

Abstract
Epidermal keratinocytes can express two types of interleukin 1 (IL-1) receptors: IL-1R1, which is active in signal transduction, and the less well characterized IL-1R2, which is incapable of transducing a signal and can be shed from cells. The binding of IL-1 in solution by IL-1R2 has been demonstrated, and it has been proposed to inhibit IL-1-mediated responses through this mechanism. We and others have reported that keratinocytes can be induced to express IL-1R2 both in vitro and in vivo, often under conditions that also favor IL-1 gene expression. We hypothesized that production of IL-1R2 by keratinocytes would be an efficient means to achieve local inhibition of IL-1-mediated responses without systemic consequences. To test this hypothesis, we have generated transgenic mice that constitutively express IL-1R2 on basal keratinocytes. Keratinocytes cultured from these animals shed the soluble form of the receptor into culture supernatants, and IL-1-inducible production of granulocyte/macrophage colony-stimulating factor was markedly inhibited. In vivo, acute cutaneous vascular leakage, as well as chronic inflammation induced by a well characterized IL-1-dependent stimulus, was significantly inhibited in IL-1R2 transgenic animals. In contrast, contact hypersensitivity was unaffected, suggesting that overexpression of IL-1R2 did not inhibit all types of inflammation globally. Finally, systemic injection of IL-1 induced equivalent levels of plasma IL-6 in IL-1R2 transgenic and nontransgenic mice, suggesting that the activity of the transgenic IL-1R2 remained predominantly local and did not influence systemic IL-1 responses. We conclude that tissue-specific production of IL-1R2 can mediate IL-1 antagonism in tissue microenvironments without systemic consequences. Our transgenic mice may be a useful tool for determining the degree to which different types of cutaneous inflammation depend on the IL-1 system.
Epidermal keratinocytes provide a useful model for studying the kinetics and regulation of interleukin 1 (IL-1)-mediated processes in a tissue accessible to visual and molecular analysis. These cells have been shown to produce agonist ligands (IL-1α and IL-1β; refs. 1–3), antagonist ligands [IL-1 receptor antagonist (IL-1ra); refs. 4–6], and both type 1 and type 2 IL-1 receptors (IL-1R1 and IL-1R2; refs. 7–10). It was recently shown that release of the primary cytokine IL-1α from basal keratinocytes in vivo is sufficient to both initiate and mediate cutaneous inflammation in vivo (11, 12). Because active species of IL-1 mediate so many pathophysiological processes relevant to inflammation (13), molecular mechanisms that limit the degree and duration of IL-1 activity have evolved. Species of IL-1α and IL-1β that are biologically active mediate their effects by binding to an 80-kDa IL-1 receptor, termed IL-1R1 (14–16). A related receptor ligand molecule, which can be made by cells in several alternatively spliced forms, is known as the IL-1 receptor antagonist or IL-1ra (4–6, 17, 18). This family of molecules binds to IL-1R1, but receptor occupancy by IL-1ra does not result in signal transduction (18). Thus, blockade of the IL-1R1 is an important means of limiting IL-1 activity (10, 16, 19), and under many circumstances, IL-1 gene expression appears to be paralleled by expression of the IL-1ra gene (5). The principal form of IL-1ra in keratinocytes is the intracellular IL-1ra, and it coexists in a cytoplasmic compartment with active preformed IL-1α in normal human and murine epidermis (6).
It has been proposed that a second pathway of IL-1 antagonism exists, in this case mediated by a cell surface receptor that does not transduce a signal and can be shed from the cell surface (10, 16, 19–21). This receptor, termed IL-1R2, maintains its capacity to bind to IL-1 species in solution and does not bind IL-1ra with high affinity (22). Stimuli that induce IL-1 gene expression in keratinocytes in vitro also strongly stimulate IL-1R2 expression (10), and inflammatory skin diseases such as psoriasis, where IL-1 has been implicated, are characterized by strong epidermal expression of IL-1R2 (9). While it is attractive to speculate that the IL-1R2 antagonizes IL-1 activity by sequestering active ligand, experimental systems designed to test this hypothesis have been imperfect, and virtually all have explored this activity of IL-1R2 in vitro rather than in vivo (10, 16, 19, 23, 24). In this study, we test the hypothesis that local production of IL-1R2, and its existence as both a cell surface and soluble ligand for IL-1, is an efficient way of down-regulating IL-1 responses in the cutaneous microenvironment without influencing systemic responses to IL-1.
We have successfully targeted IL-1R2 to basal keratinocytes using a human keratin 14 promoter (11, 12). Our results indicate that IL-1R2 expressed by keratinocytes in vivo can indeed inhibit IL-1-mediated inflammatory responses, both in epidermis and in dermis. In contrast, keratinocyte production of IL-1R2 did not lead to systemic hyporesponsiveness to IL-1. To our knowledge, these results are the first that demonstrate that IL-1R2, produced in vivo by biologically relevant cells (e.g., basal epidermal keratinocytes), can act as a powerful local IL-1 antagonist without systemic consequences. As such, these transgenic mice are useful tools to determine the role of IL-1 and related molecules in different types of cutaneous inflammation.
