Ii, and HLA-DM molecules in vivo, as has been observed in bone marrow macrophages (16) and in gut epithelial cells (17) for MHC and Ii, could lead to the display of self-peptides to which the T cell repertoire is not tolerant.
EXPERIMENTAL PROCEDURES
Cell Lines and Clones.
The phenotype of the APC used is shown in Table 1 and the reactivity of the T cell clones used is shown in Table 2. M1 cell lines were grown as adherent monolayers at 37°C in 7% CO2, in DMEM supplemented with 10% fetal calf serum (Globefarm, Cheshire, U.K.), 50 international units/ml penicillin, 50 μg/ml streptomycin, and 2 mM l-glutamine. Epstein–Barr virus-transformed B cell lines (B-LCL) were grown in RPMI 1640 tissue culture medium (Flow Laboratories) supplemented as above. T cell clones were grown in RPMI 1640 supplemented as above but with 10% human AB serum in place of fetal calf serum, and stimulated weekly with irradiated autologous peripheral blood mononuclear cells (PBMC) and recombinant interleukin 2 (rIL2; Boehringer Mannheim). The RB and ND T cell clones were raised from PBMC from two healthy volunteers (DR4,4 and DR4,6) by culturing adherent cell-depleted PBMC (106 T cells) with 3 × 105 mitomycin-C-treated M1.DR1.B7 transfectants in wells of 24-well plates. After four weekly stimulations of the bulk cultures with the transfectants, the T cells were cloned by limiting dilution and subsequently restimulated weekly with irradiated PBMC, phytohemagglutinin (PHA; Glaxo–Wellcome, Stevenage, U.K.) and rIL2; rIL2 was added 3 days after each stimulation. All clones were used for functional assays 1 week after their last stimulation.
Table 1.
APC used
| Cell line | Constitutive expression
|
Ref. | ||
|---|---|---|---|---|
| Class II | Ii | HLADM | ||
| M1* | — | − | − | 18 |
| M1.DR1 or DR3 | DR1 or DR3† | − | − | 19 |
| M1.DR1/3.Ii | DR1 or DR3† | +† | − | 19 |
| M1.DR1/3. DM | DR1 or DR3† | − | +† | |
| M1.DR1/3. Ii.DM | DR1 or DR3† | +† | +† | |
| M1.DR1.B7 | DR1† + B7.1† | − | − | 20 |
| B-LCL HOM2 | DR1 | + | + | § |
| B-LCL VAVY | DR3 | + | + | § |
| T2.DR3‡ | DR3† | + | − | 21 |
| PBMC | DR1 or DR3 | + | + | |
Inducible only for HLA-DM expression.
By transfection.
Kind gift from Peter Cresswell (Yale University, New Haven, CT).
Tenth International Histocompatibility Workshop.
Table 2.
T cells used
| T cell clone | Raised against | Allo-specificity | Responder DR type | Ref. |
|---|---|---|---|---|
| RB series | M1.DR1.B7 | DR1 | DR4,4 | |
| ND series | M1.DR1.B7 | DR1 | DR4,6 | |
| G3 and G8 | DR1 PBMC | DR1 | DR4,6 | 22 |
| L9 | DR11 PBMC | DR11/DR1* | DR5 | 22 |
| K18 | DR11 PBMC | DR11/DR1* | DR17 | 23 |
Raised against DR11, cross-reacts on DR1.
Antibodies.
The antibodies used in these experiments are described in Table 3. All these antibodies were used at saturating concentrations as judged by prior titration of binding to B-LCL.
Table 3.
Antibodies used for staining and biochemistry
| Antibody | Specificity | Isotype | Nature | Source (Ref.) |
|---|---|---|---|---|
| L243 | HLA-DRα | IgG2a | Culture supernatant | ATCC* (24) |
| 16.23 | HLA-DR3 | IgG3 | Ascites fluid | † (25) |
| VICY1 | Ii | IgG1 | Ascites fluid | ‡ (26) |
| CerCLIP.1 | CLIP | IgG1 | Ascites fluid | § (9) |
| 5C1 | HLA-DMα | IgG | Culture supernatant | § (27) |
| FITC anti- | Mouse IgG | Sheep antiserum | Sigma |
FITC, fluorescein isothiocyanate.
American Type Culture Collection, Rockville, MD.
Gift from J. Trowsdale (Imperial Cancer Research Fund, London).
Gift from W. Knapp, (University of Vienna, Vienna, Austria).
Gift from P. Cresswell.
DNA Constructs and Transfections.
