

Abstract
Human T cell clones were analyzed for their susceptibility to activation-induced cell death (AICD) in response to CD3/T cell receptor ligation. AICD was observed only in Th1 clones and was Fas-mediated, whereas Th2 clones resisted AICD. Analysis of a panel of Th0 clones, characterized by their ability to secrete both Th1 and Th2 cytokines, revealed that this subset included both AICD-sensitive (type A) and -resistant (type B) clones. Resistance to AICD by Th2 and Th0-type B clones was not due to lack of expression of either Fas receptor or its ligand. Paradoxically, the AICD-resistant clones were susceptible to apoptosis when Fas receptor was directly ligated by anti-Fas antibodies. However, prior activation of the resistant clones by monoclonal antibodies to CD3/TCR complex induced resistance against Fas-mediated apoptosis. Thus, the Fas–FasL pathway is critical for the induction of AICD in T cells, and moreover this pathway can be negatively regulated in the AICD-resistant clones by signals that are generated from ligation of the CD3/TCR complex.
T cells can undergo apoptosis under a variety of different conditions. Cytokine deprivation induces apoptosis of activated T cells (1, 2). Tumor necrosis factor α (TNF-α) can also mediate apoptosis in T cells through the p75 TNF-α receptor (3). A third form of apoptotic death is observed in T cells called activation-induced cell death (AICD) (4). AICD occurs as a consequence of repeated stimulation through the CD3/TCR (T cell receptor) of the T cells. Fas/APO-1 is a cell surface receptor belonging to the nerve growth factor receptor-TNF-α receptor family of molecules, and Fas ligand (FasL) is a member of the corresponding family of TNF-related cytokines. A role for Fas receptor (FasR) and its ligand in mediating AICD, was first drawn from studies with lpr and gld strains of mice that are deficient in functional expression of FasR and FasL, respectively (5, 6). Mature activated T cells from both lpr and gld mice are resistant to apoptosis induced by reactivation through their TCRs (5, 6). Direct evidence that AICD of mature T cells is mediated through Fas–FasL was demonstrated by several groups in T cell hybridomas, Jurkat T leukemia cells, and nontransformed preactivated T cells (7–9). All three groups reported that TCR engagement up-regulates expression of both Fas and its ligand, and that apoptosis can be inhibited by blocking either the receptor or its ligand.
In the immune system, AICD acts as a feedback mechanism for terminating an ongoing immune response (10) and serves to maintain peripheral tolerance (11, 12). Importantly, AICD may also have a major role in regulating the immune responses in disease. For example, Fas-triggered inappropriate apoptosis of peripheral T cells has been implicated in the loss of CD4+ T cells in HIV-infected individuals (13–15). T cells from individuals infected with either the Epstein–Barr virus (16, 17) or the varicella-zoster virus (17) also undergo extensive AICD in vitro. In a murine model of Schistosoma mansoni infection, a progressive increase in apoptosis of activated T cells was observed in conjunction with a decrease in T cell functions (18).
The Th1 and Th2 subsets of T cells are functionally distinct, and are defined on the basis of their cytokine profiles (19–22). Th1 cells produce interferon γ (IFN-γ), TNF-α, and interleukin 2 (IL-2), and contribute to cell-mediated immunity; Th2 cells secrete IL-4 and IL-5, and serve to help antibody responses; Th0 cells are distinguished by their ability to produce both Th1 and Th2 cytokines and are thought to be precursors to the Th1 and Th2 subsets (23). Although much is known about the functions of Th1 and Th2 cells, molecular distinctions between the two subsets are as yet poorly defined. Given the established biological significance of the delineation of T cell subsets (22, 24), it becomes important to understand whether AICD is one means of regulating subset development. Therefore, the goal of this study was to examine the regulation of AICD in antigen-specific T cells comprising the Th1, Th2, and Th0 subsets.
MATERIALS AND METHODS
T Cell Clones.
Mycobacterium leprae, tetanus-toxoid (TT), and purified protein derivative (PPD)-reactive clones were established by limiting dilution technique as previously described (25). The Th1 and Th2 clones were maintained in IL-2, with biweekly stimulation with antigen and antigen-presenting cells. To maintain their cytokine profile and provide the least amount of biasing in vitro, the Th0 clones were maintained in IL-2, with biweekly stimulation with antigen presenting cells and phytohemagglutinin. Nonspecific stimulation with PHA activates both Th1- and Th2-type cytokine gene expression and allows for maintenance of the Th0 phenotype. We have successfully used this protocol for stimulating the Th0 clones and have observed that even after several passages in vitro, the clones have retained their antigen-specificity and mixed cytokine profile. Subset-specificity of the clones was determined by measuring production of IFN-γ, IL-4, and TNF-α. For each T cell clone, 1 × 105 cells were added in 1 ml of Iscove’s media containing 10% human type AB serum in 24-well tissue culture plates, precoated with 2.5 μg of antibody to CD3 (BioSource International, Camarillo, CA). Supernatants from the stimulated cells were harvested 18–20 hr later, and cytokine quantities were measured by sandwich enzyme immunoassay using appropriate pairs of capture antibodies and biotinylated detecting antibodies (PharMingen). The levels of sensitivity for the IFN-γ, IL-4, and TNF-α ELISAs were 15 pg/ml.
Proliferation of T Cell Clones.