MATERIALS AND METHODS
DNA Construct and Transgenic Mice.
A 1.35-kb cDNA comprising the entire coding region of murine IL-1 receptors (the kind gift of J. Sims, Immunex Corp., Seattle, WA) was inserted into the BamHI site of the K14/hGH expression vector (a kind gift of E. Fuchs, University of Chicago, Chicago) by blunt end ligation. The resulting plasmid was digested with PvuI and HindIII to generate the 6.12-kb K14/IL-1R2/hGH fragment (see Fig. 1) that was used for microinjection of fertilized ova of FVB/N mice (Taconic Farms). Transgenic founders were identified by PCR and by Southern blot analysis and bred to establish lines. All animals used for experiments were F1 or F2 heterozygotes 6–12 weeks of age.
Figure 1.

DNA construct for expression of IL-1R2 in transgenic mice. Murine IL-1R2 cDNA was inserted between K14 and hGH elements at the BamHI site of the K14/hGH expression vector by blunt end ligation.
RNA Analysis.
Total RNA was isolated from tissues of transgenic and control mice by TRI REAGENT (Molecular Research Center, Cincinnati). For Northern blot analysis, 10 μg of total RNA was size-fractionated by electrophoresis on through a 1% formaldehyde/agarose gel. RNA was transferred to nylon membrane (Hybond N+ nylon membrane, Amersham) and probed with 32P-labeled cDNA for murine IL-1R2. After hybridization, blots were washed twice at 65°C in 1× standard saline citrate (SSC)/0.1% SDS, and twice in 0.1× SSC/0.1% SDS. Blots were then exposed to x-ray film at −70°C.
Cytokines and Cytokine Assay Systems.
Murine recombinant IL-1α was a kind gift of P. Lomedico (Hoffmann–LaRoche). Murine recombinant IL-1β was obtained from Endogen (Cambridge, MA). The ELISA for IL-6 was obtained from PharMingen, and the ELISA for murine granulocyte/macrophage colony-stimulating factor (GM-CSF) was obtained from Endogen.
The 35F5 antibody to the murine IL-1R1 and the 4E2 antibody to the murine IL-1R2 were the kind gifts of Richard Chizzonite (Hoffmann–La Roche). Isotype-matched control antibodies for 35F5 (rat 1gG1) and 4E2 (rat 1gG2a) were obtained from PharMingen.
Histology and Immunohistochemistry.
Hematoxylin and eosin stainings were performed by standard methods. For immunohistochemistry, ear skin biopsies from transgenic and control mice were embedded in OCT compound (Miles) and frozen in precooled isopentane.
Cryostat sections (5 μm) were fixed for 10 min in acetone at 4°C, and immunostaining of frozen sections was performed. Briefly, sections were incubated at room temperature for 1 h with rat anti-mouse IL-1R2 antibody appropriately diluted in PBS. After washing, primary antibody binding was revealed by the Vectastain Elite kit (Vector Laboratories) used according to the manufacturer’s instructions with diaminobenzidine as chromogen. Sections were lightly counterstained with methyl green and mounted.
Epidermal Cell Culture and Immunoprecipitation.
Epidermal cell suspensions were obtained by sequential dispase and trypsin digestion of split mouse ears as previously described (25, 26). Keratinocytes thus obtained were cultured on collagen-coated plates in medium containing 0.05 mM Ca2+ to allow maintenance of K14 expression. For detection of membrane-bound and soluble IL-1R2 protein, keratinocytes were cultured in methionine- and cysteine-free medium and metabolically labeled with [35S]methionine and [35S]cysteine for 4 h.
After aspiration of the conditioned medium, cells were extracted in radioimmunoprecipitation assay buffer, and aliquots of cell extract and conditioned medium were incubated with 4E2 antibody at 4°C overnight. Antibody–antigen complexes were then precipitated with protein G. Parallel samples were incubated with rat IgG as a control for nonspecific binding. After washing and boiling, samples were then subjected to electrophoresis through a 10% polyacrylamide gel. The gel was dried and exposed to film for 3 days at −70°C
Cytokine Assays.
To determine in vitro responsiveness of keratinocytes to IL-1, epidermal cells from T220 and FVB control mice were prepared and cultured overnight. The next day adherent cells were plated into 96-well plates at a density of 5 × 104 cells per well. Upon adherence cells were stimulated in duplicate with various doses of recombinant murine IL-1α for 24 h. Immunoreactive GM-CSF released into the supernatant was quantitated by ELISA.
Contact Hypersensitivity (CHS) Responses.
Mice (four to five mice per group) were shaved on the abdominal area and sensitized by applying 20 μl of 1% oxazolone (Sigma), dissolved in ethanol, to the skin. Mice were challenged 5 days later by painting 10 μl of 1% oxazolone on both sides of one ear. Ear thickness was measured before and at multiple time points after challenge with an engineer’s caliper (Dyer, Lancaster, PA). Results are expressed as the increase in ear thickness (mean ± SEM).
Vascular Permeability Assay.