The constructs used for transfecting M1 cells with DRB1*0101 and DRA1*0101 cDNAs and supertransfection with the cDNA encoding the p35 form of invariant chain have been described previously (19). M1.DR3 cells were generated in a similar manner using a DRB1*0301 cDNA (kind gift from D. Pious, University of Washington, Seattle) subcloned into the phβ-Apr-Neo expression vector. To generate M1.DR.DM transfectants, DMA and DMB cDNAs (kind gifts from A. Kelly, UMDS, Guy’s Campus, London) were subcloned into the eukaryotic expression vector pCEXV-3 (28); M1.DR transfectants were supertransfected with DM vectors and pcEXV-1-Mogpt plasmid and selected in mycophenolic acid/xanthine/hypoxanthine.
HLA-DR transfectants (M1.DR1 and M1.DR3) with high levels of cell surface expression were selected by fluorescence-activated cell sorting (EPICS cell sorter, Coulter) after staining with the anti-DRα mAb L243 and then cloned by limiting dilution. Cells supertransfected with the Ii and DM cDNAs were cloned and screened by intracytoplasmic staining with the VICY1 antibody, or by Western blotting of whole cell lysates using the DMA-specific mAb 5C1 and a rabbit anti-DMB antiserum. All transfectants were screened for appropriate constitutive and inducible expression of DR, Ii, DM, intercellular adhesion molecule 1 (ICAM-1), and lymphocyte function-associated antigen 3 (LFA-3) molecules before being used for functional and biochemical analyses.
Western Blot Analysis.
Cells (106) were pelleted, washed three times in ice-cold PBS, and lysed for 10 min on ice in lysis buffer [10 mM Tris⋅HCl, pH 7.4/150 mM NaCl/0.1% NaN3/5 mM EDTA/0.5% Nonidet P-40 (NP-40; Sigma), 0.2% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS; Sigma), with freshly added phenylmethanesulfonyl fluoride (PMSF) at 200 μg/ml] and centrifuged at 14,000 × g at 4°C. Lysates were then added to an equal volume of Laemmli sample buffer [0.0625 M Tris⋅HCl, pH 6.8/2% SDS/20% (vol/vol) glycerol] with 2% (vol/vol) 2-mercaptoethanol (2ME). Samples were either kept at room temperature or boiled for 5 min before electrophoresis in SDS/10% polyacrylamide gels and electroblotting onto nitrocellulose. The filters were then “blocked” for 1 hr at room temperature, followed by incubation with optimal concentrations of the primary antibodies for 1 hr at room temperature. After extensive washing, sheep anti-mouse IgG, F(ab)2, conjugated with horseradish peroxidase (Amersham International) was added for 1 hr at room temperature and antibody binding was revealed by using the chemiluminescent ECL system (Amersham International).
Immunofluorescence Analysis.
Cells were stained in a conventional manner in 96-well plates using saturating concentrations of primary antibodies and FITC-conjugated sheep anti-mouse IgG as the secondary antibody. After staining, cells were fixed with 1% paraformaldehyde and analyzed by flow cytometry using an Epics Profile Analyser (Coulter). In some experiments, adherent APC were split 4 days before the assay and treated with 1000 units/ml of recombinant interferon γ (rIFNγ; kindly provided by A. Meager, National Institute of Biological Standards and Control, Potters Bar, U.K.). Nontreated cells were grown in parallel for use as controls.
T Cell Proliferation Assays.
Adherent cells were trypsinized and treated with mitomycin-C (60 μg/ml) for 45 min at 37°C. B-LCL were irradiated with 120 Gy. Transfectants and B-LCL were then cultured with T cells at the cell numbers indicated in figure legends, in flat-bottomed 96-well plates in a total volume of 200 μl. After incubation for 48 hr at 37°C in a 7% CO2/93% air atmosphere, wells were pulsed with 1 μCi (37 kBq) of [3H]thymidine (Amersham International) and harvested onto glass fiber filters 16 hr later. Specific proliferation was assessed by incorporation of [3H]thymidine in the presence of T cells minus incorporation of [3H]thymidine in the absence of T cells and expressed as Δ cpm, or, when backgrounds were less than 200 cpm, as total cpm. All backgrounds were less than 1500 cpm. Standard errors were routinely less than 10%.
RESULTS
M1 Cells Do Not Constitutively Express DR, Ii, or HLA-DM Molecules but Express Conformationally Intact Dimers upon Transfection with DRA and DRB Genes.
To explore the independent influences of Ii and HLA-DM on alloantigen presentation by DR molecules, a series of transfected human fibroblast cell lines was generated expressing HLA-DR1 or -DR3 with or without Ii and HLA-DM molecules. M1 does not constitutively express DR, Ii, or HLA-DM mRNA (L.L., unpublished data; ref. 19). This was confirmed by flow cytometric analysis (data not shown and Fig. 2) and Western blotting (Fig. 1A). Transfection with the cDNA encoding the p35 form of Ii led to strong expression of Ii (Fig. 1A). The two lower molecular weight bands seen in the Ii transfectants were presumed to represent breakdown products, or altered glycosylated forms of ER-retained Ii (29).
Figure 2.