One hundred microliters of 2.5 μg/ml anti-CD3 antibody were added to each well in 96-well plates and allowed to bind for 1 hr at room temperature. The wells were rinsed thoroughly prior to addition of 1 × 104 T cell clones per well. Proliferation was measured at 72 hr by incorporation of an 8-hr pulse of [3H]-labeled thymidine.
Activation Induced Apoptosis in T Cell Subsets.
Apoptosis of all clones listed in Fig. 1 was measured by ELISA. T clones (4 × 104) were stimulated with immobilized antibodies to CD3 in 96- well tissue culture plates. Eleven to twelve hours later the cells were harvested and assayed for apoptosis using the Cell Death Assay kit (Boehringer Mannheim). Briefly, the cells were lysed in 500 μl of lysis buffer, and the cytoplasmic fractions were collected by centrifugation at 13,000 rpm. The cytoplasmic fractions were further diluted to contain 20,000 cell equivalent per ml, and presence of nucleosomes in the cytoplasm was tested by the sandwich-enzyme-immunoassay using mouse mAbs directed against DNA and histones. The cell death ELISA utilized a anti-histone capture antibody that bound to the histone component of the nucleosomes contained in the sample lysates. The bound nucleosomes were then detected by reacting with peroxidase-labeled anti-DNA antibodies. The reaction was developed with a peroxidase substrate buffer and development of color was photometrically determined by measuring absorbance values at 405 nm. Cells maintained in IL-2 served as control cells, and data are presented as percent apoptosis, an arbitrary unit calculated as 100 − (absorbance values at A405 of IL-2 cultures/absorbance values at A405 of CD3-stimulated cultures × 100).
Figure 1.

Cytokine profiles of T cell clones. Clone 59.5 is mycobacterial-specific and generated from a tuberculoid leprosy patient (21). PPD and TT-reactive clones were generated from immunized donors, and are distinguished by the prefix P and T, respectively. Clones T1, T3, T10, T14, and T15 were derived from a tetanus toxoid-reactive donor. Clones P3, P4, P7, and P2 were derived from a normal individual who is PPD-reactive. Clone P13 was generated from another PPD-reactive normal individual.
Immunofluorescent Labeling for FasR and FasL.
T cell clones were stimulated with antibodies to anti-CD3 for 4 hr. Cells to be used for FasL analysis were stimulated in the presence of 25 μM of the metalloprotease inhibitor KB8301 (Kanebo). Addition of KB8301 prevents FasL cleavage, resulting in high levels of cell surface expression of FasL (26). Stimulated cells were harvested, centrifuged, and resuspended in primary biotinylated anti-Fas or anti-FasL antibodies for 30 min at 4°C. Cells were washed and immunofluorescence was detected by reacting the cells with streptavidin-phycoerythrin for 30 min at 4°C. Flow cytometry was performed on an electronically programmable individual cell sorter Elite flow cytometer (Coulter).
Bystander Activation-Induced Cytotoxicity.
A previously described protocol (27) was utilized to measure activation-induced cytotoxicity of T cells. Jurkat T cells constitutively expressing only FasR and not FasL were labeled with [3H]thymidine (10 μCi/ml; 1 Ci = 37 GBq), and served as target cells. Effector T cells either unactivated or activated with anti-CD3 antibodies for 3 hr, were combined with 2 × 104 labeled target cells at different target to effector ratios. Eight hours later labeled unfragmented DNA with high molecular weight was harvested onto glass fiber filters and radioactivity measured in a scintillation counter. Data are expressed as percent specific cytotoxicity and calculated as [S − E/S] × 100, where S is [3H]thymidine incorporation in Jurkat T cells in the absence of effector cells; and E is incorporation in Jurkat T cells in the presence of effector cells.
RESULTS
Proliferation of T Cell Subsets to CD3 Antibodies.
We have characterized responses of 13 antigen-specific human T cell clones to CD3/TCR occupancy (Fig. 1). Three clones produce IFN-γ and TNF-α and are Th1-type. Two clones are Th2-type and secrete IL-4. The remaining eight clones are Th0-type, since they make all three cytokines. The Th1 clones (59.5, T1, and T17) proliferate weakly when stimulated by cross-linking of the CD3/TCR receptor. In contrast, the Th2 clones (T3 and P3) proliferate very well in response to CD3 stimulation alone, with cpm values of >20,000 (Fig. 2). Only three of the eight Th0 clones (T10, T14, and T15) proliferate to CD3 stimulation. We have therefore divided the Th0 clones into two subsets, Th0-type A (P1, P2, P4, P7, and P13) and Th0-type B (T10, T14, and T15).
Figure 2.

Proliferative responses of T cell clones to antibodies to CD3. T cell clones (1 × 104) were stimulated by immobilized antibodies to CD3. Proliferation was measured at 72 hr by incorporation of an 8-hr pulse of [3H]thymidine. Data are presented as counts per min (cpm).
Th2 and Th0-Type B Cells Are Resistant to AICD When Activated Through the TCR/CD3 Complex.
