

Abstract
Germline-inactivating mutations of BRCA1 result in a hereditary predisposition to breast and ovarian cancer. Truncating mutations of BRCA1 predispose to cancer and can be ascertained by protein truncation testing or sequencing. However, cancer-predisposing missense mutations of BRCA1 are difficult to distinguish from polymorphisms by genetic testing methods currently used. Here we show that expression of BRCA1 or BRCA1 fused to a GAL4 activation domain in Saccharomyces cerevesiae inhibits growth, resulting in small colonies easily distinguishable from vector-transformed controls. The growth inhibitory effect can be localized to sequences encoding the recently described BRCA1 C-terminal domains. Growth suppression by a BRCA1 fusion protein is not influenced by introduction of neutral polymorphisms but is diminished or abolished by frameshift, nonsense, or disease-associated missense mutations located in the C-terminal 305 amino acids of BRCA1. These observations may permit the functional significance of many BRCA1 sequence changes to be assessed in yeast. Additionally, the correlation of growth suppression with wild-type forms of BRCA1 suggests that the assay may be capable of detecting functionally conserved interactions between the evolutionarily conserved BRCA1 C-terminal domains and cellular elements found in both human and yeast cells.
Breast cancer is the second leading cause of cancer deaths among women in industrialized countries, and germline mutations of BRCA1 account for 2.5–5% of all cases of breast cancer (1). Women in high risk families who inherit inactivating mutations of BRCA1, a putative tumor suppressor gene, are currently estimated to have 87% and 44% lifetime risks for breast and ovarian cancer, respectively (2). Mutations of BRCA1 can be detected by direct sequencing, single-stranded conformational polymorphism analysis, and DNA chips (3). The majority of disease-associated mutations results in truncation of the ORF and may be detected by in vitro translation of cDNA (4). A minority of cancer-predisposing mutations are missense mutations, however, and may only be detected by sequence analysis (5). But sequence analysis alone cannot distinguish between disease-associated missense mutations and polymorphisms. Additional data from linkage analysis or large scale population studies are required to infer the cancer risk associated with a given missense sequence variation. Such data may not be readily available. Biologically relevant missense mutations are expected to alter gene function, so an assay that detects the functional consequences of a given missense sequence change would help distinguish rare polymorphisms from cancer-predisposing mutations (5). We have discovered a growth suppressive phenotype of human BRCA1 in yeast that may permit development of such an assay for certain missense sequence alterations.
BRCA1 encodes a protein of 1863 amino acids, the precise function of which is still unknown (6). Expression of wild-type, but not mutant, BRCA1 inhibits growth of breast and ovarian epithelial tumor cell lines (7). Two motifs are recognizable by sequence analysis: an N-terminal, zinc-coordinating RING finger domain and two, tandem BRCA1 C-terminal (BRCT) domains (8–10). The BRCT domains are targets of cancer-associated missense mutations (5) and are evolutionarily conserved (11), suggesting that they are functional regions of the gene. Several lines of evidence suggest that the BRCT domains are essential to normal function of the protein: truncations of the C terminus predispose to cancer (5), suppress the ability of BRCA1 to inhibit breast cancer cell growth (7), and abrogate the ability of this region to function as a transcriptional activation domain when fused to a heterologous DNA binding domain (12, 13). BRCT domains are also present in BARD1, a recently discovered protein partner of BRCA1 (14).
Biochemical mechanisms in humans and yeast are often similar, and many human genes, including those with no known yeast homologs (15), will function in yeast. Although there is no yeast BRCA1 homolog, the C-terminal BRCT module is conserved in several yeast proteins (8–10), including RAD9. This module, fused to a heterologous DNA binding domain, activates transcription of reporter genes in yeast (13) as well as mammalian cells (12). Therefore, we thought it might be possible to develop an assay of human BRCA1 in yeast which determines the effect of sequence changes on the function of the BRCT module. We report here an assay of human BRCA1 based on growth inhibition of yeast. The assay detects nonsense and frameshift mutations of BRCA1 and distinguishes cancer-predisposing, missense mutations located in the carboxy-terminal 305 amino acids from common polymorphisms found within the same region.
MATERIALS AND METHODS
Plasmids, BRCA1 cDNA, and Site-Directed Mutagenesis.