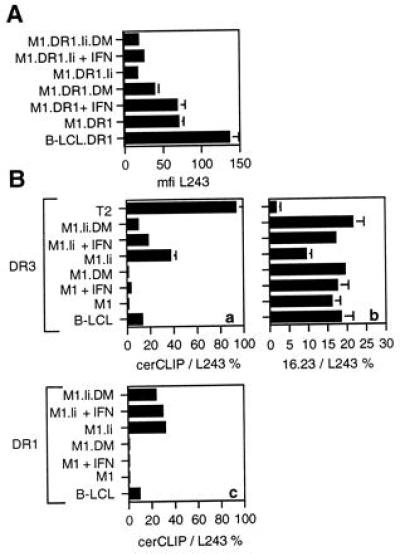
DR molecules expressed in the absence of Ii and DM display normal levels of the conformation-sensitive epitope 16.23. (A) Cells were stained with the anti-DRα mAb L243, followed by FITC-conjugated anti-mouse Ig, and analyzed by flow cytometry. The data shown give the mean fluorescence intensity (mfi) of L243 on the different cell lines from an average of 12 experiments. The error bars represent the SEM. All the cells were 100% positive for expression of DR molecules. (B) Cells were stained with cerCLIP.1 (anti-DR-bound CLIP), or 16.23 (DR3-specific conformation-dependent) followed by FITC-conjugated anti-mouse Ig. The level of expression of cerCLIP.1 (a and c) and 16.23 (b) has been normalized for the level of total DR as measured by mfi of parallel staining with L243 and expressed as a percentage of total DR. B-LCL, M1, or T2 expressed either DR3 (a and b) or DR1 (c). Where indicated the cells were induced with 1000 units/ml rIFNγ for 4 days before staining (+ IFN). The data represent the mean values from 3–16 repeated stainings depending on cell line, and error bars represent the SEM.
Figure 1.
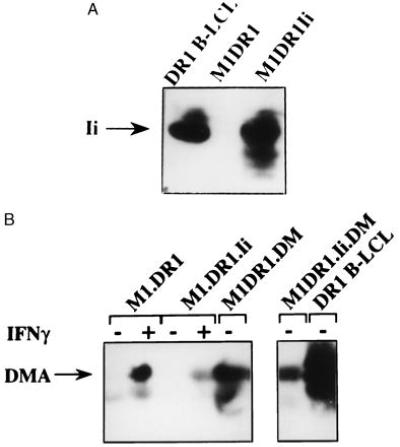
M1.DR1 is constitutively negative for expression of Ii and HLA-DM. Whole cell lysates were transferred onto nitrocellulose and probed with mAb VICY1 (A) or mAb 5C1 (B); antibody binding was detected with the ECL system (Amersham International). For B, cells were grown in the absence or presence of rIFNγ as indicated. DMA, HLA-DMα chain.
Western blotting using an anti-HLA-DM mAb, 5C1, confirmed constitutive absence of HLA-DM in M1.DR1 cells (Fig. 1B). However, treatment with rIFNγ induced HLA-DM expression to levels comparable to those in M1.DR1 cells expressing transfected HLA-DM (Fig. 1B). Western blots probed for HLA-DMA commonly reveal two bands, thought to represent different glycosylated forms (Frances Sanderson, personal communication). The transfectants expressed significantly lower levels of HLA-DMA compared with those seen in DR1 B-LCL (Fig. 1B) but this was accompanied by lower levels of cell surface DR expression. The mean levels of DR expression on the different DR1 cell lines are shown in Fig. 2A. Similar levels were seen on the DR3 cell lines. The presence of HLA-DM was also reflected by a reduction in the proportion of CLIP-occupied DR molecules, detected by the antibody cerCLIP (Fig. 2B, a and c). M1.DR transfectants showed negligible staining with cerCLIP, even following rIFNγ induction. In contrast, and as expected from the literature (e.g., ref. 9), supertransfection with Ii without HLA-DM led to a marked increase in the proportion of CLIP-occupied DR molecules at the cell surface which was reduced by co-expression of HLA-DM. The reduction was more marked in M1.DR3.Ii.DM compared with M1.DR1.Ii.DM due to the higher level of expression of HLA-DM in the former (data not shown). T2.DR3 was used as a positive control, as DR molecules in this Ii+HLA-DM− line are largely occupied with CLIP (30).