To determine why the Th1 and Th0-type A clones do not proliferate in response to CD3 stimulation, we examined the ability of the clones to undergo AICD (7–9). We have employed an ELISA that correlates with the DNA ladder technique for detecting apoptosis. We tested the panel of T cell clones described in Fig. 1 for their ability to undergo AICD following ligation of the CD3/TCR complex. All of the Th1 and Th0-type A clones underwent AICD when stimulated with antibodies to CD3 alone. In contrast, the Th2 clones and the Th0-type B clones were resistant to apoptosis (Fig. 3a). Two Th0 clones, P2 (AICD sensitive) and T15 (AICD resistant), were further tested for apoptosis as a duration of CD3 stimulation. There was a time dependent increase in apoptotic activity in clone P2 (Fig. 3b). DNA fragmentation is an early event in apoptosis, whereas cell membrane disintegration occurs later; therefore, the apparent decrease of apoptotic activity in clone P2 at 19 hr is due to complete cell lysis (Fig. 3b). No apoptosis was observed in the T15 clone at any time (Fig. 3b), ruling out a temporal delay in the induction of apoptosis in the resistant clones.
Figure 3.
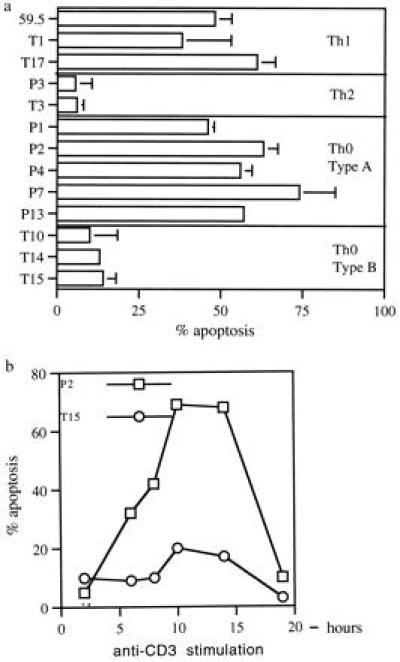
(a) AICD in T cell subsets. T clones (4 × 104) were stimulated with antibodies to CD3, and apoptosis was measured by ELISA. (b) Kinetics of apoptosis induction. At appropriate time points after CD3 stimulation, cells were harvested and assayed for apoptosis by ELISA. Maximum apoptosis occurred between 8–12 hr poststimulation.
Role of a Fas–FasL Pathway in AICD to CD3 Stimulation.
To test for a Fas–FasL pathway of AICD (7–9), induction of apoptosis was carried out in three AICD-sensitive clones (59.5, T17, and P4), in the presence or absence of a Fas–Fc fusion protein that blocks Fas–FasL interaction. Apoptosis is substantially inhibited by the presence of the fusion protein (Fig. 4a), indicating a role for a Fas–FasL pathway in the AICD-sensitive clones. It has been observed that resistance of Th2-type clones to AICD is associated with decreased expression of FasL (28–30). However, by immunofluorescence, we observed no differences in the expression of either FasR or FasL between AICD-resistant (P3, T3, T10, T14) and sensitive clones (59.5, T17, P2, and P13) (Fig. 4b). To further determine if the FasL expressed by the resistant clones is functional we carried out a bystander cytotoxicity assay. Two AICD-resistant (T3 and T15) and two-susceptible (P2 and P13) clones were either stimulated with anti-CD3 antibodies to induce FasL expression or left untreated. The clones were then tested for their ability to induce DNA fragmentation in Jurkat T cells that constitutively express only the FasR and not the FasL (data not shown). As shown in Fig. 4c all four clones, following activation, demonstrated a dose-dependent cytotoxicity for the target Jurkat cells. Unactivated cells were not cytotoxic. When Fas–Fc fusion protein was included in cultures containing a target to effector ratio of 1:4, cytotoxicity was substantially inhibited (Fig. 4c). These data indicate that the FasL expressed by the apoptosis-resistant T cell clones is functional, since it is able to ligate the FasR on the target Jurkat cells and induce their apoptosis.
Figure 4.
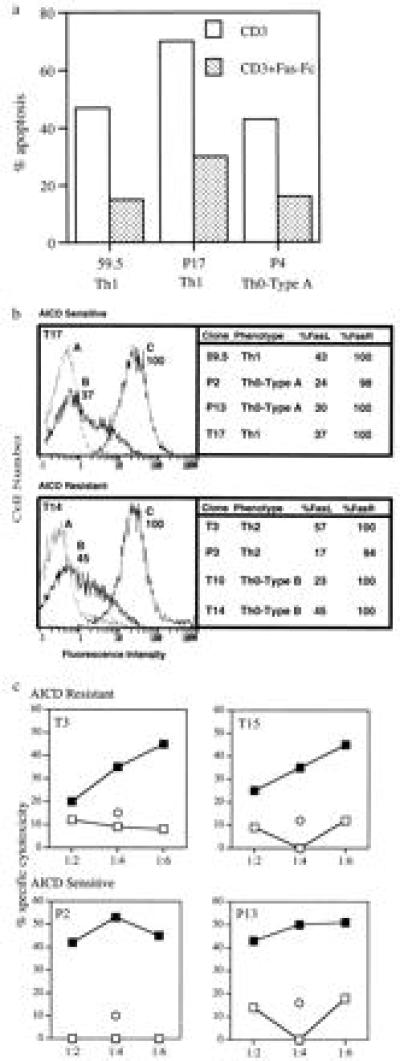
(a) AICD is inhibited by Fas–Fc fusion protein. Apoptosis-sensitive clones 59.5, P4, and T17 were stimulated with anti-CD3 antibodies in the absence (□) or presence (▪) of 10 μg/ml of Fas–Fc fusion protein and assayed for apoptosis by ELISA. Fusion protein Fas–Fc is composed of human Fas and a human immunoglobulin constant region. (b) Expression of FasR and FasL on subsets of T cell clones. For immunofluorecence, 1 × 106 T clones (59.5, T17, P3, T3, P2, P13, T10, and T14) were stimulated with anti-CD3 antibodies in the presence (for FasL) or absence (for FasR) of 25 μM of the metalloprotease inhibitor KB8301. Addition of