

Abstract
Retroviral and adeno-associated viral sequences can dramatically silence transgene expression in mice. We now report that this repression also occurs in stably infected HeLa cells when the cells are grown without selection. Expression of a transduced lacZ gene (rAAV/CMVlacZ) is silenced in greater than 90% of cells after 60 days in culture. Surprisingly, high-level expression can be reactivated by treating the cells with sodium butyrate or trichostatin A but not with 5-azacytidine. When cell clones with integrated copies of rAAV/CMVlacZ were isolated, lacZ expression was silenced in 80% of the clones; however, lacZ expression was reactivated in all of the silenced clones by treatment with butyrate or trichostatin A. The two drugs also reactivated a silenced globin gene construct (rAAV/HS2αβAS3) in stably infected K562 cells. Trichostatin A is a specific inhibitor of histone deacetylase; therefore, we propose that hyperacetylation of histones after drug treatment changes the structure of chromatin on integrated viral sequences and relieves repression of transduced genes. The reactivation of silenced, transduced genes has implications for gene therapy. Efficient viral gene transfer followed by drug treatment to relieve suppression may provide a powerful combination for treatment of various genetic and infectious diseases.
Keywords: gene transfer, adeno-associated viral vector, trichostatin A, sodium butyrate
Retroviral and adeno-associated viral (AAV) vectors are two widely used viral systems for stably transferring genes into mammalian cells. Although the transfer of genes is generally efficient with these vectors, high-level, long-term expression in primary cells has been problematic. Palmer et al. (1) demonstrated efficient transfer of genes into primary skin fibroblasts with retroviral vectors, but expression was gradually suppressed over a period of 1 month, and a number of groups have observed inactivation of transferred β-globin genes after transduced bone marrow cells are transplanted into mice (Michel Sadelain, personal communication). These results are similar to the inhibition of retroviral expression observed by Jaenisch and colleagues after infection of preimplantation mouse embryos (2, 3). We recently demonstrated that retroviral long terminal repeat (LTR) sequences completely suppress β-globin gene expression in transgenic mice even in the presence of locus control region DNase I hypersensitive site 2 (HS2) sequences that normally direct high-level, position-independent expression (4). Severe inhibition is also observed when LTR HS3 β constructs are tested in transgenic mice (James Ellis, personal communication). To localize sequences responsible for suppression, we inserted five separate subfragments of the retroviral LTR upstream of HS2β and tested these constructs for expression in mice. Surprisingly, four of the five fragments inhibited expression of the transgene (unpublished work); the only LTR sequence that did not inhibit expression was a fragment containing the retroviral enhancer and promoter.
We recently tested AAV inverted terminal repeat (ITR) sequences in the assay described above and demonstrated that AAV ITRs also severely inhibit β-globin gene expression in transgenic mice, even in the presence of locus control region sequences (unpublished work). These results suggest a general host mechanism for silencing viral genes and cellular genes inserted in viral vectors. Expression of genes stably transduced with AAV vectors has been successful (5–7); however, in all of these experiments, dominant marker genes were included in the constructs, and stable transductants were isolated after drug selection. In a more recent set of experiments in which no drug selection was used, human bone marrow CD34+ cells were infected at high efficiency with recombinant AAV (rAAV) containing CMV/lacZ, but no lacZ message could be detected in burst-forming unit-erythroid colonies (8). Subsequently, Miller et al. (9) demonstrated γ-globin gene expression in burst-forming unit-erythroid colonies after infection with rAAV containing the human γ-globin gene without a selectable marker; however, expression in this case may have resulted from viral genomes that were not yet integrated into the host genome (Arthur Nienhuis, personal communication).
As described below, we infected HeLa cells with recombinant AAV containing a CMV/lacZ gene and found that the number of blue cells observed after 5-bromo-4-chloro-3-indolyl β-d-galactoside (X-Gal) staining decreased dramatically over a period of 60 days. Interestingly, lacZ expression could be efficiently reactivated by adenovirus infection (unpublished work). To determine whether drug treatment could mimic the effects of adenoviral infection, we tested a large number of drugs for the ability to derepress transduced sequences. Surprisingly, butyrate and trichostatin A but not 5-azacytidine reactivated expression to high levels. Butyrate and trichostatin A also reactivated a silenced globin gene construct that was stably transduced into K562 cells.
MATERIALS AND METHODS
Construction of rAAV Vectors and Production of Recombinant Virions.
The rAAV/CMVlacZ vector was constructed, and the viral lysate was prepared and titered as described (10); the titer was 1 × 108 per ml. The lysate was heated at 56°C for 15 min to inactivate wild-type adenovirus.
The anti-sickling gene AS3 is similar to the anti-sickling gene AS2 described in ref. 11 but contains an additional modification and has stronger anti-sickling effects (unpublished work). HS2αβAS3 contains a 1.5-kb KpnI–BglII HS2 fragment, a −335 α-globin gene promoter, and a 2.4-kb NcoI–AvrII β-globin gene fragment. HS2αβAS3 is subcloned into the SalI site of the rAAV cloning vector pMAV53 (unpublished work). The rAAV/HS2αβAS3 vector was then packaged in parallel with rAAV/CMVlacZ using the same conditions.
HeLa Cell Infection and Drug Treatment.