Cell surface staining with broadly reactive anti-DR antibodies such as L243, DA6.231, and 4C7 revealed no differences between the DR1 and DR3 molecules expressed by M1.DR transfectants in the presence or absence of Ii or HLA-DM, and those expressed by B-LCL (data not shown). The epitope recognized by the 16.23 mAb depends upon DR3 molecules acquiring a mature conformation, and is lost in DM-negative B-LCL (10, 11, 31, 32) as illustrated for T2-DR3 in Fig. 2Bb. Similarly, the proportion of DR3 molecules expressing 16.23 was reduced in M1.DR3.Ii compared with normal B-LCL and was restored by induction of or transfection with HLA-DM (Fig. 2Bb). However, the M1.DR3 transfectants, despite the absence of HLA-DM, displayed levels of the 16.23 epitope comparable with those on normal DR3+ B-LCL (Fig. 2Bb), and these were not influenced by coexpression of HLA-DM. This suggested that cell surface DR3 molecules expressed in M1.DR3 acquired a mature conformation, despite the absence of both Ii and HLA-DM, and that the 16.23 epitope is HLA-DM dependent only when Ii is present. Further, stable DR dimers, of heterogeneous molecular weight, were detectable on Western blotting and cell surface iodination (data not shown).
These serological data suggest that the cell surface DR molecules expressed by the transfected cell lines were conformationally intact. To determine whether the absence of Ii resulted in the display of an altered set of peptides at the cell surface and whether these would be influenced by the presence of HLA-DM, the patterns of alloreactivity of T cell clones raised against DR+Ii−DM− and DR+Ii+DM+ APC were defined.
Most Alloreactive T Cell Clones Raised Against DR1+ PBMC Stimulators Failed to Recognize M1.DR1 Cells.
Two sets of anti-DR1 alloreactive T cell clones were used as tools to address the influence of Ii on peptide occupancy of MHC class II molecules in intact cells. The first was generated against allogeneic, DR1+Ii+HLA-DM+ PBMC (22, 23). The majority of these clones (4/6) were poorly stimulated by M1.DR1 and none were stimulated by M1.DR1.Ii (clones L9, K18, G3, and G8, Figs. 3, 5, and 6). The lower level of DR expression on the M1.DR1 cells, coupled with the absence of B7 costimulatory molecules on the transfectants, may have contributed to the failure of some clones to respond to the M1.DR1 cells. However, as shown in Fig. 3, two clones, L9 and K18, proliferated when stimulated by M1.DR1 (results shown are representative of at least four experiments). Furthermore, despite the lower level of DR expression on M1.DR1 compared with B-LCL (Fig. 2A), K18 was consistently stimulated at least as well by M1.DR1 as by B-LCL, suggesting an increased display of a peptide that was expressed in limiting amounts by the DR molecules on B-LCL. However, these experiments do not indicate whether a different set of peptides is presented in the absence of invariant chain. This was addressed directly by raising T cell clones against Ii− transfectants.
Figure 3.
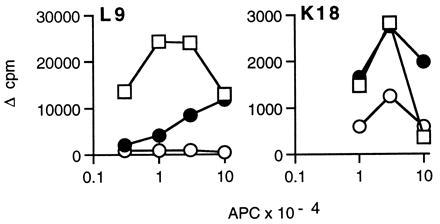
Comparison of presentation by M1.DR1, M1.DR1.Ii, and DR1.B-LCL to two alloreactive T cell clones raised against PBMC. Titrations of mitomycin-C-treated M1.DR1 (•), M1.DR1.Ii (○), or irradiated DR1.B-LCL (□) were used to stimulate 104 alloreactive T cells. After 48 hr, 1 μCi of [3H]thymidine was added to each well. The cultures were harvested 16 hr later and radioactivity was measured by liquid scintillation spectroscopy. Results are expressed as mean Δ cpm from triplicate cultures. SEM were all less than 10%.
Figure 5.
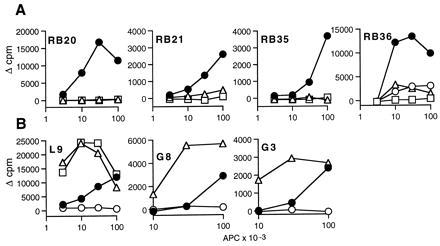
Expression of HLA-DM in M1.DR1.Ii inhibits presentation to alloreactive T cell clones raised against M1.DR1.B7 but augments presentation to alloreactive T cell clones raised against PBMC. The ability of M1.DR1.Ii.DM (▵) to stimulate T cell clones raised against M1.DR1.B7 (A) and PBMC (B) was compared with that of M1.DR1 (•), DR1.B-LCL (□) and M1.DR1.Ii (○). The assays were as described in the legends of Figs. 3 and 4 in the text. SEM were all less than 10%. Data for presentation by M1.DR1.Ii are not included for clones RB20, RB21, and RB35 as it fails to stimulate and comparable data are shown for two of these clones in Fig. 4. For clarity, the B-LCL curves have been omitted for the G3 and G8 assays. Maximal stimulation by B-LCL was 27,456 Δ cpm for G3 and 19,149 Δ cpm for G8.
Figure 6.