KB8301 prevents FasL cleavage, resulting in high levels of cell surface expression of FasL After 3 hr the cells were washed in PBS containing 2% serum and reacted with biotinylated antibody to FasR (C) or to the FasL (B) (PharMingen) or no antibody (A) for 45 min at 4°C. Cells were washed and developed with streptavidin-conjugated phycoerythrin (PE) for 30 min at 4°C. Immunofluorescence was measured by an electronically programmable individual cell sorter flow cytometer. Data are presented as percentage of T cells that are positive for FasR and FasL expression. The Figure includes representative fluorescence-activated cell sorter profiles of an AICD-resistant and -susceptible clone. (c) Bystander activation-induced cytotoxicity. Target cells (Jurkat T cells expressing Fas constitutively) were labeled with [3H]thymidine (10 μCi/ml). Anti-CD3-activated (filled symbols) or untreated (open symbols) effector cells (P2, T3, P13 and T15) were combined with 2 × 104 labeled targets at target to effector ratios of 1:2, 1:4, and 1:6, respectively. Eight hours later unfragmented-high-molecular-weight DNA was harvested and radioactivity was measured in a scintillation counter. Data are expressed as percent specific cytotoxicity and are representative of one of two individual experiments. Cytotoxicity for Jurkat T cells by effector cells was also carried out in the presence of 10 μg/ml of Fas–Fc fusion protein at the target to effector ratio of 1:4 (represented as ○)
Both AICD-Susceptible and -Resistant Clones Undergo Apoptosis When FasR Is Directly Ligated.
Since AICD is Fas-mediated in this system, we hypothesized that the AICD-resistant clones would also be resistant to apoptosis when their FasR was directly ligated by anti-Fas antibodies. Contrary to what we expected, the Th2 and Th0-type B clones were susceptible to apoptosis when the FasR was directly ligated by soluble anti-Fas antibodies. As shown in Fig. 5a, clones T3 (Th2-type) and T15 (Th0-type B) were resistant to apoptosis when stimulated with anti-CD3 antibodies. However, in the presence of anti-Fas antibodies alone, both these AICD-resistant clones underwent apoptosis. In parallel, we examined apoptotic responses of two AICD-sensitive Th0-type A clones (P2 and P13) to anti-CD3 and anti-Fas antibodies. As expected, both clones exhibited high apoptotic activity when stimulated with either anti-CD3 or anti-Fas antibodies.
Figure 5.
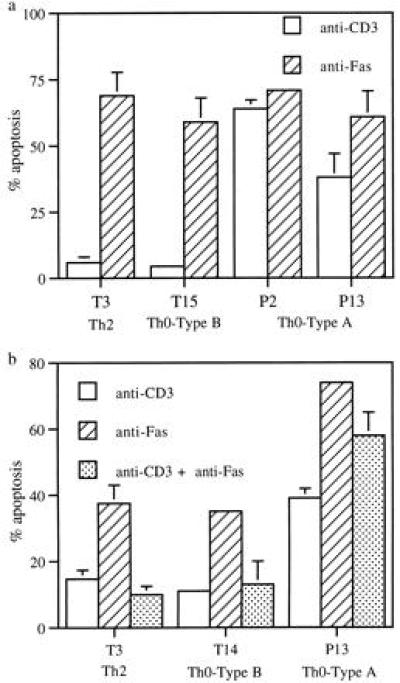
(a) Antibodies to Fas induces apoptosis in both AICD-resistant and -susceptible clones. T cell clones (5 × 104) were cultured for 4 hr in the absence and presence of varying dilutions of soluble anti-Fas mAb (AM01L from Calbiochem). Cells were harvested, and apoptosis was measured by ELISA as described earlier. (b) Activation by anti-CD3 mAb induced resistance to Fas-mediated apoptosis in AICD-resistant clones. T clones (5 × 104) were cultured for 30 min in 96-well plates coated with anti-CD3 antibody, before addition of 100 ng of soluble Fas mAb to the appropriate wells. Four hours later cells were harvested and apoptosis was measured by previously described ELISA.
Activation Through TCR/CD3 Complex-Induced Resistance of Th2 Cells to Fas-Mediated Apoptosis.
We next tested if signals generated through the TCR/CD3 receptor were affecting the sensitivity of the Th2 and Th0-type B cells to FasR ligation. Stimulation of Th2 and Th0-type B clones with anti-CD3 for 30 min prior to addition of soluble anti-Fas antibody induced resistance to apoptosis. As shown in Fig. 5b, anti-CD3 stimulation alone did not induce apoptosis in clones T3 and T14. In the presence of Fas antibodies significant apoptosis was observed in both clones. However, prior stimulation with anti-CD3 antibodies abrogated apoptosis induced by the Fas antibodies (Fig. 5b). In the AICD-sensitive clone P13, CD3-stimulation was not able to induce resistance to apoptosis by anti-Fas antibodies (Fig. 5b).