This clone of BRCA1 has been published (3). We used pACT2 (CLONTECH) containing new restriction sites in the polylinker for subcloning of BRCA1. pAD–BRCA1–Δ1 was created by subcloning in-frame at the EcoRI site of BRCA1 into the modified pACT2 vector. pAD–BRCA1–Δ2 constructs were created by subclone of EcoRI–BamHI fragments from vectors kindly provided prepublication by A. N. A. Montiero, A. August, and H. Hanafusa. pAD–BRCA1–Δ3 was created by an in-frame subclone of the BRCA1 3′ end at the NcoI site. Site-directed mutagenesis was performed by standard PCR techniques (16), and all constructs were verified by sequencing. BRCA1 was directionally subcloned into pVT-U100 and pYES2 using 5′ HindIII and 3′ XhoI sites. pVT-U100 (17) is a 2-μ yeast expression vector with an alcohol dehydrogenase promotor. pYES2 is a 2-μ yeast expression vector with a GAL1-inducible promotor (Invitrogen).
Sequencing Analysis.
All mutations were confirmed by direct sequencing by the Taq DyeDiDeoxy Terminator Cycle Sequencing method (Applied Biosystems) using an Applied Biosystems model 373A automated sequencer.
Yeast Strains and Media.
The following strains were used: HF7c [MATa, ura3–52, trp1–901, leu2–3, his3–200, lys2–801, ade2–101, 112, gal4–542, gal80–538, LYS2::Gal1-HIS3, URA3::(GAL4 17-mers)3-CYC1-lacZ], SFY526 [MATa, ura3–52, trp1–901, leu2–3, his3–200, lys2–801, ade2–101, 112, canr, gal4–542, gal80–538, URA3::GAL1-lacZ], and YPH 499a (18) [MATa, ura3–52, lys2–801(amber), ade2–101(ochre), trp1–Δ63, his3–Δ200 leu2–Δ1]. HF7c and SFY526 were obtained from CLONTECH. Basic methods for yeast manipulations were carried out as described (19). Reagents for preparation of agar plates for prototrophic selection of yeast and for demonstration of growth inhibition were obtained from BIO-101, Vista, CA; media were prepared according to manufacturer’s specification. For large experiments, “drop-out” plates were purchased (Bioplates, Gaithersburg, MD). The agar contained 1.7% yeast nitrogen base, 2% dextrose, 0.5% ammonium sulfate, and 1.7% agar with amino acids minus those used for selection (SD-leu for selection of pAD vectors and SD-ura for selection of pVT-U100 and pYES2 vectors).
Transformation of Yeast.
All strains of yeast demonstrated the small colony phenotype when transformed with pAD–BRCA1, but the phenotype was most pronounced for Hf7c, which was therefore used for experiments in this study whenever possible. Experiments using the URA3 plasmids pVT-U100 and pYES2 were performed in YPH 499a. Yeast were transformed as follows: 50 ml of yeast extract/peptone/dextrose (YPD) was inoculated with a fresh colony of yeast and shaken at 225 rpm for 16–18 h at 30°C. Saturated culture was diluted the next morning to an OD600 of 0.2 in 300 ml of YPD. The culture was incubated shaking 225 rpm at 30°C for an additional 3 h to an OD600 of 0.6–1.0. The cells were pelleted, the supernatant was decanted, and the pellet was resuspended in 1.5 ml of LiOAc solution (0.1 M lithium acetate/10 mM Tris⋅HCl/1 mM EDTA Na2, pH 7.5); 50-μl aliquots were distributed to microcentrifuge tubes containing supercoiled plasmid and 50 μg of denatured salmon sperm as carrier, followed by addition of 300 μl of LiOAc solution containing 40% polyethylene glycol 3500–4000. After vortexing to mix, the tube was shaken 225 rpm at 30°C for 30 min. DMSO, 35 μl per sample, was added and mixed, and the tubes were incubated for 15 min in a 42°C water bath before a 2-min incubation on ice. The cells were pelleted by centrifugation and resuspended in 1 ml of water, and 200 μl of sample/plate was spread onto the appropriate selection plates. These primary incubation plates were incubated for 60 h at 30°C before photography or quantitation.
For yeast growth curves, single transformed colonies were grown to saturation in selective media. Cultures were centrifuged, and pelleted yeast were diluted to an OD600 of 0.2 in YPD, then shaken together at 250 rpm at 30°C and sampled hourly for OD600 determination.