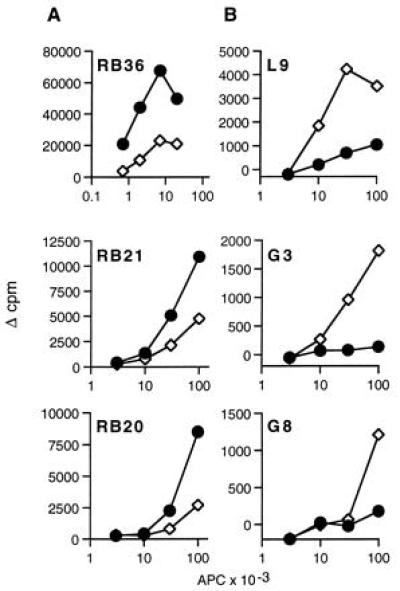
In the absence of Ii, expression of HLA-DM augments presentation by M1.DR1 to alloreactive T cell clones generated against allogeneic PBMC but inhibits presentation to clones raised against M1.DR1.B7. Mitomycin-C-treated APC, M1.DR1 (•), and M1.DR1.DM (⋄) were used as stimulators for alloreactive T cell clones (104 per well) raised against M1.DR1.B7 (A) or PBMC (B). The assays were as described in the legends of Figs. 3 and 4 and in the text. SEM were less than 10%.
T Cell Clones Raised Against M1.DR1 Failed to Recognize M1.DR1. Ii or DR1.B-LCL.
To determine whether a distinct set of peptides was displayed by class II molecules expressed in the absence of the Ii chain, M1.DR1, supertransfected with B7.1, which is required for the initiation of a primary alloresponse (20), were used to generate anti-DR1 clones from peripheral blood T cells. M1.DR1.B7 cells were potent stimulators, and a panel of 14 anti-DR1 clones was generated from short-term T cell lines. Of these 14 clones, 13 were relatively B7 independent (data not shown) and proliferated in response to M1.DR1 but failed to proliferate or proliferated only weakly in response to DR1.B-LCL, despite the significantly higher level of DR1 expression by the B cells. Further, M1.DR.Ii stimulated little or no response. The results are shown for six representative clones in Fig. 4.
Figure 4.
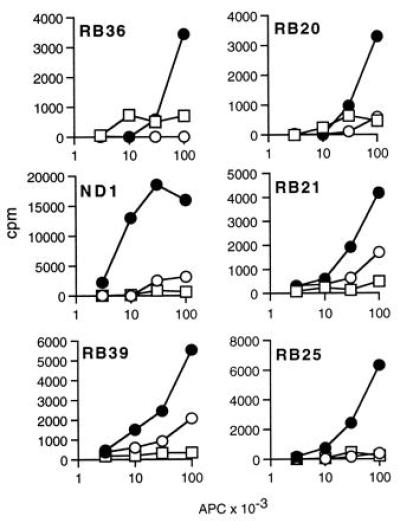
Comparison of presentation by M1.DR1, M1.DR1.Ii, and DR1.B-LCL to six alloreactive T cell clones raised against M1.DR1.B7. Titrations of mitomycin-C-treated M1.DR1 (•), M1.DR1.Ii (○), or irradiated DR1.B-LCL (□) were used to stimulate alloreactive T cells (104 cells per well). The assays were undertaken as described in the legend of Fig. 3. SEM were all less than 10%.
The Alloreactive Clones Raised Against M1.DR1 Do Not Recognize a Lineage-Specific Epitope Loaded in the Conventional Class II Pathway.
To address the possibility that the anti-DR1 clones raised against M1.DR1 were not simply specific for lineage-specific peptides loaded in the classical class II pathway and absent from B-LCL, the clones were tested against M1.DR1.Ii.DM, since coexpression of Ii and HLA-DM should optimize the presentation of peptides loaded in the endocytic pathway. However, as illustrated for four clones in Fig. 5A, M1.DR1.Ii.DM failed to stimulate the RB clones tested, making this a highly unlikely explanation. The lack of response was not simply attributable to the low levels of DR expressed at the cell surface because clones raised against PBMC were better stimulated by M1.DR1.Ii.DM than by M1.DR1 or M1.DR1.Ii (illustrated for three clones in Fig. 5B).
HLA-DM Influences T Cell Allorecognition of HLA-DR1 Independently of Ii.
The role of HLA-DM in liberating the CLIP peptide from MHC class II molecules and facilitating the loading of endosomally derived peptides is well established. It is also clear from in vitro systems using soluble molecules that HLA-DM can facilitate peptide exchange by class II molecules, involving the release of peptides other than CLIP (33). The two panels of anti-DR1 clones were used to determine whether HLA-DM has a more general effect on the array of peptides displayed by MHC class II molecules. As shown in Fig. 6B, the responses of three conventionally raised clones, L9, G3, and G8, to the DR1-expressing transfectants were markedly enhanced when HLA-DM was coexpressed. In marked contrast, proliferation of the alloreactive T cell clones raised against M1.DR1.B7—i.e., Ii−DM− APC, was inhibited by the presence of HLA-DM in M1.DR1 (Fig. 6A).