Mice were shaved on the dorsal surface 1 day before the assay, and those in the anagen phase of hair growth were excluded. Various doses of phorbol-12-myristate 13-acetate (PMA) dissolved in 100 ml of 100% ethanol were applied to a 1.5 cm2 area on the shaved surface. After 6 h, 8.3 μl/g body weight of 1% Evan’s blue dye solution was injected through the tail vein. Exactly 30 min after injection, a 6-mm-diameter biopsy was taken from the PMA-treated area and minced in 0.3 ml of 0.5% Na2SO4. Evan’s blue dye in the tissue was extracted in acetone overnight and measured in solution by absorbance at 620 nm (Pharmacia LKB UltrospecIII).
To control for variations in injected dose of dye, liver tissue was also excised, weighed, extracted, and analyzed. For blocking experiments, 25 μg of either 35F5 or rat IgG was injected intradermally 15 min before PMA application. The mice were then treated as described above. Control tissue was taken from PMA-treated but noninjected adjacent skin. Four to five mice were used within each experimental group, and each experiment was repeated three times. Results are expressed as the mean ± SD.
Chronic PMA-Induced Inflammation.
PMA (1 μg) dissolved in 20 μl of 100% ethanol was applied on one mouse ear three times over a period of 1 week. The inflammatory skin response was assessed by measuring ear thickness using an engineer’s caliper (Dyer) and by histology. Biopsies were taken 24 h after the third PMA application and stained by hematoxylin and eosin. Each experimental group consisted of four to five animals, and experiments were repeated three times. All data are expressed as the mean difference in ear thickness due to PMA treatment (in micrometers ± SD).
RESULTS
Generation of Transgenic Mice.
Transgenic mice were generated using the construct shown in Fig. 1. Two of 20 live-born mice were identified as transgenic founders by PCR and Southern blot analysis. They were bred to establish independent lines designated T2 20 and T2 30. All experiments were performed on lines that were heterozygous for the transgene, along with nontransgenic littermate controls.
RNA Analysis.
To determine relative levels of transgene mRNA expression, skin RNA from T2 20, T2 30, and control mice was analyzed for expression of IL-1R2 by Northern blotting. High level expression of transgene RNA was found in both lines, and densitometric analysis revealed 30- and 8-fold overexpression in T2 20 and T2 30 mice, respectively, compared with endogenous IL-1R2 expression in skin of FVB control mice (data not shown).
Keratinocytes from T2 20 Transgenic Mice Express Membrane-Bound and Soluble Forms of IL-1R2 Both in Vivo and in Vitro.
Immunohistochemical analysis of ear skin stained with the anti-muIL-1R2 specific antibody 4E2 (Fig. 2) demonstrated high level expression of the transgene product in basal keratinocytes and in keratinocytes of the outer root sheath. The pattern of immunoreactivity is consistent with cell surface expression on basal keratinocytes. No staining was observed in control animals. Because of recent data suggesting that the IL-1R2 is shed from the cell surface, it was important to determine whether T2 20 keratinocytes released the soluble form of the receptor.
Figure 2.

IL-1R2 is expressed in the basal layer of epidermis in T2 20 mice. Frozen sections of ear skin from T2 20 (A) and FVB control (B) mice were stained for IL-1R2 with 4E2 antibody. No reactivity is shown in wild-type epidermis (B).
Therefore, cultured transgenic and control keratinocytes were metabolically labeled with [35S]methionine, and a monoclonal antibody to the murine IL-1R2 (4E2) was used for immunoprecipitation of both cell pellets and conditioned media. A significant immunoreactive species with an apparent molecular weight of ≈65 kDa could be precipitated only by 4E2, and only from T2 20 keratinocytes, but not from control keratinocytes (Fig. 3A). An isotype-matched irrelevant antibody did not immunoprecipitate a comparable 65-kDa species from T2 20 keratinocytes.
Figure 3.

T2 20 keratinocytes produce cell associated (A) and soluble (B) IL-1R2. Cell pellets from, and media conditioned by, transgenic, FVB, and PMA-stimulated FVB keratinocytes were immunoprecipitated with antibody for IL-1R2 (4E2) and isotope-matched control antibody (rat IgG2a). Note that a strong band at ≈65 kDa (A, lane 3) and ≈45 kDa (B, lane 3) is only present in transgenic cell pellets and media conditioned by transgenic keratinocytes that were immunoprecipitated with 4E2 antibody.
Similarly, shed IL-1R2 could only be detected in medium conditioned by transgenic keratinocytes (Fig. 3B). The molecular weight of the shed IL-1R2 species (≈45 kDa) is consistent with published reports (19). No soluble IL-1R2 was present in unstimulated control keratinocyte supernatants; low levels could be detected in PMA-stimulated keratinocyte supernatants from control mice after prolonged exposure of the blot (data not shown).
IL-1-Induced GM-CSF Production Is Diminished in T2 20 Transgenic Mice.