Cells (2 × 105) were infected with 10 μl of viral lysate. In the case of hydroxyurea pretreatment, cells were incubated with 4 mM hydroxyurea for 24 h before infection. At days 6, 10, 20, 30, 40, 50, and 60 after infection, 2 × 105 cells per well were plated into 12-well plates. After overnight culture, the medium was replaced with fresh medium (control) or with medium containing 3 μM 5-azacytidine, 50 mM sodium butyrate, or 3 μM trichostatin A. After 24 h of treatment with these drugs, cells were fixed and stained with X-Gal as described (12). Blue cells were examined with an inverted microscope and counted.
Cloning, PCR Screening, and Southern Blot Analysis of rAAV/CMVlacZ-Infected HeLa Cells.
HeLa cells (1 × 104) pretreated with hydroxyurea were infected with 100 μl of heat-inactivated viral lysate in a total volume of 250 μl of medium for 24 h. After 21 days of culture, rAAV/CMVlacZ-infected cells were cloned by limiting dilution. Single colony clones were trypsinized and half of the cells from each clone were transferred to 1 well of a 24-well plate; the other half of the cells was placed into an Eppendorf tube for PCR analysis of lacZ DNA. PCR positive clones were expanded to 100-mm plates, and DNA was extracted for Southern blot hybridization (12). Some of the cells were plated into 12-well plates for drug treatment and X-Gal staining.
Cloning, Screening, and Southern Blot Analysis of rAAV/HS2αβAS3-Infected K562 Cells.
Cells (5 × 103) were infected with 50 μl of heat-inactivated rAAV/HS2αβAS3 lysate in a total volume of 200 μl in a 96-well plate. After 24 h of infection, cells were transferred to an Eppendorf tube, pelleted, washed twice with fresh medium, plated, and grown for 30 days. Cells were then cloned by limiting dilution as described above and analyzed by PCR and Southern blot hybridization.
Reverse Transcription (RT)-PCR Analysis of rAAV/HS2αβAS3-Transduced K562 Clones.
Cells (7.5 × 106) from each clone were split into 3 wells of a 6-well plate. Medium (2×) containing sodium butyrate or trichostatin A was added at final concentrations of 50 mM and 3 μM, respectively. Normal medium was added to the third well as a control. After 24 h, cells were collected by centrifugation, and total RNA was extracted with RNA STAT60 (Tel-Test, Friendswood, TX). Approximately 1 μg of total RNA of each sample was reverse transcribed using the 1st Strand cDNA Synthesis Kit (Boehringer Mannheim), and 2 μl (1/10 volume) of cDNA was used for PCR. A 25-cycle PCR (92°C for 30 s and 68°C for 90 s) was used to achieve linear amplification, and 0.5 μl of [α-32P]dCTP (3,000 Ci/mmol; 1 Ci = 37 GBq; NEN) was included in each reaction. Three primers were used together to amplify both endogenous α-globin cDNA and αβAS3 cDNA. The forward primer (5′-ACT CTT CTG GTC CCC ACA GA-3′) from the α-globin 5′ untranslated region was common to both endogenous α-globin mRNA and αβAS3 mRNA. One reverse primer (5′-GTT GGG CAT GTC GTC CAC GT-3′) was specific for endogenous α-globin exon 2, and the other reverse primer (5′-TCA CTA AAG GCA CCG AGC AC-3′) was specific for αβAS3 exon 2. PCR products (5 μl) were mixed with 20 μl of formamide loading dye, denatured at 70°C, and electrophoresed on 5% denaturing polyacrylamide gels. The intensity of PCR bands was quantitated on a PhosphorImager (Molecular Dynamics). The level of butyrate and trichostatin A induction of αβAS3 was calculated by the following formula: [counts per minute of αβAS3 from butyrate or trichostatin A samples/counts per minute of α from butyrate or trichostatin A samples]/[counts per minute of αβAS3 from control samples/counts per minute of α from control samples].
RESULTS
Treatment with Butyrate and Trichostatin A Reactivates lacZ Gene Expression.
Russell et al. (13) recently demonstrated that treatment of cells with hydroxyurea before AAV infection significantly increased viral integration efficiency; therefore, we treated HeLa cells with 4 mM hydroxyurea for 24 h before infection with rAAV/CMVlacZ. After infection at a multiplicity of infection (moi) of 5, the cells were plated and examined for lacZ expression at 6, 10, 20, 30, 40, 50, and 60 days after infection. Interestingly, the number of lacZ-expressing cells decreased progressively with time until few cells expressed the transduced gene at 60 days (Table 1). The same silencing of lacZ expression was observed with cells that were not pretreated with hydroxyurea (Table 2). In an attempt to reactivate lacZ expression, we treated the cells with 5-azacytidine, which has been used to reactivate endogenous genes whose promoters or enhancers are methylated (14–16). Interestingly, 5-azacytidine did not reactivate lacZ expression (Tables 1 and 2) even when a broad range of concentrations were tested (data not shown). In contrast, treatment with sodium butyrate dramatically reactivated lacZ expression. Even after 60 days, when lacZ expression was silenced in 99% of the cells, sodium butyrate treatment reactivated expression to high levels. Sodium butyrate inhibits histone deacetylases but also has a number of other activities (17). To determine whether the inhibition of histone deacetylation was the major cause of lacZ reactivation, cells were treated with trichostatin A, which is a specific inhibitor of histone deacetylase (18). The data in Table 1 and 2, and Fig. 2 demonstrate that trichostatin A also induces lacZ expression to high levels. These results suggest that histone deacetylation is integrally involved in the silencing of transduced genes in this system and that inhibition of histone deacetylases is a powerful method for reactivating gene expression.