DISCUSSION
The results presented here suggest that in the absence of Ii, MHC class II molecules display an altered array of self-peptides, and that this is influenced by HLA-DM even in the absence of Ii. On the basis of these observations, if circumstances arise in vivo in which the Ii chain becomes limiting, DR molecules might present novel self-peptides to which the T cell repertoire has not been rendered tolerant. In addition, these findings support the hypothesis that HLA-DM has a broader role in antigen presentation by MHC class II molecules than merely that of facilitating the removal of CLIP.
Three possible mechanisms that could account for the altered patterns of alloreactivity to the DR+ cells with or without Ii and with or without HLA-DM were addressed experimentally. No evidence was obtained for any conformational difference between the DR1 molecules expressed at the surface of the M1.DR1 cells and those on B-LCL. Furthermore, the M1.DR1 cells have been shown previously to be fully competent at presenting peptide to DR1-restricted T cells (19). The data did not support the suggestion that the T cell clones raised against the M1.DR1 cells required fibroblast-specific peptides presented in the classical endocytic pathway, in that coexpression of Ii and DM that should have optimized the presentation of such peptides reduced the clones’ reactivity. The data therefore favor the third explanation, namely that, in the absence of Ii, MHC class II molecules become occupied by a distinct set of naturally processed peptides. This may result from the association of DR molecules with unfolded proteins or polypeptides within the ER. Previously it has been argued that, in the absence of Ii, peptide loading in the ER does not occur because class II assembly and maturation are severely disrupted and little class II egresses to the cell surface, as best illustrated in Ii knockout mice (12, 13). Others demonstrated that in the absence of Ii, class II molecules associated with other polypeptides in the ER, including chaperone molecules (34, 35) thought to be involved in retaining misfolded class II. However, using an in vitro translation system, it was shown that nascent class II molecules could bind peptide in microsomes and that this precluded binding with Ii (36). We have made parallel observations in DR-transfected insect and HeLa cells in which high molecular weight MHC class II-containing complexes are seen. Protease digestion of these complexes revealed that they contained SDS-stable αβ dimers (G.A., L. Karlsson, M. Vestberg, L. Teyton, R.L., and P. A. Peterson, unpublished results). Similar results have been reported by Busch et al. (37) for HeLa cells. The pattern of heterogeneous DR dimers seen on Western blots of M1.DR1 cells is very similar (data not shown). Busch et al. (37) showed that such class II–protein complexes were transported out of the ER in Ii− HeLa cells and that mutations within the peptide binding groove impaired the association with polypeptides. Our data with the alloreactive T cell clones suggest that the binding of peptides or proteins in the ER is functionally significant in that it leads to the display of a distinct set of cell surface class II–peptide complexes.
HLA-DM-mediated inhibition of DR1 allorecognition by T cell clones raised against M1.DR1.B7 fibroblasts is consistent with pre-endosomal loading of DR molecules in Ii− cells with peptides that are subsequently exchanged in the endosomal compartment when HLA-DM is present. HLA-DM presumably facilitates exchange of low-affinity peptides for those that bind DR1 molecules more efficiently. In the absence of Ii, DR molecules may route to the endosomes via the cell surface or directly from the ER by using the DRβ chain dileucine endosomal targeting motif (38). The ability to enter the endosomal pathway in the absence of Ii appears to be cell type specific (37, 39). Two very recent in vivo studies in mice have supported the concept that altered routing of class II (38) or the absence of Ii (40) generates cell surface class II–peptide complexes that differ from those on conventional APC and alter T cell antigen- and allo-reactivity. The recently published data from Katz et al. (41) are consistent with our results and contain some complementary data. In that study, all the T cells used were generated in a conventional manner, against Ii+ and H-2M+ stimulator cells. Surprisingly, they demonstrated that 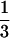 of these hybridomas showed reduced responses when stimulated by H-2M+ APC, suggesting that HLA-DM led to a reduction in surface expression of relevant class II–peptide complexes. In our study, the expression of HLA-DM enhanced the responses of all conventionally raised clones tested. We have taken the analysis further by also raising T cell clones against Ii− and HLA-DM− stimulator cells, to test the hypothesis that the absence of Ii and HLA-DM would lead to altered peptide display. The differing specificities of the M1.DR1-raised clones and the inhibitory effects of the presence of HLA-DM in the absence of Ii on presentation to such clones provide compelling data to support our hypothesis.
of these hybridomas showed reduced responses when stimulated by H-2M+ APC, suggesting that HLA-DM led to a reduction in surface expression of relevant class II–peptide complexes. In our study, the expression of HLA-DM enhanced the responses of all conventionally raised clones tested. We have taken the analysis further by also raising T cell clones against Ii− and HLA-DM− stimulator cells, to test the hypothesis that the absence of Ii and HLA-DM would lead to altered peptide display. The differing specificities of the M1.DR1-raised clones and the inhibitory effects of the presence of HLA-DM in the absence of Ii on presentation to such clones provide compelling data to support our hypothesis.