Galactose Induction of BRCA1 Expression.
YPH 499a were transformed by the galactose-inducible yeast expression plasmid, pYES2, as above; 200 μl/plate of cell transformation mix was spread onto Minimal SD-ura, 2% Raffinose, and 1.7% agar plates (BIO-101). An equal aliquot was spread onto Minimal SD-ura, 2% galactose, and 1% raffinose agar plates. Colony size was visually assessed after 90 h of incubation at 30°C.
Western Blot Analysis.
Transformed yeast were grown in selective media to saturation. Cells were pelleted, resuspended at OD600 0.2 in YPD, and grown to mid-log phase. Rapidly cooled, pelleted cells were resuspended in 300 μl of 1.85 M NaOH/1% 2-mercaptoethanol per unit of OD600 and incubated for 10 min at 4°C before addition of an equal volume of 50% trichloroacetic acid. After 30 min at 4°C, precipitates were boiled 5 min in 25 μl/OD600 of 2 × SDS + 10% vol/vol 1 M Tris base. Equal amounts of crude lysate were separated by SDS/PAGE, transferred to nitrocellulose, and probed with an mAb to the influenza virus hemagglutination antigen (HA) epitope (HA.11, Babco) or an mAb to amino acids 1–302 of BRCA1 (MS110, a gift of R. Scully, Dana Farber Cancer Center, Cambridge, MA) as appropriate. Blots were developed by enhanced chemiluminescence techniques or by iodinated anti-mouse antibody before quantitation on a phosphorimager.
RESULTS
A Fusion Protein Containing Human BRCA1 Inhibits Growth of Yeast.
In the course of two-hybrid experiments, the GAL4 transcriptional activation domain and the nuclear localization signal from the simian virus 40 large T antigen were fused to the BRCA1 cDNA in a yeast 2-μ plasmid, pACT2, to create pAD–BRCA1. Saccharomyces cerevesiae transformed by pAD–BRCA1 formed colonies that were considerably smaller than controls after incubation at 30°C for 60 h (Fig. 1A). Colonies of yeast were resuspended in water, and the number of cells per colony was determined by counting. pAD–BRCA1-transformed colonies contained 30-fold fewer cells per colony than controls (Fig. 2C), and small colony formation correlated with slow growth in liquid culture (Fig. 1B).
Figure 1.
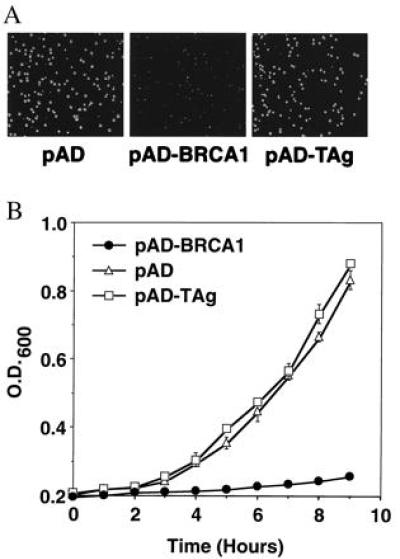
Small colony formation by yeast transformed with pAD–BRCA1. (A) Photograph of S. cerevesiae HF7c colonies after transformation with the indicated plasmids. pAD, pACT2 control vector; pAD–BRCA1, BRCA1 in pACT2; pAD–TAg, simian virus 40 large T antigen in pACT2. (B) Yeast growth curves in YPD media. Each point represents a mean of three cultures ± SEM.
Figure 2.