To determine whether overexpression of IL-1R2 on keratinocytes inhibited their response to IL-1 in vitro, we asked whether secondary (IL-1-inducible) cytokine production was altered in T2 20 transgenic mice. Cultured T2 20 and control keratinocytes (four mice per group) were stimulated with various doses of IL-1α, and GM-CSF production was measured after 24 h using a specific ELISA (Fig. 4). Whereas nontransgenic keratinocytes showed a dose-dependent response to increasing extracellular IL-1α, the response of transgenic keratinocytes was markedly inhibited, even at the highest concentrations of extracellular IL-1. Therefore, overexpression of IL-1R2 by keratinocytes significantly blunts their response to IL-1 in vitro.
Figure 4.
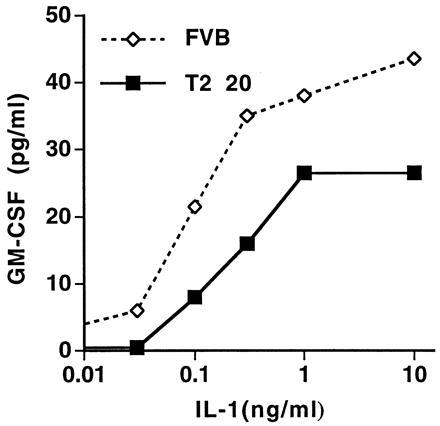
GM-CSF response to IL-1α. Cultured keratinocytes from T2 20 and control FVB mice were stimulated with various doses of murine IL-1α for 24 h. Immunoreactive GM-CSF released into the supernatant was quantitated by ELISA. The results are representative of two experiments showing virtually identical outcomes.
PMA-Induced Vascular Permeability Depends on Local Release of IL-1 and Is Significantly Reduced in IL-1R2 Transgenic Mice.
To assess the responses of T2 20 mice to IL-1-mediated inflammation in vivo, we chose to study their response to the topical application of phorbol ester, a well characterized inflammatory model in which IL-1 plays a prominent role. IL-1α gene expression in skin of FVB mice is rapidly induced by topical application of the phorbol ester PMA (12), and two independent groups have demonstrated that phorbol ester induced inflammatory cell infiltration, epidermal hyperplasia, and vascular leakage, all of which can be blocked by systemic or local administration of anti-IL-1 antibodies (27, 28). Endothelial cell leakage was induced by topical application of PMA on the dorsal surface of mice and subsequent Evan’s blue dye i.v. injection. This protocol results in extravasation of dye at PMA-treated sites, and control animals showed an obvious blue staining of PMA-treated skin.
This qualitative difference can be assessed quantitatively by extracting Evan’s blue dye from excised tissue in acetone and measuring absorbance spectrophotometrically. In Fig. 5A, the extravasation of Evan’s blue dye was inhibited significantly by local injection of a blocking antibody to the IL-1R1, whereas an isotope-matched control antibody had no effect. This experiment extends the results from other laboratories (27–29) regarding the IL-1 dependence of this phenomena. In Fig. 5B, control mice were compared with T2 20 mice. Significantly less dye extravasated into PMA-treated skin of T2 20 mice, indicating that there was a substantial inhibition of this acute response in vivo, compared with age-matched nontransgenic FVB mice.
Figure 5.

Spectrophotometic evaluation of absorbed Evan’s blue dye at PMA-treated sites. Areas measuring 1.5 cm2 were treated with PMA, and 8.3 μl/g mouse of a 1% Evan’s blue solution was injected i.v. 6 h later. After 30 min, skin biopsies were taken, and the dye was extracted overnight from the tissue and measured in a spectrophotometer at 620 nm. Data represent the mean absorbance at 620 nm ± SD. (A) Diminished PMA induced extravasation of Evan’s blue dye by local injection of 35F5, a blocking antibody to IL-1R1. An isotype-matched control antibody had no effect. ∗, P < 0.02 compared with control skin (Student’s t test).
Both transgenic and nontransgenic mice had equivalent baseline extravasation of Evan’s blue dye into liver, indicating that similar amounts of dye were injected and that there were no systemic effects on vascular permeability attributable to epidermal transgene expression (data not shown).
Reduced Chronic Inflammatory Response to PMA in IL-1R2 Transgenic Mice.
To further assess the response of T2 20 mice to PMA, we employed a more chronic model in which PMA was painted on ears of transgenic and control mice three times within 1 week (12). The inflammatory response was assessed quantitatively by measurement of ear thickness and qualitatively by measuring corresponding histological changes. Both groups showed increased ear swelling after each of the three PMA applications, but there was significant inhibition of the response in transgenic mice, most marked after the third application of PMA (Fig. 6A). Histologically, there was also an obvious difference, with less pronounced epidermal hyper proliferation and dermal inflammation in transgenic mice compared with controls (not shown).
Figure 6.

(A) Ear swelling response to PMA treatment. PMA (1 μg) was applied topically on each side of the ear three times on days 0, 3, and 6. The increase in ear thickness relative to the untreated ear was determined 24 h after each PMA application and reported as the mean ± SD. (B) Significant reduction in the magnitude of ear swelling.
IL-1-Independent Inflammatory Responses in Skin Are Not Inhibited in IL-1R2 Transgenic Mice.