Our conclusion that the discriminatory responses of the T cells reflects differences in peptide display is inferential. Clearly, direct proof will be gained only by defining the peptides recognized by the M1.DR1-raised alloreactive T cell clones, which will require the reconstitution of T cell responses with eluted, HPLC-fractionated, naturally processed peptides.
These observations have potentially important implications for the role of discordant regulation of Ii and HLA-DM in antigen presentation, and they suggest that HLA-DM may influence peptide occupancy of MHC class II molecules more broadly than merely by catalyzing the removal of CLIP. M1 demonstrates clear discordant regulation of HLA-DM, Ii, and class II gene expression. Up-regulation of class II molecules but not Ii has been reported in bone marrow macrophages (16) and in gut epithelial cells (17). Limited comparisons have been made of the levels of induction of MHC class II, Ii, and HLA-DM in specialized APC and in other cell types in which MHC class II molecule expression is induced by agents such as IFNγ. If the ratios of class II, Ii, and HLA-DM molecules vary in vivo, particularly in the context of an inflammatory response, these data suggest that this would lead to the display of an altered set of self-peptides. Such altered display might predispose to the induction of an autoimmune response, the characteristics of which would be determined by the cell type on which class II expression occurred.
Acknowledgments
We thank Liz Simpson and Ragnar Lindstedt for critical reading of the manuscript and for valuable discussions. L.L. was supported by a Medical Research Council Clinician Scientist Fellowship.
ABBREVIATIONS
- MHC
major histocompatibility complex
- ER
endoplasmic reticulum
- Ii
invariant chain
- APC
antigen-presenting cells
- PBMC
peripheral blood mononuclear cells
- FITC
fluorescein isothiocyanate
- rIFNγ
recombinant interferon γ
References
- 1.Newcomb J R, Cresswell P. J Immunol. 1993;150:499–507. [PubMed] [Google Scholar]
- 2.Germain R. Semin Immunol. 1995;7:361–372. doi: 10.1006/smim.1995.0041. [DOI] [PubMed] [Google Scholar]
- 3.Ghosh P, Amaya M, Mellins E, Wiley D C. Nature (London) 1995;378:457–462. doi: 10.1038/378457a0. [DOI] [PubMed] [Google Scholar]
- 4.Malcherek G, Gnau V, Jung G, Rammensee H G, Melms A. J Exp Med. 1995;181:527–536. doi: 10.1084/jem.181.2.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sette A, Southwood S, Miller J, Appella E. J Exp Med. 1995;181:677–683. doi: 10.1084/jem.181.2.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miller J. Immunol Res. 1994;13:244–252. doi: 10.1007/BF02935616. [DOI] [PubMed] [Google Scholar]
- 7.Romagnoli P, Layet C, Yewdell J, Bakke O, Germain R N. J Exp Med. 1993;177:583–596. doi: 10.1084/jem.177.3.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Avva R R, Cresswell P. Immunity. 1994;1:763–774. doi: 10.1016/s1074-7613(94)80018-9. [DOI] [PubMed] [Google Scholar]
- 9.Denzin L K, Robbins N F, Carboy Newcomb C, Cresswell P. Immunity. 1994;1:595–606. doi: 10.1016/1074-7613(94)90049-3. [DOI] [PubMed] [Google Scholar]
- 10.Fling S P, Arp B, Pious D. Nature (London) 1994;368:554–558. doi: 10.1038/368554a0. [DOI] [PubMed] [Google Scholar]
- 11.Morris P, Shaman J, Attaya M, Amaya M, Goodman S, Bergman C, Monaco J J, Mellins E. Nature (London) 1994;368:551–554. doi: 10.1038/368551a0. [DOI] [PubMed] [Google Scholar]
- 12.Bikoff E K, Huang L Y, Episkopou V, van Meerwick J, Germain R N, Robertson E J. J Exp Med. 1993;177:1699–1712. doi: 10.1084/jem.177.6.1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Viville S, Neefjes J, Lotteau V, Dierich A, Lemeur M, Ploegh H, Benoist C, Mathis D. Cell. 1993;72:635–648. doi: 10.1016/0092-8674(93)90081-z. [DOI] [PubMed] [Google Scholar]
- 14.Bodmer H, Viville S, Benoist C, Mathis D. Science. 1994;263:1284–1286. doi: 10.1126/science.7510069. [DOI] [PubMed] [Google Scholar]