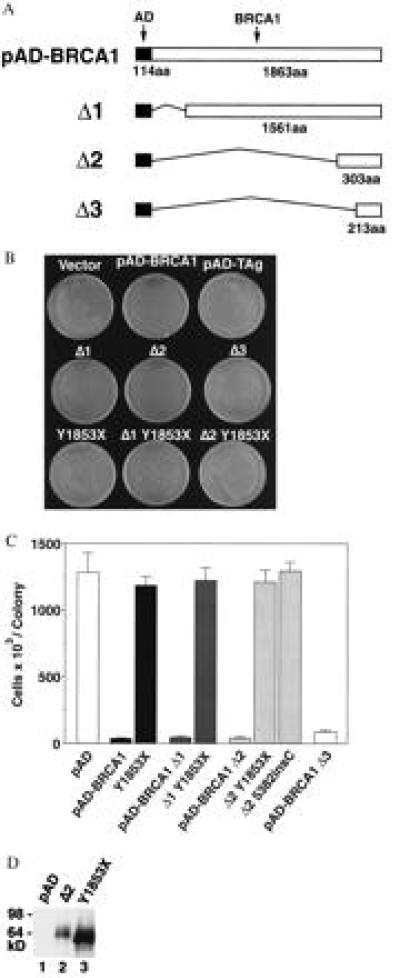
The C-terminal BRCA1 fragment (fused to the GAL4 activation domain and the nuclear localization signal of simian virus 40 large T antigen) is sufficient for small colony formation. Truncating mutations revert the small colony phenotype. (A) Schematic representation of pAD–BRCA1, pAD–BRCA1–Δ1, pAD–BRCA1–Δ2, and pAD–BRCA1–Δ3. Filled bar indicates GAL4 activation domain fused to the nuclear localization signal of simian virus 40 large T antigen; open bar indicates BRCA1 sequences. Thin connecting lines represent regions deleted. Numbers represent number of amino acids (aa). (B) Photograph of HF7c colonies formed after transformation with the indicated plasmids. Y1853X, pAD–BRCA1 Y1853X; Δ1–Y1853X, pAD–BRCA1–Δ1 modified by Y1853X nonsense mutation, and so forth. (C) Number of cells (× 103) per colony after transformation with the plasmids indicated. Single, transformed colonies were resuspended in water and counted on a hemocytometer. Each bar represents the mean of six cell counts ± SEM. All counts were performed by an individual blinded to the vector used. Δ1–Y1853X, pAD–BRCA1–Δ1 modified by Y1853X nonsense mutation, and so forth. (D) Expression of pAD–BRCA1–Δ2 and pAD–BRCA1Δ2 Y1853X in yeast. Anti-HA Western blot detection of pAD–BRCA1–Δ2 (lane 2) and pAD–BRCA1–Δ2 Y1853X (lane 3) in crude yeast lysate from colonies transformed with corresponding vectors. All AD fusion constructs in this study contain an HA epitope detectable by Western blot.
We sought to localize by deletion analysis the region of BRCA1 in the fusion protein necessary for growth inhibition. pAD–BRCA1–Δ1 (deleted for BRCA1 codons 1–302) and pAD–BRCA1–Δ2 (deleted for BRCA1 codons 1–1559) inhibited growth of yeast in the assay (Fig. 2 A-C). Thus the RING domain (residues 20–68) was not required to inhibit colony growth. Likewise, the nuclear localization signal of BRCA1 was not required. However, a partial deletion of the BRCT domains (10) in pAD–BRCA1–Δ3 (deleted for BRCA1 codons 1–1650) resulted in a two-fold increase in the number of cells per colony as compared with pAD–BRCA1 transformed yeast (Fig. 2 A-C).
Cancer-predisposing mutations of BRCA1 are thought to inactivate gene function in humans (20). To evaluate the capability of this assay to distinguish functionally significant mutations of BRCA1 from polymorphisms, we tested several sequence variants (Table 1). To determine the effect of nonsense, frameshift, or other truncating mutations on the activity of pAD–BRCA1 in this assay, we performed site-directed mutagenesis to introduce a cancer-predisposing nonsense mutation, Y1853X (21), that truncates the BRCA1 ORF by 11 codons. Yeast transformed with Y1853X mutations of pAD–BRCA1, pAD–BRCA1–Δ1, or pAD–BRCA1–Δ2 all formed colonies of normal size (Fig. 2, B and C). A cancer-associated frameshift mutation, nucleotide 5382insC (resulting in Q1756C+ to Stop1829; ref. 6), similarly abrogated the activity of pAD–BRCA1–Δ2 in the assay (Fig. 2C), and truncations of the BRCT domain created randomly during PCR mutagenesis of pAD–BRCA1 resulted in colonies of normal size. Western blot analysis demonstrated similar levels of expression for all constructs (Fig. 2D and data not shown).
Table 1.