Table 1.
Reactivation of lacZ gene expression in rAAV/CMVlacZ infected HeLa cells by treatment with sodium butyrate or trichostatin A
| Day | Control | 5-Azacytidine | Butyrate | Trichostatin A |
|---|---|---|---|---|
| 6 | 447 | 375 | 994 | 1,341 |
| 10 | 136 | 111 | 893 | 1,168 |
| 20 | 130 | 136 | 1,112 | 1,428 |
| 30 | 37 | 50 | 795 | 1,001 |
| 40 | 34 | 26 | 835 | 1,115 |
| 50 | 15 | 19 | 771 | 1,076 |
| 60 | 23 | 24 | 934 | 1,138 |
HeLa cells were treated for 24 h with 4 mM hydroxyurea, washed, and then infected for 24 h with rAAV/CMVlacZ at an moi of 5. After infection, cells were washed three times with PBS and cultured for the days indicated. At the days shown in the table, cells were trypsinized and plated in 12-well plates. After overnight culture, cells were treated with various drugs for 24 h and then stained with X-Gal. The numbers shown in the table represent the average number of blue cells in each well that were treated with (i) no drug (control), (ii) 3 μM 5-azacytidine, (iii) 50 mM sodium butyrate, or (iv) 3 μM trichostatin A.
Table 2.
Reactivation of lacZ gene expression in non-pretreated rAAV/CMVlacZ infected HeLa cells after posttreatment of cells with sodium butyrate or trichostatin A
| Day | Control | 5-Azacytidine | Butyrate | Trichostatin A |
|---|---|---|---|---|
| 6 | 40 | 26 | 264 | 281 |
| 10 | 13 | 18 | 258 | 304 |
| 20 | 5 | 8 | 185 | 250 |
| 30 | 6 | 3 | 132 | 248 |
| 40 | 3 | 2 | 132 | 174 |
| 50 | 1 | 1 | 123 | 178 |
| 60 | 1 | 1 | 131 | 220 |
HeLa cells without hydroxyurea pretreatment were infected overnight with rAAV/CMVlacZ at an moi of 5 and treated in the same way as described in the legend of Table 1.
Figure 2.
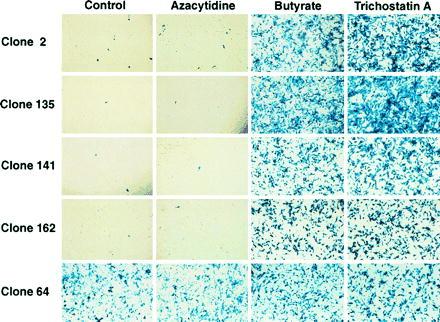
X-Gal staining of five representative HeLa cell clones containing integrated rAAV/CMVlacZ. Cells from each clone were assayed for lacZ expression with or without treatment with 5-azacytidine, sodium butyrate, and trichostatin A. Few cells in four of the five untreated controls expressed lacZ; however, both sodium butyrate and trichostatin A dramatically activated lacZ expression. These results are representative of the results obtained with all 25 clones analyzed. One of the 5 clones (clone 64) and 5 of 25 clones overall (data not shown) contained a constitutively active lacZ gene.
All Silenced Vector Integration Sites Are Responsive to Butyrate and Trichostatin A Treatments.
Wild-type AAV can integrate at a specific site on chromosome 19 (19, 20); however, recombinant AAV vectors that do not contain the rep gene normally intergate at random sites in the host genome (6). To determine the percentage of rAAV/CMVlacZ integration sites that are responsive to butyrate or trichostatin A treatment, HeLa cells were infected with rAAV/CMVlacZ (moi = 1,000) and cloned by limiting dilution. Stably transduced cells were identified by PCR and confirmed by Southern blot hybridization. Twenty-five clones were analyzed, and the Southern blot data on five representative clones are illustrated in Fig. 1. Fig. 1 A and B depict the results observed with ScaI and XbaI digestion, respectively. ScaI does not cut in rAAV/CMVlacZ; therefore, if the construct is integrated into the HeLa cell genome, a DNA fragment that is larger than unit length (5.0 kb) should hybridize with the lacZ probe. Fig. 1A demonstrates that all five clones contain copies of rAAV/CMVlacZ in large ScaI fragments (greater than 20 kb). XbaI cuts rAAV/CMVlacZ once; therefore, digestion with this enzyme and hybridization with the lacZ probe should produce a junction fragment that is different for each integration site. A unit length fragment would be produced if head-to-tail copies are present. Fig. 1B demonstrates that all five clones contain different rAAV/CMVlacZ junction fragments and that clones 141 and 162 also contain unit length fragments that hybridize with the probe. These results strongly suggest that the clones contain stably integrated copies of rAAV/CMVlacZ.
Figure 1.