- 15.Lechler R I, Lombardi G, Batchelor J R, Reinsmoen N, Bach F H. Immunol Today. 1990;11:83–88. doi: 10.1016/0167-5699(90)90033-6. [DOI] [PubMed] [Google Scholar]
- 16.Schneider H-J, Opel H, Ballhausen W, Henkes W, Steinlein P, Reske K. Eur J Immunol. 1987;17:1235–1242. doi: 10.1002/eji.1830170904. [DOI] [PubMed] [Google Scholar]
- 17.Vidal K, Samarut C, Magaud J P, Revillard J P, Kaiserlian D. J Immunol. 1993;151:4642–4650. [PubMed] [Google Scholar]
- 18.Royer-Pakora B, Peterson W D, Jr, Haseltine W A. Exp Cell Res. 1984;151:408–420. doi: 10.1016/0014-4827(84)90391-4. [DOI] [PubMed] [Google Scholar]
- 19.Dodi A I, Brett S, Nordeng T, Sidhu S, Batchelor R J, Lombardi G, Bakke O, Lechler R I. Eur J Immunol. 1994;24:1632–1639. doi: 10.1002/eji.1830240727. [DOI] [PubMed] [Google Scholar]
- 20.Hargreaves R, Logiou V, Lechler R. Int Immunol. 1995;7:1505–1513. doi: 10.1093/intimm/7.9.1505. [DOI] [PubMed] [Google Scholar]
- 21.Riberdy J M, Cresswell P. J Immunol. 1992;148:2586–2590. [PubMed] [Google Scholar]
- 22.Lombardi G, Sidhu S, Lamb J R, Batchelor J R, Lechler R I. J Immunol. 1989;142:753–759. [PubMed] [Google Scholar]
- 23.George A, Dazzi F, Lynch J, Sidhu S, Marelli F, Batchelor R J, Lombardi G, Lechler R I. Int Immunol. 1994;6:1785–1790. doi: 10.1093/intimm/6.11.1785. [DOI] [PubMed] [Google Scholar]
- 24.Lampson L A, Levy R. J Immunol. 1980;125:293–299. [PubMed] [Google Scholar]
- 25.Johnson J P, Meo T, Riethmuller G, Schendel D J, Wank R. J Exp Med. 1982;156:104–111. doi: 10.1084/jem.156.1.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Quaranta V, Majdic O, Stingl G, Liszka K, Honigsmann H, Knapp W. J Immunol. 1984;132:1900–1905. [PubMed] [Google Scholar]
- 27.Sanderson F, Thomas C, Neefjes J, Trowsdale J. Immunity. 1996;4:87–96. doi: 10.1016/s1074-7613(00)80301-5. [DOI] [PubMed] [Google Scholar]
- 28.Miller J, Germain R. J Exp Med. 1986;164:1478–1489. doi: 10.1084/jem.164.5.1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schutze M P, Peterson P A, Jackson M R. EMBO J. 1994;13:1696–1705. doi: 10.1002/j.1460-2075.1994.tb06434.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Riberdy J M, Newcomb J R, Surman M J, Barbosa J A, Cresswell P. Nature (London) 1992;360:474–477. doi: 10.1038/360474a0. [DOI] [PubMed] [Google Scholar]
- 31.Pious D, Dixon L, Levine F, Cotner T, Johnson R. J Exp Med. 1985;162:1193–1207. doi: 10.1084/jem.162.4.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mellins E, Kempin S, Smith L, Monji T, Pious D. J Exp Med. 1991;174:1607–1615. doi: 10.1084/jem.174.6.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Denzin L K, Cresswell P. Cell. 1995;82:155–165. doi: 10.1016/0092-8674(95)90061-6. [DOI] [PubMed] [Google Scholar]
- 34.Busch R, Vturina I Y, Drexler J, Momburg F, Hammerling G J. Eur J Immunol. 1995;25:48–53. doi: 10.1002/eji.1830250110. [DOI] [PubMed] [Google Scholar]
- 35.Schaiff W, Hruska K, Bono C, Shuman S, Schwartz B. J Exp Med. 1992;176:657–666. doi: 10.1084/jem.176.3.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bijlmakers M J, Benaroch P, Ploegh H L. EMBO J. 1994;13:2699–2707. doi: 10.1002/j.1460-2075.1994.tb06560.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Busch R, Cloutier I, Sekaly R-P, Hammerling G J. EMBO J. 1996;15:418–428. [PMC free article] [PubMed] [Google Scholar]
- 38.Smiley S, Rudensky A Y, Glimcher L H, Grusby M J. Proc Natl Acad Sci USA. 1996;93:241–244. doi: 10.1073/pnas.93.1.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Simonsen A, Momburg F, Drexler J, Hammerling G J, Bakke O. Int Immunol. 1993;5:903–917. doi: 10.1093/intimm/5.8.903. [DOI] [PubMed] [Google Scholar]
- 40.Ignatowicz L, Kappler J, Marrack P. Cell. 1996;84:521–529. doi: 10.1016/s0092-8674(00)81028-4. [DOI] [PubMed] [Google Scholar]
- 41.Katz J F, Stebbins C, Appella E, Sant A J. J Exp Med. 1996;184:1747–1753. doi: 10.1084/jem.184.5.1747. [DOI] [PMC free article] [PubMed] [Google Scholar]