BRCA1 mutations studied
| Disease-associated mutations | ||
|---|---|---|
| Y1853X | Nonsense | Tyr to Stop |
| 5382insC | Frameshift | Q1756C+ |
| A1708E | Missense | Ala to Glu |
| M1775R | Missense | Met to Arg |
| P1749R | Missense | Pro to Arg |
| C61G | Missense | Cys to Gly |
| Polymorphisms | ||
| S1613G | Missense | Ser to Gly |
| M16521M16521 | Missense | Met to Ile |
| Unclassified sequence variants | ||
| V1713A | Missense | Val to Ala |
| P1637L | Missense | Pro to Leu |
Missense mutations that predispose to cancer are presumed to inactivate tumor suppressor gene function (22). We therefore sought to test the effect of BRCA1 missense mutations on the activity of pAD–BRCA1–Δ2 in this assay. Yeast transformed by pAD–BRCA1–Δ2 modified by A1708E, a missense mutation predisposing to cancer (23), formed large colonies equal in size to vector controls (Fig. 3 B and C). Two other mutations, M1775R (6, 23) and P1749R (24), each partially inactivated the growth inhibitory function of pAD–BRCA1–Δ2 (Fig. 3, B and C). The results correlate with published data that A1708E, M1775R, and P1749R abrogate the transcriptional activity of BRCA1 fusion proteins in other assays (12, 13). Protein expression levels were similar for all constructs by Western blotting.
Figure 3.
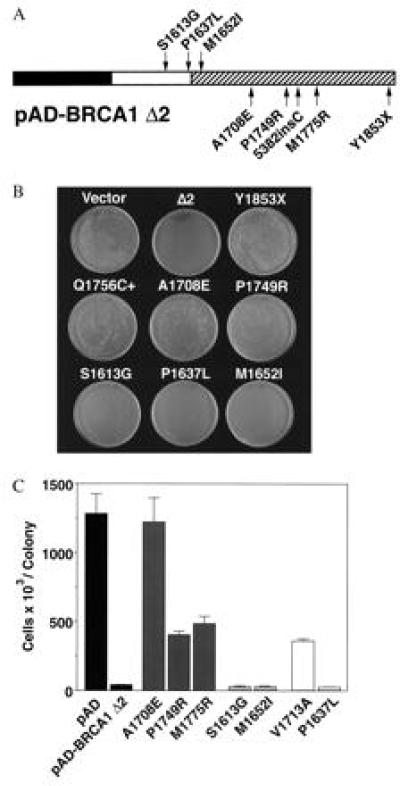
Cancer-associated missense mutations, but not polymorphisms, revert the small colony phenotype. (A) Schematic of pAD–BRCA1–Δ2 showing location of missense mutations used in this figure. Polymorphisms are drawn above the line; disease-associated missense mutations are drawn below the line. V1713A is not represented. Hatched region indicates BRCT domains. (B) Photograph of HF7c colonies formed after transformation with the indicated plasmids. Y1853X, pAD–BRCA1 Y1853X; Δ1 Y1853X, pAD–BRCA1–Δ1 modified by Y1853X nonsense mutation, and so forth. (C) Number of cells (× 103) per colony after transformation with the plasmids indicated. Single, transformed colonies of HF7c were resuspended in water and counted on a hemocytometer. Each bar represents the mean of six cell counts ± SEM. All counts were performed by an individual blinded to the vector used. Counts are for pAD–BRCA1–Δ2 modified by cancer-predisposing missense mutations (A1708E, P1749R, and M1775R; solid black), polymorphisms (S1613G and M1652I; grey), or misclassified/unclassified sequence variants (V1713A, P1637L; white) (see text). Disease-associated mutations diminished or abolished growth inhibition, and polymorphisms maximally inhibited growth. Mean ± SEM cell counts (× 103) for pAD–BRCA1–Δ2 or modified by S1613G, M1652I, or P1637L were 38.25 ± 6.75, 27 ± 6.75, 30 ± 4.5, and 20.25 ± 6.75, respectively.
In contrast to the tumorigenic mutations examined above, polymorphisms of BRCA1 do not predispose to cancer and are thought to have no influence on gene function. S1613G and M1652I are common polymorphisms (refs. 21 and 25 and M. Luce, personal communication), and M1652I alters a conserved residue (11) of the first BRCT domain (8). Yeast transformed by pAD–BRCA1–Δ2 modified by S1613G or M1652I formed small colonies identical in size and number of cells per colony to pAD–BRCA1–Δ2 transformants (Fig. 3 B and C). Protein expression levels were similar for all constructs by Western blotting. Thus, S1613G and M1652I had no effect on growth inhibition, consistent with their classification as polymorphisms.