DISCUSSION
Our study suggests that fundamental regulatory differences exist between Th1- and Th2-type cells in response to CD3-receptor ligation. Th1 clones undergo AICD when stimulated with antibodies to CD3. In contrast, Th2 clones resist AICD. Several in vivo studies have demonstrated that after encounter with either superantigens (31–33) or specific antigens (34, 35), the majority of activated T cells are deleted by AICD. However, it has been consistently observed in these systems that the deletion is never complete. Relevant to our observations, the residual cells that are not deleted after in vivo activation express a high level of Th2-type cytokines, indicating that Th2 cells are resistant to AICD even in vivo (36).
The Th0 clones tested include both AICD-sensitive and -resistant clones, raising an intriguing possibility that acquisition of an AICD-resistant or -susceptible phenotype precedes commitment to the Th1 or Th2 subset. Th1 cytokines, including TNF-α (3) and IFN-γ (37, 38), induce apoptosis in T cells. That all the Th0 clones that we have tested produce TNF-α and IFN-γ and can nevertheless be subdivided into susceptible and resistant phenotypes, indicate that AICD in our system is mediated by neither TNF-α nor IFN-γ. Using Fas–Fc fusion protein that prevents the ligation of FasL to FasR, we then demonstrated that in Th1 and Th0-type A cells AICD was mediated via Fas–FasL interaction.
There is some evidence to suggest that Th1 cells express elevated levels of FasL and are sensitive to AICD, whereas Th2 cells do not express FasL and are AICD resistant (28). However, other reports demonstrate the expression of FasL on both Th1 and Th2 cells (39). In our system the resistance of Th2 and Th0-type B clones to AICD is not due to lack of expression of either FasR or FasL, since both the apoptosis sensitive and resistant clones express equivalent amounts of both FasR and FasL. In addition, we also show that the FasL expressed by the resistant clones is functional. Consequently, we hypothesized that the Fas signal transduction pathway was impaired in the AICD-resistant clones. However, direct ligation of the FasR induced apoptosis in the AICD-resistant clones. These results were initially surprising, since it appeared paradoxical that AICD-resistant clones would be susceptible to Fas-mediated apoptosis. We then made the observation that activation through TCR/CD3 complex induced resistance in the Th2 and Th0-type B clones to Fas-mediated apoptosis. These data indicate that in AICD-susceptible clones the signal for apoptosis is generated on ligation of the FasR. However, in the resistant clones (Th2 and Th0-type B) the death signal that is transmitted following ligation of Fas to its receptor is prevented when the cells are simultaneously activated through their TCR/CD3 complex. Consistent with our observations, apoptosis of naive spleen cells induced by soluble Fas ligand can also be prevented by prior activation of the cells with mAbs to CD3/TCR complex (40). In Th1 and Th2 cells, CD3/TCR complex selectively engages different intracellular biochemical signaling pathways (41). This may explain why CD3 stimulation provides resistance to Fas-mediated apoptosis only in Th2 and Th0-type B clones.
The FasR-mediated signal transduction pathway leading to apoptosis is initiated by trimerization of the receptor (42). This is followed by sequential recruitment of two proteins to the receptor, a Fas-associating protein with death domain (FADD) and a cytosolic protease designated FLICE (43). Activation of FLICE leads to the characteristic structural changes in the cell leading to apoptosis. Bcl-2 and Bcl-xL proteins are potent inhibitors of apoptotic cell death induced by several stimuli, including FasR (44–46). Only recently has the molecular link between the Bcl-2 family of anti-apoptotic proteins and FLICE been described. Bcl-2 interacts with and modulates CED-4 (or its mammalian equivalent) which in turn binds FLICE. In the absence of Bcl-2 binding, CED-4 can activate FLICE resulting in apoptosis (47, 48). Based on the above model of the FasR signal transduction pathway, several testable predictions can be made for the ability of a subset of T cells to resist AICD. In AICD-resistant clones, CD3 stimulation could prevent oligomerization of the FasR, or inhibit recruitment of either FADD or FLICE to the FasR. Alternatively, CD3 stimulation may differentially up-regulate Bcl-2 and Bcl-xL genes in the AICD-resistant and susceptible clones (44, 45). FasR ligation also results in the hydrolysis of sphingomyelin to generate ceramides (49, 50) that is associated with activation of proteases. CD3 stimulation could interfere in the generation of ceramides in the AICD-resistant clones.
The differentiation and maintenance of T cell subsets is influenced by the strength of the activation signal that is generated from the antigen-presenting cell in association with its costimulatory molecules (51). Several experiments have suggested that both Th1 and Th2 cells require costimulation, during their differentiation (52, 53), but once established Th2 cells are less dependent on costimulation than Th1 cells (54). Thus, altering the signals generated through CD3/TCR ligation, either with different doses of anti-CD3 antibodies, or by coligating the CD28 molecule could alter the responses of Th1 and Th2 cells to AICD. Intracellular pathogens modify accessory cell functions and it is conceivable that antigen presentation by these modified macrophages alters the strength of the activation signal. For example, lipoarabinomanan, a mycobacterial-specific lipid that is secreted in large amounts by the bacteria can down-regulate antigen presentation functions of macrophages (55). Macrophages derived from the granulomas of schistosome-infected mice express reduced amounts of the costimulatory molecule B7, and specifically induce anergy in cells of the Th1-type (56). Thus, absence or presence of reduced costimulation could lead to preferential anergy and apoptosis of Th1 cells.