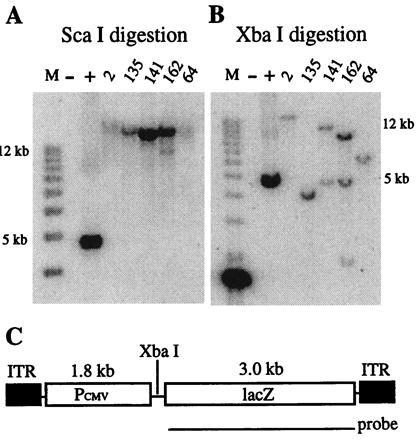
Southern blot analysis of HeLa cell clones after rAAV/CMVlacZ infection. HeLa cells were infected with rAAV/CMVlacZ, grown for 3 weeks, and then plated at limiting dilution. Individual cells were expanded for 4 weeks, and DNA was extracted for analysis. Genomic DNA from uninfected HeLa cells was used as negative control, and a 5.0-kb fragment from the rAAV/CMVlacZ vector was spiked into HeLa cell DNA for a positive control. A 3.0-kb lacZ DNA fragment was used as the probe. (A) Genomic DNAs were digested with ScaI, which does not cut rAAV/CMVlacZ. (B) Genomic DNAs were digested with XbaI, which cuts once inside the vector as shown in C. (C) Map of the rAAV/CMVlacZ virus.
Cells from each clone were assayed for lacZ expression with or without treatment with 5-azacytidine, sodium butyrate, and trichostatin A (Fig. 2). Few cells in four of the five untreated controls expressed lacZ; however, both sodium butyrate and trichostatin A dramatically activated lacZ expression. These results are representative of the results obtained with all 25 clones analyzed. As indicated above, trichostatin A is a specific inhibitor of histone deacetylase; therefore, the hyperacetylation of histones that occurs in the presence of this drug appears to be responsible for lacZ reactivation. Interestingly, one of the 5 clones illustrated in Fig. 2 and 5 of the 25 clones overall (data not shown) contained a constitutively active lacZ gene. These results suggest that 20% of the AAV integration sites is not silenced in HeLa cells.
Reactivation of Gene Expression by Histone Deacetylase Inhibitors Is Not Promoter- or Cell-Type-Specific.
To determine whether reactivation of silenced, virally transduced genes by butyrate and trichostatin A is applicable to other promoters and cell types, a human β-globin gene vector was constructed. This vector contains the human β-globin locus control region HS2 sequence linked to a human α-globin promoter driving an anti-sickling β-globin gene. The human β-globin HS2 sequence was shown previously to direct high-level, erythroid-specific expression of α- and β-globin genes in transgenic mice (21–26) and in cultured erythroid cells (24, 27–29). The human α-globin promoter was chosen because this promoter is less susceptible than the β-globin promoter to silencing induced by plasmid sequences (ref. 30; T. M. R. Ryan and T.M.T., unpublished work), retroviral LTR sequences, and AAV ITR sequences (ref. 4; unpublished work). The α-globin promoter has been used successfully by other investigators in AAV vectors (7). Fig. 3C illustrates the rAAV/HS2αβAS3 construct. Human erythroleukemia cells (K562) were infected with the virus, and 14 clones of stably transduced cells were obtained by limiting dilution and PCR analysis. Southern blots of six representative clones are illustrated in Fig. 3 A and B. Digestion of genomic DNA with ScaI, which does not cut rAAV/HS2αβAS3, demonstrated that the viral sequences were incorporated into high molecular weight DNA (greater than 25 kb) and, therefore, were not present as episomal vectors (Fig. 3A). Digestion with EcoRI, which cuts only once in rAAV/HS2αβAS3, demonstrated single junction fragments indicative of single copy, random integrants (Fig. 3B). Head-to-tail tandem arrays would produce a unit length band of 4.5 kb, but this band was not observed. Thirteen of the 14 clones analyzed contained an intact copy of the transduced construct.
Figure 3.
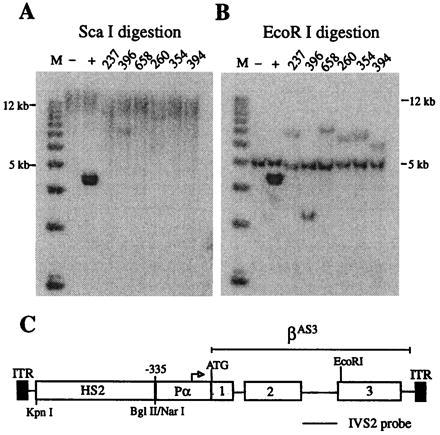
Southern blot analysis of rAAV/HS2αβAS3-transduced K562 cell clones. Cells were infected, grown for 30 days, and plated at limiting dilution. Individual cells were expanded for 4 weeks, and DNA was extracted and analyzed as described for HeLa cell clones, except that genomic DNA from uninfected K562 cells was used as a negative control and a 4.5-kb fragment of the rAAV/HS2αβAS3 vector was spiked into K562 cell DNA for a positive control. A 790-bp HinfI–HinfI fragment of the β-globin gene IVS2 was used as a probe. (A) Genomic DNAs were digested with ScaI, which does not cut in rAAV/HS2αβAS3. Hybridization of the probe to high molecular weight bands (greater than 25 kb) demonstrate that viral sequences are not present as episomal vectors. (B) Genomic DNAs were digested with EcoRI, which cuts once in the β-globin gene. The single junction fragments that hybridize to the probe demonstrate that all six clones contain single copy integrants of the transduced gene. The endogenous β-globin gene fragment (5.5 kb) ran slightly faster than expected in this gel. (C) Map of rAAV/HS2αβAS3.