The role of IL-1 in CHS is not straightforward, with some studies implicating IL-1α and IL-1β in this process (30, 31), and others suggesting that CHS is not IL-1-dependent (32). Two independent studies on IL-1β null mice both agree, however, that the response to the contact sensitizer oxazolone is not diminished in this system. A standard CHS response to a conventional sensitizing dose of oxazolone in FVB mice revealed that the presence of the transgene does not affect the magnitude or the duration of this response (not shown). The same phenomenon was seen when lower sensitizing doses were used. This result indicates that not all cutaneous inflammatory responses are inhibited in T2 20 transgenic mice, and is consistent with the interpretation that the oxazolone-induced CHS response is not strictly dependent on IL-1 activity.
Systemic Responses to IL-1 Are Intact in T2 20 Mice.
A somewhat trivial explanation of the data shown in Figs. 5 and 6 would be that high levels of soluble IL-1R2 released by basal keratinocytes into plasma lead to systemic inhibition of IL-1-mediated responses. To test this possibility, IL-1α and IL-1β were injected i.v. into T2 20 and nontransgenic mice, and plasma IL-6 levels were measured (Table 1). This analysis confirmed the IL-1 dependence of this event and indicated that T2 20 animals were indistinguishable from nontransgenic controls with regard to IL-6 levels. These data indicate that systemic responsiveness to IL-1 was not suppressed in T2 20 mice.
Table 1.
IL-6 plasma levels
| IL-1 stimulation | FVB at 3 h | T2 20 at 3 h | FVB at 5 h | T2 20 at 5 h |
|---|---|---|---|---|
| IL-1α (0.5 μg) | 417 ± 9.6 | 408 ± 9 | 396 ± 18.6 | 393 ± 15.6 |
| IL-1α (0.05 μg) | 45 ± 7.7 | 72 ± 10.8 | 36 ± 11.7 | 39 ± 21 |
| IL-1β (0.5 μg) | 297 ± 126 | 279 ± 75 | 174 ± 46.3 | 198 ± 54 |
| IL-1β (0.05 μg) | 45 ± 9.9 | 42 ± 7.7 | 57 ± 6 | 117 ± 69 |
Both FVB and T2 20 animals were injected i.v. with two different doses of IL-1α or IL-1β. IL-6 plasma levels were determined by ELISA 3 and 5 h after IL injection. No significant difference in IL-6 plasma levels was found between FVB and T2 20 mice under any of the above conditions. All values are the mean ± SD of four animals per group and are expressed as picograms per milliliter.
DISCUSSION
Cytokines, including IL-1α stored in keratinocytes, are emergency molecules, designed to be active for limited periods of time in local microenvironments such as skin (33–35). We have postulated previously that keratinocyte expression of IL-1R2 could be a mechanism by which IL-1-mediated cutaneous responses are negatively regulated (10, 11). The data presented in this report represent the first in vivo evidence of inhibition of IL-1 inhibition mediated by IL-1R2 in cutaneous microenvironment, and indicates that this local inhibition can proceed without systemic antagonism of IL-1.
IL-1R2 has long been suspected of having IL-1 antagonist activity (10, 16, 19–21). In vitro studies have suggested that blockade of IL-1R2 may enhance cellular responses to suboptimal levels of IL-1 (16), and soluble shed IL-1R2 inhibits IL-1-induced collagenase and PGE2 production (23). A recent study examined cells transfected with IL-1R2 and concluded that in addition to the inhibitory effects of shed IL-1R2, cell surface IL-1R2 can inhibit IL-1 responses in vitro. However, while there is one report that describes diminished delayed hypersensitivity skin reaction with a soluble form of IL-1R1 administered to patients (36), we were unable to find any report that demonstrated that elevated expression of IL-1R2 in vivo inhibits IL-1-mediated inflammation.
The present study was designed to test the capacity of constitutively expressed IL-1R2 to modify acute and chronic local IL-1-mediated biological events in vivo. Epidermis is an accessible tissue in which all elements of the IL-1 system are represented (33, 34) and was, therefore, an ideal organ in which to address this issue (33). We successfully generated two independent lines of transgenic mice that overexpress IL-1R2 in basal epidermis. The expression of the transgene at the level of protein and mRNA was confirmed, and the higher expressing line (T2 20) was selected for extensive analysis. Keratinocytes derived from transgenic mice were markedly hyporesponsive to IL-1α in vitro, as evidenced by their diminished production of GM-CSF. Interestingly, the maximal response to IL-1 in the T2 20 cells never approached the maximal response in nontransgenic cells. This is reminiscent of recent data reported on IL-1R2 transfected cells (24) and appears to represent inhibition of the IL-1 response by cell surface as well as soluble IL-1R2. Consistent with this interpretation, immunoprecipitation with antibodies to IL-1R2 demonstrated high levels of both cell-bound and free IL-1R2 in the transgenic keratinocyte cultures; in contrast, this protein was undetectable in control keratinocyte cultures. Because of this clear-cut IL-1 inhibitory activity in vitro, together with RNA and immunohistochemical findings confirming high level tissue-specific expression of the transgene in vivo, we felt that the T2 20 mice represented a valid model to test of the hypothesis that IL-1R2 could function to inhibit IL-1-mediated cutaneous responses in vivo.