The correlation between disease-associated mutations of BRCA1 and abrogation of growth inhibition suggested that this assay may be used to assess the functional significance of unclassified BRCA1 mutations. We therefore tested two mutations of BRCA1 that have been difficult to classify because of an uncertain correlation with cancer predisposition. V1713A was found in the germline of an individual with breast and ovarian cancer from a family with 15 cases of breast or ovarian cancer (26) and was not found in 180 control chromosomes (26) or in a separate database of 3000 chromosomes (M. Luce, personal communication). However, the mutation alters a residue that is isoleucine in murine BRCA1, and no other cancer-affected relatives were available to verify segregation of the V1713A sequence change with disease, creating ambiguity about the relationship between V1713A and cancer predisposition. In this assay, yeast transformed with pAD–BRCA1–Δ2 modified by V1713A formed easily visible colonies comparable in size and number of cells per colony to M1775R and P1749R cancer-predisposing controls (Fig. 3C), suggesting that V1713A inactivates a function that protects against cancer and is consistent with detection of this sequence change in the germline of an individual with a high probability of hereditary breast–ovarian cancer.
P1637L, like V1713A, was detected in the germline and tumor DNA of an individual with early onset ovarian cancer from a family with a history of breast and ovarian cancer (23). This sequence change alters a conserved residue (11) and was not detected in 162 control chromosomes (23). Accordingly, the P1637L sequence change was reported as a cancer-associated mutation. However, subsequent to publication, additional sequence analysis revealed a frameshift mutation (nucleotide 2575delC) present in the same allele as P1637L (ref. 27; A. Futreal, personal communication). A second chromosome from an unrelated patient also has been discovered to contain both the frameshift mutation and P1637L, implying that P1637L is a rare polymorphism in linkage disequilibrium with the 2575delC frameshift mutation (ref. 27; A. Futreal, personal communication). Modification of pAD–BRCA1–Δ2 by P1637L had no effect on the formation of small colonies (Fig. 3 B and C), consistent with evidence that this sequence change is a rare polymorphism.
We sought to correlate our results with an alternate yeast assay of BRCA1 activity (13). In this assay, BRCA1 fused to the GAL4 DNA binding domain activated a HIS3 reporter gene. We fused BRCA1 codons 303-1863 to the GAL4 DNA binding domain in a 2-μ yeast expression plasmid to create pBD–BRCA1 and transformed HF7c yeast containing a HIS3 reporter gene under transcriptional control of a GAL4 response element. HF7c transformed by pBD–BRCA1, but not control vector, grew on plates lacking histidine, compatible with HIS3 transactivation by a BRCA1 fusion protein as reported. HF7c transformed by pBD–BRCA1 modified by Y1853X did not grow on plates lacking histidine, consistent with the link of this mutation to cancer (21), results in our growth assay, and published results (13).
P1749R is a cancer-associated mutation that was discovered in the germline of an ovarian cancer family (24), that alters a conserved residue (8), and that inactivated growth inhibition by pAD–BRCA1–Δ2 in this study. Although this mutation has been shown to inactivate transcriptional activity of a BRCA1 fusion protein in mammalian cells (12), we found that HF7c transformed by pBD–BRCA1 modified by P1749R grew on plates lacking histidine, suggesting P1749R had no noticeable impact on HIS3 gene activation. This suggests that the HIS3 reporter gene assay as performed here may be less sensitive in detecting functional effects of mutations than the yeast assay based on growth inhibition.
High Level Expression of Full Length BRCA1 Alone Inhibits Growth.
To determine whether the GAL4 activation domain is necessary for the small colony phenotype, we expressed full length BRCA1 alone from pVT-U100 (17), a 2-μ vector with an alcohol dehydrogenase promoter. This had no effect on cell growth rates or colony size despite levels of BRCA1 protein expression higher than in yeast transformed by pAD–BRCA1. However, when even higher levels of BRCA1 expression were achieved using the yeast vector pYES2 containing a GAL1 promoter, a small colony phenotype was seen upon induction with galactose. Introduction of the Y1853X mutation abrogated growth inhibition. However, introduction of the cancer-predisposing mutation C61G (6), which alters the penultimate zinc-coordinating cysteine of the RING finger domain, did not affect the small colony phenotype. This result is consistent with our finding that the RING finger domain is not required for growth inhibition and suggests that the assay is unable to detect missense mutations of this domain. Thus, BRCA1 alone is capable of producing the small colony phenotype at sufficiently high levels of expression although fusion of BRCA1 to the GAL4 activation domain and nuclear localization signal of simian virus 40 large T antigen inhibited cell growth at much lower levels of expression.