These 13 clones were tested for αβAS3 expression by RT-PCR as described in Materials and Methods. The endogenous human α-globin gene was chosen as an internal control because previous studies demonstrated that synthesis of the adult globins in K562 cells is not significantly induced or suppressed by butyrate treatment (31–33). Fig. 4 illustrates RT-PCR analysis of the same six clones that were analyzed in Fig. 3. In three of the clones, expression of αβAS3 was dramatically induced by butyrate and trichostatin A. The level of butyrate induction was 8.1-fold for clone 237, 33-fold for clone 396, and 9-fold for clone 658. The level of trichostatin A induction was 10.2-fold for clone 237, 19-fold for clone 396, and 14-fold for clone 658. Overall, αβAS3 expression was inducible in 6 of the 13 clones, and the average level of induction was 10.6-fold and 9.0-fold for butyrate and trichostatin A, respectively. αβAS3 expression in the other seven clones was constitutive, and three of these clones (260, 354, and 394) are illustrated in Fig. 4.
Figure 4.
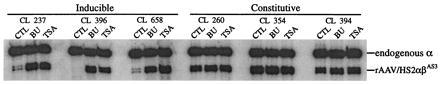
RT-PCR analysis of six representative K562 clones containing integrated rAAV/HS2αβAS3. K562 cell clones were treated with sodium butyrate (BU) or trichostatin A (TSA) for 24 h; untreated cells were included as controls (CTL). Total RNA was extracted with RNA STAT60. RT-PCR of endogenous α-globin mRNA served as an internal control. A dramatic induction of αβAS3 gene expression was observed in half of the clones. The level of butyrate induction was 8.1-fold for clone 237, 33-fold for clone 396, and 9-fold for clone 658. The level of trichostatin A induction was 10.2-fold for clone 237, 19-fold for clone 396, and 14-fold for clone 658. Overall, βAS3 expression was inducible in 6 of the 13 clones, and the average level of induction was 10.6-fold and 9.0-fold for butyrate and trichostatin A, respectively. Expression of αβAS3 in the other half of the samples was constitutive as illustrated by clones 260, 354, and 394.
DISCUSSION
The results described above demonstrate that expression of virally transduced genes is silenced in 80% of rAAV/CMVlacZ-transduced HeLa cell clones and approximately 50% of rAAV/HS2αβAS3-transduced K562 clones that contain stably integrated copies of the transgenes. Surprisingly, treatment of the cells with sodium butyrate or trichostatin A dramatically reactivates expression of silenced constructs. The specificity of trichostatin A, an inhibitor of histone deacetylase, strongly suggests that hypoacetylation of histones is integrally involved in the silencing of virally transduced genes.
Our initial screen for drugs that would reactivate expression of rAAV/CMVlacZ included the DNA demethylation agent 5-azacytidine; the ribonucleoside diphosphate reductase inhibitor hydroxyurea; DNA polymerase inhibitor aphidicolin; RNA polymerase inhibitor actinomycin D; protein synthesis inhibitor cycloheximide; topoisomerase I inhibitor camptothecin; topoisomerase II inhibitors novobiocin, etoposide, and amsacrine; and sodium butyrate. Only sodium butyrate rescued rAAV/CMVlacZ expression. Sodium butyrate reactivated lacZ expression at concentrations as low as 0.5 mM; however, maximal reactivation was observed at 50 mM.
Sodium butyrate induces a number of cellular and metabolic changes in mammalian cells and activates the expression of several endogenous genes by mechanisms that are not clearly understood (17). Recently, butyrate and its metabolic derivatives phenylbutyrate and phenylacetate have been used in clinical trials to increase fetal hemoglobin (HbF) expression in patients with thalassemia or sickle cell disease (34–38), and the short chain fatty acid propionic acid has also been reported to stimulate fetal globin gene expression (39). When phenylbutyrate, phenylacetate, and propionate were tested for reactivation of rAAV/CMVlacZ expression in HeLa cells, phenylbutyrate and propionate induced lacZ expression, although less effectively than butyrate, but phenylacetate had no effect (data not shown). These results suggest that the mechanism for reactivation of viral transgenes may be different from the activation of some endogenous genes.
Histone hyperacetylation is one of many cellular changes induced by butyrate (17, 40). To determine whether histone acetylation was specifically involved in reactivation of rAAV/CMVlacZ expression, we tested the potent histone deacetylase inhibitor trichostatin A (18). Our results demonstrate that trichostatin A is a powerful inducer of rAAV/CMVlacZ expression and, therefore, suggest that reactivation of the viral transgene is most likely due to hyperacetylation of histones. The drug activates expression at concentrations as low as 30 nM, and maximal effects are observed at 3 μM. This concentration of trichostatin A (3 μM) is more than 10,000 times lower than the concentration of butyrate (50 mM) required to induce an equivalent effect.
To determine whether the effects of butyrate and trichostatin A were specific for the cytomegalovirus (CMV) promoter and/or for HeLa cells, we stably transduced human erythroleukemia cells (K562) with a human globin gene construct (rAAV/HS2αβAS3). αβAS3 expression was severely suppressed in half of the K562 clones examined. All of the silenced clones were reactivated by butyrate and trichostatin A. These results suggest that histone deacetylases may bind directly or indirectly to viral sequences and inhibit globin gene expression. Host sequences that flank the integrated, recombinant virus must influence deacetylase binding because transduced genes are constitutively active in some clones. Also, the human α-globin gene promoter appears to be less susceptible to ITR-induced suppression than the CMV promoter. This result is consistent with our unpublished observation that rAAV/HS2αβAS3 transgenes containing the α-globin promoter are expressed at higher levels than rAAV/HS2βAS3 transgenes containing the β-globin promoter in mice.