Application of PMA to mouse skin results in an inflammatory reaction characterized acutely by increased vascular permeability, epidermal hyperproliferation, and the recruitment of a mixed leukocytic infiltrate (12, 27, 28). These responses have been extensively studied, and keratinocyte IL-1α has been shown to be an important mediator of phorbol ester-induced effects in mouse skin (27–29). Transgenic mice that express high levels of the IL-1R1 on basal keratinocytes show exaggerated inflammation in response to PMA, also confirming that IL-1 is strongly induced in epidermis by topical PMA application (12). Not only are phorbol esters potent inducers of IL-1α mRNA in skin (12, 27, 29), but both acute and delayed inflammatory effects can be inhibited by in vivo administration of blocking IL-1α antibodies (27, 28). Furthermore, we have shown in this study that acute PMA-induced vascular leakage is inhibited by intradermal injection of anti-IL-1R1 monoclonal antibody (Fig. 6A). Thus, blockade of IL-1, at either the ligand or the receptor level, results in a diminished cutaneous response to PMA.
Both immediate and delayed cutaneous responses to PMA were inhibited in the T2 20 mice as compared with littermate controls. PMA-induced changes in vascular leakage were assessed by i.v. injection of Evan’s blue dye. In control animals, there was marked leakage of dye in skin at the site of PMA application but not in skin treated with vehicle lacking PMA. In the T2 20 mice, equivalent applications of PMA were followed by significantly less extravasation of Evan’s blue dye. These data are consistent with the interpretation that keratinocyte IL-1R2 is antagonizing the PMA-induced, IL-1-mediated increase in vascular permeability that occurs 6 h after PMA application. Presumably, this is occurring at the level of binding and sequestering newly synthesized and/or released IL-1α by the transgenically expressed IL-1R2.
More chronic effects of PMA were also inhibited in T2 20 mice; these animals exhibited a highly significant reduction in ear swelling after repetitive PMA application. Histological analysis revealed an obvious reduction in both epidermal hyperplasia and dermal inflammatory cell infiltration that correlated well with the quantitative ear swelling data. It is clear from previous studies that the degree of PMA-induced inflammation is affected by secondary cytokines produced by keratinocytes (12), so that the mechanism of the chronic inflammatory inhibition could also be at the level of reduction of keratinocyte secondary IL-1-inducible cytokine production.
Because of the pleiotropic inflammatory effects of IL-1 and its ability to activate numerous cell types the necessity for evolution of mechanisms to control excessive IL-1 activity is easy to understand. The first such mechanism to be identified was the IL-1 receptor antagonist (4–6, 17, 18), a factor that exists in at least three alternatively spliced forms and blocks binding of IL-1 to IL-1R1. Our data, taken together with data from in vitro systems, would suggest that IL-1R2 clearly fulfills a similar role. However, whereas IL-1ra competes with IL-1 for binding to its signaling receptor and thus antagonizes its actions, IL-1R2 may negatively regulate the IL-1 system by binding IL-1 and thereby preventing subsequent NF-κB-mediated signal transduction via IL-1R1 (24). It would make good biological sense for the principal IL-1-producing cells (such as macrophages and keratinocytes) to possess both of these negative regulatory mechanisms to minimize the possibility of unwanted autocrine stimulation. Indeed, this appears to be the case. An elegant control on this system is evidenced by the relative inability of IL-1R2 to bind IL-1ra with high affinity (22). Therefore, the two independent mechanisms of negative regulation, IL-1ra at the receptor level and IL-1R2 at the ligand level, appear to inhibit IL-1-mediated events synergistically (23).
An important feature of our model is that the inhibition of inflammation was specific for IL-1. Whereas PMA-mediated inflammation was inhibited, inflammation induced by the immune response to the contact sensitizer oxazolone was not affected. Although the role of IL-1 in CHS in general remains controversial, two independent groups have shown that CHS to oxazolone is independent of the IL-1 axis in IL-1β null mice (30, 32). We therefore chose oxazolone as an IL-1-independent means of inducing inflammation. Our results show consistently that, unlike PMA-mediated inflammation, oxazolone-induced inflammation was indistinguishable in T2 20 mice and controls. Therefore, a potential unanticipated benefit of these transgenic mice is their utility in determining which inflammatory mediators in skin involve the IL-1 axis and which do not.
Because cytokines are likely to be most important physiologically in tissue microenvironments, limiting their activity locally but not systematically would be advantageous to the host. Tissue-specific inhibition of IL-1 responses may be best achieved by a molecule such as IL-1R2, which can be expressed on the cell surface as well as shed into solution. Our data show clearly that local inhibition of IL-1-mediated responses in T2 20 mice can be significant in the absence of systemic antagonism of IL-1. IL-6 responses to systematically administered IL-1 were indistinguishable in T 220 and control mice. It is likely that sufficiently high expression of IL-1R2, with attendant shedding from cells in vivo, would result in systemic leakage of soluble IL-1R2 and systemic suppression of inflammation. However, our model clearly shows that there are conditions under which primary cytokine inhibition can remain localized to skin. Certainly, the converse has been appreciated for decades; cutaneous inflammation can proceed without systemic consequences.