The biological relevance of the differential ability of Th1 and Th2 subsets of T cells to undergo AICD is underscored by the ability of the T cell subsets to influence disease outcome (22, 24). Th1 response is associated with protection from intracellular pathogens such as leishmania, while Th2 response results in exacerbation of disease (22). In contrast, preferential activation of Th1 response is central to the pathogenesis of many autoimmune diseases (57, 58) and Th2 immune response is associated with disease resistance (59). Many recent studies implicate a role for Fas–FasL pathway in the peripheral deletion of autoimmune T cells (60, 61). The data presented here suggest that increased susceptibility of Th1 type cells to AICD may be a protective mechanism of the host to delete potentially autoreactive T cells. On the other hand in chronic infections parasites exploit this to preferentially eliminate Th1 cells and switch a protective Th1 immune response to a pathogenic Th2 response. Thus, AICD may play a significant role in the suppression of Th1-dependent effector functions, thereby providing a new paradigm to explain the switch from Th1 to Th2-type cytokines, that is associated with many chronic infections (18, 21, 62–64). We have established a framework to further examine the molecular mechanisms regulating subset-specific AICD, that in the future may provide targets for therapeutic intervention in diseases where T cell apoptosis is dysregulated.
Acknowledgments
KB8301 is a generous gift from Kanebo, Ltd. (Osaka). We thank Douglas Green (La Jolla Institute of Allergy and Immunology, La Jolla, CA) for the Fas–Fc fusion protein; Marc Monestier and David Mosser (Temple University) for review of the manuscript; and John Gibas for help with flow cytometry. P.S. is grateful to Barry Bloom for helpful discussions.
ABBREVIATIONS
- AICD
activation-induced cell death
- TCR
T cell receptor
- FasR
Fas receptor
- FasL
Fas ligand
- TNF
tumor necrosis factor
- IFN
interferon, IL, interleukin
- FADD
Fas-associating protein with death domain
- PPD
purified protein derivative
- TT
tetanus-toxoid
References
- 1.Cohen J J. Immunol Today. 1993;14:126–130. doi: 10.1016/0167-5699(93)90214-6. [DOI] [PubMed] [Google Scholar]
- 2.Akbar A N, Borthwick N J, Wickremasinghe R G, Panayiotidis P, Pilling D, Bofill M, Krajewski S, Reed J C, Salmon M. Eur J Immunol. 1996;26:294–299. doi: 10.1002/eji.1830260204. [DOI] [PubMed] [Google Scholar]
- 3.Zheng L, Fisher G, Miller R E, Peschon J, Lynch D H, Lenardo M J. Nature (London) 1995;377:377–351. doi: 10.1038/377348a0. [DOI] [PubMed] [Google Scholar]
- 4.Green D R, Scott D W. Curr Opin Immunol. 1994;6:476–487. doi: 10.1016/0952-7915(94)90130-9. [DOI] [PubMed] [Google Scholar]
- 5.Itoh N, Yonehara S, Ishii A, Yonehara M, Mizushima S, Sameshima M, Hase A, Seto Y, Nagata S. Cell. 1991;66:223–243. doi: 10.1016/0092-8674(91)90614-5. [DOI] [PubMed] [Google Scholar]
- 6.Russel J H, Rush B, Weaver C, Wang R. Proc Natl Acad Sci USA. 1993;90:4409–4413. doi: 10.1073/pnas.90.10.4409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dhein J, Walczak H, Baumler C, Debatin K-M, Krammer P H. Nature (London) 1995;373:438–441. doi: 10.1038/373438a0. [DOI] [PubMed] [Google Scholar]
- 8.Brunner T, Mogil R J, LaFace D, Yoo N J, Mahboubi A, Echeverri F, Martin S J, Force W R, Lynch D H, Ware C F, Green D R. Nature (London) 1995;373:441–444. doi: 10.1038/373441a0. [DOI] [PubMed] [Google Scholar]
- 9.Ju S-T, Panka D J, Cui H, Ettinger R, El-Khatib M, Sherr D H, Stanger B Z, Harshak-Rothstein A. Nature (London) 1995;373:444–448. doi: 10.1038/373444a0. [DOI] [PubMed] [Google Scholar]
- 10.Kabelitz D, Pohl T, Pechhold K. Immunol Today. 1993;14:338–339. doi: 10.1016/0167-5699(93)90231-9. [DOI] [PubMed] [Google Scholar]
- 11.Fisher G H, Rosenberg F J, Straus S E, Dale J K, Middelton L A, Lin A Y, Strober W, Lenardo M J, Puck M J. Cell. 1995;81:935–946. doi: 10.1016/0092-8674(95)90013-6. [DOI] [PubMed] [Google Scholar]