Histone acetylation and deacetylation have been implicated in the regulation of gene expression in many systems. In Drosophila, histone deacetylation is involved in the establishment of heterochromatin, and histone acetylation is involved in male X chromosome hyperactive transcription (41). In yeast, the acetylation of histone H3 and H4 N termini is inhibited by SIR3 and SIR4 proteins that promote heterochromatin formation at telomeres and centromeres and contribute to silencing at the mating type locus (42). In Xenopus, histone hyperacetylation plays a positive role in transcription factor access to nucleosomal DNA and influences both gene expression and development (43, 44). Recently, Brownell et al. (45) cloned the tetrahymena histone acetyltransferase A gene and demonstrated that this gene encodes a protein strikingly homologous to yeast Gcn5p, which is a transcriptional transactivator. This result strongly suggests that acetyltransferases can be directly involved in gene activation. More recently, Taunton et al. (46) cloned the first human histone deacetylase gene and demonstrated that this gene encodes a protein strikingly homologous to the yeast transcriptional repressor Rpd3p. Interestingly, Taunton et al. (46) used a trichostatin A analogue to purify the deacetylase by affinity chromatography. Homology between this human histone deacetylase and Rpd3p strongly suggests that histone deacetylation can be directly involved in gene silencing in mammalian cells.
Although the mechanisms responsible for modification of histones at specific chromosomal sites are unknown, some insights can be gained from the activities of Gcn5p and Rpd3p. Neither of these transcription factors bind DNA, but they are part of protein complexes that bind to specific DNA sequences. Presumably, once the complex is bound to specific target sites, Gcn5p and Rpd3p enzymatically modify nearby histones. The resulting local changes in nucleosome structure may either enhance or silence gene expression (47, 48). This is further supported by recent discoveries that Gcn5p can acetylate histone H3 and H4 at specific lysines (49), and many important transcriptional factors, P/CAF (48), CBP (50), and TAF250 (51), have intrinsic histone acetyltransferase activity.
Fig. 5 illustrates a model for silencing virally transduced genes. A host protein or protein complex binds to viral sequences (AAV ITRs or retroviral LTRs) and recruits a histone deacetylase to the site through protein–protein interactions. The enzyme deacetylates histone H3 and H4 N-terminal tails in the region, and the resulting change in chromatin structure inhibits adjacent promoters. Treatment with trichostatin A specifically inhibits the deacetylase. Subsequent acetylation of histones produces a chromatin structure that allows transcription factors to bind to nearby promoters and activate gene expression.
Figure 5.
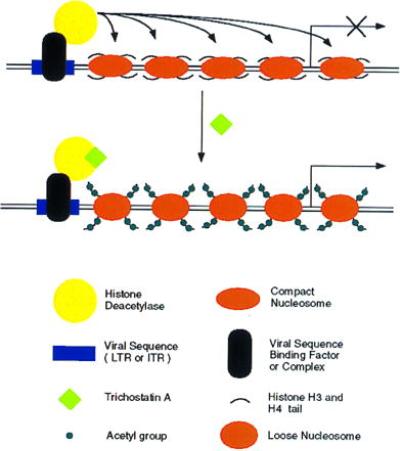
Model for silencing and reactivation of recombinant viral genes. A host protein or protein complex binds to viral sequences (AAV ITRs or retroviral LTRs) and recruits a histone deacetylase to the site through protein–protein interactions. The enzyme deacetylates histone H3 and H4 N-terminal tails, and the resulting change in chromatin structure inhibits expression from adjacent promoters. Treatment with trichostatin A specifically inhibits histone deacetylases. Subsequent acetylation of histones produces a chromatin structure that allows transcription factors to bind to nearby promoters and activate gene expression.
In summary, expression of AAV transduced CMVlacZ genes is silenced in 80% of HeLa cell clones and HS2αβAS3 is suppressed in 50% of K562 cell clones containing intact, integrated copies of the recombinant viral vector. Treatment of the cells with butyrate or trichostatin A dramatically reactivates transgene expression. Trichostatin A is a specific inhibitor of histone deacetylase; therefore, hyperacetylation of histones appears to be integrally involved in reactivating virally transduced genes. The reactivation of silenced, virally transduced genes has implications for gene therapy. Efficient viral gene transfer followed by drug treatment to relieve suppression may provide a powerful combination for treatment of various genetic and infectious diseases.
Acknowledgments
We thank Dr. Minoru Yoshida for the generous gift of trichostatin A and Jinxiang Ren for excellent technical assistance in packaging rAAV. This work was supported in part by Grants R01 HL51649 and HL 35559 from the National Institutes of Health.