Other authors have suggested that anti-inflammatory stimuli, such as IL-4 and glucocorticosteroids, are principal regulators of IL-1R2 (19). Although this is certainly the case, we have found that in keratinocytes, proinflammatory stimuli, such as PMA and interferon γ, are also potent stimuli of IL-1R2 gene expression (10). Similarly, high levels of IL-1R2 are expressed on basal keratinocytes in the inflammatory skin disease psoriasis (9). We interpret these findings as consistent with the idea that IL-1-mediated inflammation induces IL-1R2 expression as a compensatory response to down-regulate the inflammatory process. Thus, elevated IL-1R2 in psoriasis may represent the “smoking gun” that implicates prior IL-1-mediated initiation of inflammation. Patients with psoriasis do not exhibit systemic inhibition of inflammatory responses occurring outside of skin, despite the abundance of IL-1R2 in their lesional psoriatic skin. It is likely that in these patients, the biological rationale for epidermal IL-1R2 expression is local, not systemic, control of inflammation.
Taken together, our data provide the first conclusive evidence to our knowledge that IL-1R2 may antagonize IL-1-mediated events in vivo. That this antagonism is specific for IL-1 and restricted to tissues in which the IL-1R2 is expressed is also novel. These activities of the IL-1R2 make it attractive as a candidate for genetically based therapeutic approaches to chronic inflammation limited to specific tissues. These studies also confirm and extend the utility of tissue-specific transgenic approaches to defining the in vivo potential of both pro- and anti-inflammatory biological response modifiers.
ABBREVIATIONS
- IL-1
interleukin 1
- IL-1ra
IL-1 receptor antagonist
- IL-1R1 and IL-1R2
type 1 and type 2 IL-1 receptors
- PMA
phorbol-12-myristate 13-acetate
- CHS
contact hypersensitivity
- GM-CSF
granulocyte/macrophage colony-stimulating factor
References
- 1.Kupper T S, Ballard D W, Chua A O, McGuire J, Flood P M, Horowitz M C, Langdon R C, Lightfoot L, Gubler U. J Exp Med. 1986;164:2095–2101. doi: 10.1084/jem.164.6.2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hauser C, Saurat J-H, Schmitt A, Jaunin F, Dayer J-M. J Immunol. 1986;136:3317–3323. [PubMed] [Google Scholar]
- 3.Mizutani H, Black R, Kupper T S. J Clin Invest. 1991;87:1066–1071. doi: 10.1172/JCI115067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bigler C F, Norris D A, Weston W L, Arend W P. J Invest Dermatol. 1992;98:38–44. doi: 10.1111/1523-1747.ep12494196. [DOI] [PubMed] [Google Scholar]
- 5.Hammerberg C, Arend W P, Fisher G, Chan L S, Berger A E, Haskill J S, Voorhees J J, Cooper K D. J Clin Invest. 1992;90:571–583. doi: 10.1172/JCI115896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haskill S, Martin G, Van Le L, Morris J, Peace A, Bigler C F, Jaffe G J, Hammerberg C, Spron S A, Fong S, Arend W P, Ralph P. Proc Natl Acad Sci USA. 1991;88:3681–3685. doi: 10.1073/pnas.88.9.3681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kupper T S, Lee F, Birchall N, Clark S, Dower S. J Clin Invest. 1988;82:1787–1792. doi: 10.1172/JCI113792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blanton R A, Kupper T S, McDougall J K, Dower S K. Proc Natl Acad Sci USA. 1989;86:1273–1277. doi: 10.1073/pnas.86.4.1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Groves R W, Sherman L, Mizutani H, Dower S K, Kupper T S. Am J Pathol. 1994;145:1048–1056. [PMC free article] [PubMed] [Google Scholar]
- 10.Groves R W, Giri J G, Sims J E, Dower S K, Kupper T S. J Immunol. 1995;154:4065–4071. [PubMed] [Google Scholar]
- 11.Groves R W, Mizutani H, Kieffer J D, Kupper T S. Proc Natl Acad Sci USA. 1995;92:11874–11878. doi: 10.1073/pnas.92.25.11874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Groves R W, Rauschmayr T, Nakamura K, Sarkar S, Williams I R, Kupper T S. J Clin Invest. 1996;98:336–344. doi: 10.1172/JCI118797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dinarello C A. Blood. 1996;87:2095–2147. [PubMed] [Google Scholar]