- 12.Rieux-Laucat F, Le Diest F, Hivroz C, Roberts I A G, Debatin K M, Fisher A, de Villartay J P. Science. 1995;268:1347–1349. doi: 10.1126/science.7539157. [DOI] [PubMed] [Google Scholar]
- 13.Groux H, Torpier G, Monte D, Mouton Y, Capron A, Ameisen J C. J Exp Med. 1992;175:331–340. doi: 10.1084/jem.175.2.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Estaquier J, Idziorek T, Weiping Z, Emilie D, Farber C-M, Bourez J-M, Ameisen J C. J Exp Med. 1995;182:1759–1767. doi: 10.1084/jem.182.6.1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Katsikis P D, Wunderlich E S, Smith C A, Herzenberg L A, Herzenberg L A. J Exp Med. 1995;181:2029–2036. doi: 10.1084/jem.181.6.2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Uehara T, Miyawaki T, Ohta K, Tamaru Y, Yokoi T, Nakamura S, Taniguchi N. Blood. 1992;80:452–458. [PubMed] [Google Scholar]
- 17.Akbar A N, Borthwick N, Salmon M, Gombert W, Bofill M, Shamsadeen N, Pilling D, Pett S, Grundy J E, Janossy G. J Exp Med. 1993;178:427–438. doi: 10.1084/jem.178.2.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ameisen J C, Estaquier J, Idziorek T. Immunol Rev. 1994;142:9–51. doi: 10.1111/j.1600-065x.1994.tb00882.x. [DOI] [PubMed] [Google Scholar]
- 19.Mosmann T R, Coffman R L. Annu Rev Immunol. 1989;7:145–173. doi: 10.1146/annurev.iy.07.040189.001045. [DOI] [PubMed] [Google Scholar]
- 20.Janeway C R J, Carding S, Jones B, Murray J, Portoles P, Rasmussen R, Rojo J, Saizawa K, West J, Bottomly K. Immunol Rev. 1988;101:39–80. doi: 10.1111/j.1600-065x.1988.tb00732.x. [DOI] [PubMed] [Google Scholar]
- 21.Salgame P, Abrams J, Clayberger C, Goldstein H, Convit J, Modlin R, Bloom B R. Science. 1991;254:279–282. doi: 10.1126/science.254.5029.279. [DOI] [PubMed] [Google Scholar]
- 22.Romagnani S. Annu Rev Immunol. 1994;12:227–257. doi: 10.1146/annurev.iy.12.040194.001303. [DOI] [PubMed] [Google Scholar]
- 23.Seder R A, Paul W E. Annu Rev Immunol. 1994;12:635–673. doi: 10.1146/annurev.iy.12.040194.003223. [DOI] [PubMed] [Google Scholar]
- 24.Bloom B R, Modlin R L, Salgame P. Annu Rev Immunol. 1992;10:453–488. doi: 10.1146/annurev.iy.10.040192.002321. [DOI] [PubMed] [Google Scholar]
- 25.Salgame P R, Modlin R L, Bloom B R. Int Immunol. 1989;1:121–129. doi: 10.1093/intimm/1.2.121. [DOI] [PubMed] [Google Scholar]
- 26.Kayagaki N, Kawasaki A, Ebata T, Ohmoto H, Ikeda S, Inoue S, Yoshino K, Okumura K, Yagita H. J Exp Med. 1995;182:1777–1783. doi: 10.1084/jem.182.6.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matzinger P. J Immunol Methods. 1991;145:185–192. doi: 10.1016/0022-1759(91)90325-a. [DOI] [PubMed] [Google Scholar]
- 28.Ramsdell F, Seaman M S, Miller R E, Picha K S, Kennedy M K, Lynch D H. Int Immunol. 1994;6:1545–1553. doi: 10.1093/intimm/6.10.1545. [DOI] [PubMed] [Google Scholar]
- 29.Ashany D, Song X, Lacy E, Nikolic-Zugic J, Friedman S M. Proc Natl Acad Sci USA. 1995;92:11225–11229. doi: 10.1073/pnas.92.24.11225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ju S-T, Ciu H, Panka D J, Ettinger R, Marshak-Rothstein A. Proc Natl Acad Sci USA. 1994;91:4185–4189. doi: 10.1073/pnas.91.10.4185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Webb S, Morris C, Sprent J. Cell. 1990;63:1249–1256. doi: 10.1016/0092-8674(90)90420-j. [DOI] [PubMed] [Google Scholar]
- 32.MacDonald H R, Baschieri S, Lees R K. Eur J Immunol. 1991;21:1963–1966. doi: 10.1002/eji.1830210827. [DOI] [PubMed] [Google Scholar]
- 33.Kawabe Y, Ochi A. Nature (London) 1991;349:245–248. doi: 10.1038/349245a0. [DOI] [PubMed] [Google Scholar]
- 34.Kearney E R, Pape K A, Loh D Y, Jenkins M K. Immunity. 1994;1:327–339. doi: 10.1016/1074-7613(94)90084-1. [DOI] [PubMed] [Google Scholar]
- 35.Zhang L, Shannon J, Sheldon J, Teh H S, Mak T W, Miller R G. J Immunol. 1994;152:2222–2228. [PubMed] [Google Scholar]