ABBREVIATIONS
- LTR
long terminal repeat
- HS2
hypersensitive site 2
- AAV
adeno-associated viral
- rAAV
recombinant AAV
- ITR
inverted terminal repeat
- X-Gal
5-bromo-4-chloro-3-indolyl β-d-galactoside
- RT
reverse transcription
- moi
multiplicity of infection
References
- 1.Palmer T D, Rosman G J, Osborne W R A, Miller D A. Proc Natl Acad Sci USA. 1991;88:1330–1334. doi: 10.1073/pnas.88.4.1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harbers K, Jahner D, Jaenisch R. Nature (London) 1981;293:540–542. doi: 10.1038/293540a0. [DOI] [PubMed] [Google Scholar]
- 3.Jahner D, Stuhlmann H, Stewart C L, Harbers K, Lohler J, Simon I, Jaenisch R. Nature (London) 1982;298:623–628. doi: 10.1038/298623a0. [DOI] [PubMed] [Google Scholar]
- 4.McCune S L, Townes T M. Nucleic Acids Res. 1994;22:4477–4481. doi: 10.1093/nar/22.21.4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Samulski R J, Chang L-S, Shenk T. J Virol. 1989;63:3822–3828. doi: 10.1128/jvi.63.9.3822-3828.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Walsh C E, Liu J M, Xiao X, Young N S, Nienhuis A W, Samulski R J. Proc Natl Acad Sci USA. 1992;89:7257–7261. doi: 10.1073/pnas.89.15.7257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ponnazhagan S, Nallari M L, Srivastava A. J Exp Med. 1994;179:733–738. doi: 10.1084/jem.179.2.733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goodman S, Xiao X, Donahue R E, Moulton A, Miller J, Walsh C, Young N S, Samulski R J, Nienhuis A W. Blood. 1994;84:1492–1500. [PubMed] [Google Scholar]
- 9.Miller J L, Donahue R D, Sellers S E, Samulski R J, Young N S, Nienhuis A W. Proc Natl Acad Sci USA. 1994;91:10183–10187. doi: 10.1073/pnas.91.21.10183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luhovy M, McCune S, Dong J Y, Prchal J F, Townes T M, Prchal J T. Biol Blood Marrow Transplant. 1996;2:24–30. [PubMed] [Google Scholar]
- 11.McCune S L, Reilly M, Chomo M J, Asakura T, Townes T M. Proc Natl Acad Sci USA. 1994;91:9852–9856. doi: 10.1073/pnas.91.21.9852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ausubel F M, Brent R, Kingston R E, Moore D D, Seidman J G, Smith J A, Struhl K. Current Protocols in Molecular Biology. New York: Wiley; 1988. [Google Scholar]
- 13.Russell D W, Alexander I E, Miller A D. Proc Natl Acad Sci USA. 1995;92:5719–5723. doi: 10.1073/pnas.92.12.5719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ferguson A T, Lapidus R G, Baylin S B, Davidson N E. Cancer Res. 1995;55:2279–2283. [PubMed] [Google Scholar]
- 15.Lee Y W, Klein C B, Kargacin B, Salnikow K, Kitahara J, Dowjat K, Zhitkovich A, Christie N T, Costa M. Mol Cell Biol. 1995;15:2547–2557. doi: 10.1128/mcb.15.5.2547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yoshiura K, Kanai Y, Ochiai A, Shimoyama Y, Sugimura T, Hirohashi S. Proc Natl Acad Sci USA. 1995;92:7416–7419. doi: 10.1073/pnas.92.16.7416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kruh J. Mol Cell Biochem. 1982;42:65–82. doi: 10.1007/BF00222695. [DOI] [PubMed] [Google Scholar]
- 18.Yoshida M, Kijima M, Akita M, Beppu T. J Biol Chem. 1990;265:17174–17179. [PubMed] [Google Scholar]
- 19.Kotin R M, Siniscalco M, Samulski R J, Zhu X, Hunter L, Laughlin C A, McLaughlin S, Muzyczka N, Rocchi M, Berns K I. Proc Natl Acad Sci USA. 1990;87:2211–2215. doi: 10.1073/pnas.87.6.2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Samulski R J, Zhu X, Xiao X, Brook J D, Housman D E, Epstein N, Hunter L A. EMBO J. 1991;10:3941–3950. doi: 10.1002/j.1460-2075.1991.tb04964.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Curtin P T, Liu D, Liu W, Chang J C, Kan Y W. Proc Natl Acad Sci USA. 1989;86:7082–7086. doi: 10.1073/pnas.86.18.7082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ryan T M, Behringer R R, Martin N C, Townes T M, Palmiter R D, Brinster R L. Genes Dev. 1989;3:314–323. doi: 10.1101/gad.3.3.314. [DOI] [PubMed] [Google Scholar]