- 14.Sims J E, March C J, Cosman D, Widmer M B, MacDonald H R, McMahan C, Grubin C E, Wignall J M, Jackson J, Call S M, Friend D, Alpert A, Gillis S, Urdal D L, Dower S K. Science. 1988;241:585–589. doi: 10.1126/science.2969618. [DOI] [PubMed] [Google Scholar]
- 15.Curtis B M, Gallis B, Overell R W, McMahan C, DeRoos P, Ireland R, Eisenman J, Dower S K, Sims J E. Proc Natl Acad Sci USA. 1989;86:3045–3049. doi: 10.1073/pnas.86.9.3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sims J E, Gayle M, Slack J L, Alderson M, Bird T, Giri J G, Colotta F, Mantovani A, Shanebeck K, Grabstein K, Dower S K. Proc Natl Acad Sci USA. 1993;90:6155–6159. doi: 10.1073/pnas.90.13.6155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carter D B, Deibel M R, Dunn C J, Tomich C-S C, Labore A L, et al. Nature (London) 1990;344:633–638. doi: 10.1038/344633a0. [DOI] [PubMed] [Google Scholar]
- 18.Hannum C H, Wilcox C J, Arend W P, Joslin F G, Dripps D J, Heimdal P L, Armes L G, Sommer A, Eisenberg S P, Thompson R C. Nature (London) 1990;343:336–340. doi: 10.1038/343336a0. [DOI] [PubMed] [Google Scholar]
- 19.Colotta F, Re F, Muzio M, Bertini R, Polentarutti N, Sironi M, Giri J G, Dower S K, Sims J E, Mantovani A. Science. 1993;261:472–475. doi: 10.1126/science.8332913. [DOI] [PubMed] [Google Scholar]
- 20.Slack J L, McMahan C, Waugh S, Schooley K, Spriggs M K, Sims J E, Dower S K. J Biol Chem. 1993;268:2513–2524. [PubMed] [Google Scholar]
- 21.Giri J G, Robb R, Wong W L, Horuk R. Cytokine. 1992;4:18–23. doi: 10.1016/1043-4666(92)90031-l. [DOI] [PubMed] [Google Scholar]
- 22.Symons J A, Young P R, Duff G W. Proc Natl Acad Sci USA. 1995;92:1714–1718. doi: 10.1073/pnas.92.5.1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burger D, Chicheportiche R, Giri J G, Dayer J M. J Clin Invest. 1995;96:38–41. doi: 10.1172/JCI118045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Re F, Sironi M, Muzio M, Matteucci C, Introna M, Orlando S, Penton-Ro I G, Dower S K, Sims J E, Colotta F, Mantovani A. J Exp Med. 1996;183:1841–1850. doi: 10.1084/jem.183.4.1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sullivan S, Bergstresser P R, Tigelaar R E, Streilein J W. J Invest Dermatol. 1985;84:491–495. doi: 10.1111/1523-1747.ep12273454. [DOI] [PubMed] [Google Scholar]
- 26.Kilgus O, Payer E, Schreiber S, Elbe A, Strohal R, Stingl G. J Invest Dermatol. 1993;100:674–680. doi: 10.1111/1523-1747.ep12472339. [DOI] [PubMed] [Google Scholar]
- 27.Oberyszyn T M, Sabourin C L K, Bijur G N, Oberyszyn A S, Boros L G, Robertson F M. Mol Carcinog. 1993;7:238–248. doi: 10.1002/mc.2940070406. [DOI] [PubMed] [Google Scholar]
- 28.Lee W Y, Lockniskar M F, Fischer S M. FASEB J. 1994;8:1081–1087. [PubMed] [Google Scholar]
- 29.Lee W Y, Fischer S M, Butler A P, Lockniskar M F. Mol Carcinog. 1993;7:26–35. doi: 10.1002/mc.2940070106. [DOI] [PubMed] [Google Scholar]
- 30.Shornick L P, de Togni P, Mariathasan S, Goellner J, Strauss-Schoenberg J, Karr R W, Ferguson T A, Chaplin D D. J Exp Med. 1996;183:1427–1436. doi: 10.1084/jem.183.4.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Enk A H, Angeloni V L, Udey M C, Katz S I. J Immunol. 1993;150:3698–3704. [PubMed] [Google Scholar]
- 32.Zheng H, Fletcher D, Kozak W, Jiang M, Hofmann K J, Conn C A, Soszynski D, Grabiec C, Trumbauer M E, Shaw A, Kostura M J, Stevens K, Rosen H, North R J, Chen H Y, Tocci M J, Kluger M J, Van der Ploeg L H T. Immunity. 1995;3:9–19. doi: 10.1016/1074-7613(95)90154-x. [DOI] [PubMed] [Google Scholar]
- 33.Kupper T S. J Clin Invest. 1990;86:1783–1789. doi: 10.1172/JCI114907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kupper T S, Groves R W. J Invest Dermatol. 1995;105:62S–66S. doi: 10.1111/1523-1747.ep12316087. [DOI] [PubMed] [Google Scholar]
- 35.Williams I R, Kupper T S. Life Sci. 1996;58:1485–1507. doi: 10.1016/0024-3205(96)00042-2. [DOI] [PubMed] [Google Scholar]
- 36.Dower S K, Fanslow W, Jacobs C, Waugh S, Sims J E, Widmer M B. Ther Immunol. 1994;1:113–122. [PubMed] [Google Scholar]