- 36.Zhang L, Miller R G, Zhang J. J Exp Med. 1996;183:2065–2073. doi: 10.1084/jem.183.5.2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu Y, Janeway J, C A. J Exp Med. 1990;172:1735–1739. doi: 10.1084/jem.172.6.1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Groux H, Monte D, Plouvier B, Capron D, Ameisen J C. Eur J Immunol. 1993;23:1623–1629. doi: 10.1002/eji.1830230734. [DOI] [PubMed] [Google Scholar]
- 39.Suda T, Okazaki T, Naito Y, Yokoto T, Arai N, Ozaki S, Nakao K, Nagata S. J Immunol. 1995;154:3806–3813. [PubMed] [Google Scholar]
- 40.Suda T, Tanaka M, Miwa K, Nagata S. J Immunol. 1996;157:3918–3924. [PubMed] [Google Scholar]
- 41.Fitch F W, McKisic M D, Lancki D W, Gajewski T F. Annu Rev Immunol. 1993;11:29–48. doi: 10.1146/annurev.iy.11.040193.000333. [DOI] [PubMed] [Google Scholar]
- 42.Dhein J, Daniel P T, Trauth B C, Oehm A, Moller P, Krammer P H. J Immunol. 1992;149:3166–3173. [PubMed] [Google Scholar]
- 43.Muzio M, Chinnaiyan A M, Kischkel F C, O’Rourke K, Shevchenko A, Ni J, Scaffidi C, Bretz J D, Zhang M, Gentz R, Mann M, Krammer P H, Peter M E, Dixit V M. Cell. 1996;85:817–827. doi: 10.1016/s0092-8674(00)81266-0. [DOI] [PubMed] [Google Scholar]
- 44.Itoh N, Tsujimoto Y, Nagata S. J Immunol. 1993;151:621–627. [PubMed] [Google Scholar]
- 45.Boise L H, Min A J, Noel P J, June C H, Accavitti M A, Lindsten T, Thompson C B. Immunity. 1995;3:87–98. doi: 10.1016/1074-7613(95)90161-2. [DOI] [PubMed] [Google Scholar]
- 46.Memon S A, Moreno M B, Petrak D, Zacharchuk C M. J Immunol. 1995;155:4644–4652. [PubMed] [Google Scholar]
- 47.Chinnaiyan A M, O’Rourke K, Lane B R, Dixit V M. Science. 1997;275:1122–1126. doi: 10.1126/science.275.5303.1122. [DOI] [PubMed] [Google Scholar]
- 48.Golstein P. Science. 1997;275:1081–1082. doi: 10.1126/science.275.5303.1081. [DOI] [PubMed] [Google Scholar]
- 49.Nagata S, Golstein P. Science. 1995;267:1449–1456. doi: 10.1126/science.7533326. [DOI] [PubMed] [Google Scholar]
- 50.Gulbins E, Bissonnette R, Mahboubi A, Martin S, Nishioka W, Brunner T, Baier G, Baier-Bitterlich G, Byrd C, Lang F, Kolesnick R, Altman A, Green D. Immunity. 1995;2:341–351. doi: 10.1016/1074-7613(95)90142-6. [DOI] [PubMed] [Google Scholar]
- 51.Thompson C B. Cell. 1995;81:979–982. doi: 10.1016/s0092-8674(05)80001-7. [DOI] [PubMed] [Google Scholar]
- 52.Kuchroo V K, Das M P, Brown J A, Ranger A M, Zamvil S S, Sobel R A, Weiner H L, Nabavi N, Glimcher L H. Cell. 1995;80:707–718. doi: 10.1016/0092-8674(95)90349-6. [DOI] [PubMed] [Google Scholar]
- 53.Allison J P. Curr Opin Immunol. 1994;6:414–419. doi: 10.1016/0952-7915(94)90120-1. [DOI] [PubMed] [Google Scholar]
- 54.McKnight A J, Perez V L, Shea C M, Gray G S, Abbas A K. J Immunol. 1994;152:5220–5225. [PubMed] [Google Scholar]
- 55.Moreno C, Mehlert A, Lamb J. Clin Exp Immunol. 1988;74:206. [PMC free article] [PubMed] [Google Scholar]
- 56.Villanueva P O F, Reiser H, Stadecker M J. J Immunol. 1994;153:5190–5199. [PubMed] [Google Scholar]
- 57.Liblau R S, Singer S M, McDevitt H O. Immunol Today. 1995;16:34–38. doi: 10.1016/0167-5699(95)80068-9. [DOI] [PubMed] [Google Scholar]
- 58.Simon A K, Seipelt E, Sieper J. Proc Natl Acad Sci USA. 1994;91:8562–8566. doi: 10.1073/pnas.91.18.8562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen C, Nabavi N. Immunity. 1994;1:147–154. doi: 10.1016/1074-7613(94)90108-2. [DOI] [PubMed] [Google Scholar]
- 60.Singer G G, Abbas A K. Immunity. 1994;1:365–371. doi: 10.1016/1074-7613(94)90067-1. [DOI] [PubMed] [Google Scholar]
- 61.Singer G G, Carrera A C, Marshak-Rothstein A, Martinez C, Abbas A K. Curr Opin Immunol. 1994;6:913–920. doi: 10.1016/0952-7915(94)90013-2. [DOI] [PubMed] [Google Scholar]
- 62.Yamamura M, Uyemura K, Deans R J, Weinberg K, Rea T R, Bloom B R, Modlin R L. Science. 1991;254:277–279. doi: 10.1126/science.254.5029.277. [DOI] [PubMed] [Google Scholar]
- 63.Heinzel F P, Schoenhaut D S, Rerko R M, Rosser L E, Gately M K. J Exp Med. 1993;177:1505–1509. doi: 10.1084/jem.177.5.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sher A, Coffman R L. Annu Rev Immunol. 1992;10:385–409. doi: 10.1146/annurev.iy.10.040192.002125. [DOI] [PubMed] [Google Scholar]