- 23.Ryan T M, Behringer R R, Townes T M, Palmiter R D, Brinster R L. Proc Natl Acad Sci USA. 1989;86:37–41. doi: 10.1073/pnas.86.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fraser P, Hurst J, Grosveld F. Nucleic Acids Res. 1990;18:3503–3508. doi: 10.1093/nar/18.12.3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Caterina J J, Ryan T M, Pawlik K M, Palmiter R D, Brinster R L, Behringer R R, Townes T M. Proc Natl Acad Sci USA. 1991;88:1626–1630. doi: 10.1073/pnas.88.5.1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Caterina J J, Ciavatta D J, Donze D, Behringer R R, Townes T M. Nucleic Acids Res. 1994;22:1006–1011. doi: 10.1093/nar/22.6.1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tuan D Y H, Solomon W B, London I M, Lee D P. Proc Natl Acad Sci USA. 1989;86:2554–2558. doi: 10.1073/pnas.86.8.2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ney P A, Sorrentino B P, Lowrey C H, Nienhuis A W. Nucleic Acids Res. 1990;18:6011–6017. doi: 10.1093/nar/18.20.6011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ney P A, Sorrentino B P, McDonagh K T, Nienhuis A W. Genes Dev. 1990;4:993–1006. doi: 10.1101/gad.4.6.993. [DOI] [PubMed] [Google Scholar]
- 30.Townes T M, Lingrel J B, Chen H Y, Brinster R L, Palmiter R D. EMBO J. 1985;4:1715–1723. doi: 10.1002/j.1460-2075.1985.tb03841.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lozzio C B, Lozzio B B, Machado E A, Fuhr J E, Lair S V, Bamberger E G. Nature (London) 1979;281:709–780. doi: 10.1038/281709b0. [DOI] [PubMed] [Google Scholar]
- 32.Cioe L, McNab A, Hubbell H R, Meo P, Curtis P, Roevera G. Cancer Res. 1981;41:237–243. [PubMed] [Google Scholar]
- 33.Guerrasio A, Vainchenker J, Breton-Gorius J, Testa U, Rosa R, Thomopoulos P, Titeux M, Guichard J, Beuzard Y. Blood Cells. 1981;7:165–176. [PubMed] [Google Scholar]
- 34.Fibach E, Prasanna P, Rodger G, Samid D. Blood. 1993;82:2203–2209. [PubMed] [Google Scholar]
- 35.Perrine S P, Ginder G D, Faller D V, Dover G J, Ikuta T, Witkowska H E, Cai S-P, Vichinsky E P, Olivieri N F. N Engl J Med. 1993;328:81–86. doi: 10.1056/NEJM199301143280202. [DOI] [PubMed] [Google Scholar]
- 36.Dover G J, Brusilow S, Charache S. Blood. 1994;84:339–343. [PubMed] [Google Scholar]
- 37.Perrine S P, Olivieri N F, Faller D V, Vichinsky E P, Dover G J, Ginder G D. Am J Pediatr Hematol Oncol. 1994;16:67–71. [PubMed] [Google Scholar]
- 38.Collins A F, Pearson H A, Giardina P, McDonagh K T, Brusilow S W, Dover G. Blood. 1995;85:43–49. [PubMed] [Google Scholar]
- 39.Little J A, Dempsey N J, Tuchmlan M, Ginder G D. Blood. 1995;85:1712–1718. [PubMed] [Google Scholar]
- 40.Riggs M G, Whittaker R G, Neumann J R, Ingram V M. Nature (London) 1977;268:462–464. doi: 10.1038/268462a0. [DOI] [PubMed] [Google Scholar]
- 41.Turner B M, Birley A J, Lavender J. Cell. 1992;69:375–384. doi: 10.1016/0092-8674(92)90417-b. [DOI] [PubMed] [Google Scholar]
- 42.Hecht A, Laroche T, Strahl-Bolsinger S, Gasser S M, Grunstein M. Cell. 1995;80:583–592. doi: 10.1016/0092-8674(95)90512-x. [DOI] [PubMed] [Google Scholar]
- 43.Lee D Y, Hayes J J, Pruss D, Wolffe A P. Cell. 1993;72:73–84. doi: 10.1016/0092-8674(93)90051-q. [DOI] [PubMed] [Google Scholar]
- 44.Almouzni G, Khochbin S, Dimitrov S, Wolffe A P. Dev Biol. 1994;165:654–669. doi: 10.1006/dbio.1994.1283. [DOI] [PubMed] [Google Scholar]
- 45.Brownell J E, Zhou J, Ranalli T, Kobayashi R, Edmondson D G, Roth S Y, Allis D C. Cell. 1996;84:843–851. doi: 10.1016/s0092-8674(00)81063-6. [DOI] [PubMed] [Google Scholar]
- 46.Taunton J, Hassig C A, Schreiber S L. Science. 1996;272:408–411. doi: 10.1126/science.272.5260.408. [DOI] [PubMed] [Google Scholar]
- 47.Wolffe A P. Science. 1996;272:371–372. doi: 10.1126/science.272.5260.371. [DOI] [PubMed] [Google Scholar]
- 48.Yang X J, Ogryzko V V, Nishikawa J, Howard B H, Nakatani Y. Nature (London) 1996;382:319–324. doi: 10.1038/382319a0. [DOI] [PubMed] [Google Scholar]
- 49.Kuo M H, Brownell J E, Sobel R E, Ranalli T A, Cook R G, Edmondson D G, Roth S Y, Allis D C. Nature (London) 1996;383:269–272. doi: 10.1038/383269a0. [DOI] [PubMed] [Google Scholar]
- 50.Bannister A J, Kouzarides T. Nature (London) 1996;384:641–643. doi: 10.1038/384641a0. [DOI] [PubMed] [Google Scholar]
- 51.Mizzen C A, Yang X J, Kokubo T, Brownell J E, Bannister A J, Owen-Hughes T, Workman J, Wang L, Berger S L, Kouzarides T, Nakatani Y, Allis D C. Cell. 1996;87:1261–1270. doi: 10.1016/s0092-8674(00)81821-8. [DOI] [PubMed] [Google Scholar]