DISCUSSION
Many genes controlling predisposition to human diseases have been cloned in the past 10 years, and the identification of many more is anticipated. It is likely that most individuals will carry medically relevant genetic variations. The identification of BRCA1 has received considerable attention because of its role in breast cancer, the most common malignancy of women, and there is a strong public interest in testing for BRCA1 mutations. However, genetic testing of BRCA1 is plagued by numerous technical obstacles, which include the large size of the gene and the difficulty of distinguishing polymorphisms from functionally relevant missense mutations (28). A functional assay of human BRCA1 in yeast, such as has been devised for p53 (29), might address many of the technical difficulties.
We have described here findings that human BRCA1, alone or as a fusion protein, inhibits growth of yeast by an uncharacterized mechanism. The BRCT domains of the C terminus of BRCA1, which are necessary for the cancer-protective function of BRCA1 in humans, are necessary for this activity in yeast. Neutral polymorphisms of the C terminus, including one affecting a conserved residue within the first BRCT domain (M1652I), have no effect on the small colony phenotype. Nonsense and frameshift mutations and cancer-predisposing missense mutations of the C terminus revert the phenotype. This suggests that the assay may potentially be useful in detection of truncation mutations of BRCA1 and in classification of missense mutations of the BRCT domains.
These data also raise the hypothesis that the C terminus of BRCA1 inhibits growth of yeast by a mechanism analogous to the means by which BRCA1 suppresses cancer formation in humans. This hypothesis is indirectly supported by the correlation of growth suppressive function in yeast with cancer-protective function in humans for multiple alleles of BRCA1. Recently, physical association and partial subcellular colocalization of BRCA1 with RAD51 has been described, suggesting a possible role for BRCA1 in cell cycle control and/or DNA repair (30). Determining the molecular mechanisms of BRCA1-mediated growth inhibition in yeast may provide important insights into BRCA1-mediated tumor suppression in humans.
By complementing available population data, this assay may be useful in the future classification of missense sequence variants of the BRCT domains. The BRCA1 polymorphisms tested had no effect on growth inhibition. In contrast, one cancer-associated missense mutation, A1708E, completely reverted growth inhibition, and two other missense mutations, M1775R and P1749R, partially reverted growth suppression. The results for M1775R and P1749R suggest that some mutations will have intermediate effects on growth suppression that may be difficult to interpret, emphasizing the need to evaluate data from this or any functional assay of BRCA1 in the context of data from the human population. The reason for incomplete reversion of growth inhibition by the M1775R and P1749R mutations may become clear once the molecular mechanism of BRCA1-dependent growth suppression is better understood, and it remains possible that the differing effects of missense mutations in this assay may ultimately correlate with differing phenotypes in the human population.
This assay did not detect a cancer-predisposing missense mutation of the RING finger, and only the C-terminal 305 amino acids were required for maximal activity. This suggests that the assay assesses the integrity of only the BRCT domains of BRCA1. Eighty-seven percent of reported mutations disrupt the BRCT domains by truncation (5), and an additional number disrupt the domains by missense mutation. Although we cannot rule out the possibility that certain truncating mutations of pAD–BRCA1 might give a small colony phenotype, the assay may ultimately provide a means to detect most cancer-predisposing mutations. The association of BRCA1 with BARD1, recently identified by a two-hybrid assay (14), may provide a means in yeast to detect missense mutations of the RING finger domain not detected by this assay.
In summary, the data presented here suggest that the functional consequence of certain alterations in BRCA1 sequence can be assessed in this yeast expression assay. We have shown that wild-type and common polymorphisms of BRCA1 are distinguishable from several types of cancer-predisposing mutations by virtue of their effects on yeast colony size. The correlation between known inactivating mutations altering the BRCT domains and abrogation of growth inhibition in this assay suggests that it may be used to predict the presence of functionally relevant alterations in BRCA1. Additionally, this strong correlation suggests that the assay may be capable of detecting functionally conserved interactions between BRCA1 and cellular elements found in both human and yeast cells.
Acknowledgments
We thank Deborah J. Kruep and C. Robbins for technical assistance. We thank L. Samelson, J. Struewing, J. Bonifacino, T. Rouault, and R. Humphrey for their comments on the manuscript. J.S.H. thanks W. M. Linehan for continued support.
ABBREVIATIONS
- BRCT domain
BRCA1 C-terminal domain
- YPD
yeast extract/peptone/dextrose
- HA
influenza virus hemagglutination antigen
References
- 1.Easton D F, Bishop D T, Ford D, Crockford G P the Breast Cancer Linkage Consortium. Am J Hum Genet. 1993;56:678–701. [PMC free article] [PubMed] [Google Scholar]
- 2.Ford D, Easton D F, Bishop D T, Narod S A, Goldgar D E. Lancet. 1994;343:692–695. doi: 10.1016/s0140-6736(94)91578-4. [DOI] [PubMed] [Google Scholar]
- 3.Hacia J G, Brody L C, Chee M S, Fodor S P, Collins F S. Nat Genet. 1996;14:441–447. doi: 10.1038/ng1296-441. [DOI] [PubMed] [Google Scholar]
- 4.Hogervorst F B L, Cornelis R S, Bout M, van Vliet M, Oosterwijk J D, Olmer R, Bakker B, Klijn J G M, Vasen H F A, Meijers-Heijboer H, Menko F H, Cornelisse C J, den Dunnen J T, Devilee P, van Ommen G-J B. Nat Genet. 1995;10:208–212. doi: 10.1038/ng0695-208. [DOI] [PubMed] [Google Scholar]
- 5.Couch F J, Weber B L. Hum Mutat. 1996;8:8–18. doi: 10.1002/humu.1380080102. [DOI] [PubMed] [Google Scholar]
- 6.Miki Y, Swensen J, Shattuck-Eidens D, Futreal P A, Harshman K, et al. Science. 1994;266:66–71. doi: 10.1126/science.7545954. [DOI] [PubMed] [Google Scholar]
- 7.Holt J T, Thompson M E, Szabo C, Robinson-Benion C, Arteaga C L, King M C, Jensen R A. Nat Genet. 1996;12:298–302. doi: 10.1038/ng0396-298. [DOI] [PubMed] [Google Scholar]
- 8.Koonin E V, Altschul S F, Bork P. Nat Genet. 1996;13:266–268. doi: 10.1038/ng0796-266. [DOI] [PubMed] [Google Scholar]
- 9.Bork P, Hofmann D, Bucher P, Neuwald A F, Altschul S F, Koonin E V. FASEB J. 1997;11:68–76. [PubMed] [Google Scholar]
- 10.Callebaut I, Mornon J-P. FEBS Lett. 1997;400:25–30. doi: 10.1016/s0014-5793(96)01312-9. [DOI] [PubMed] [Google Scholar]
- 11.Szabo C I, Wagner L A, Francisco L V, Roach J C, Argonza R, Ostrander E A. Hum Mol Genet. 1996;5:1289–1298. doi: 10.1093/hmg/5.9.1289. [DOI] [PubMed] [Google Scholar]
- 12.Chapman M S, Verma I M. Nature (London) 1996;382:678–679. doi: 10.1038/382678a0. [DOI] [PubMed] [Google Scholar]
- 13.Monteiro A N A, August A, Hanafusa H. Proc Natl Acad Sci USA. 1996;93:13595–13599. doi: 10.1073/pnas.93.24.13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu L C, Wang Z W, Tsan J T, Spillman M A, Phung A, Xu X L, Yang M C, Hwang L Y, Bowcock A M, Baer R. Nat Genet. 1996;14:430–440. doi: 10.1038/ng1296-430. [DOI] [PubMed] [Google Scholar]
- 15.Schärer E, Iggo R. Nucleic Acids Res. 1992;20:1539–1545. doi: 10.1093/nar/20.7.1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ausubel F M, Brent R, Kingston R E, Moore D D, Seidman J G, Smith J A, Struhl K. Current Protocols in Molecular Biology. New York: Wiley; 1995. [Google Scholar]
- 17.Vernet T, Dignard D, Thomas D Y. Gene. 1987;52:225–233. doi: 10.1016/0378-1119(87)90049-7. [DOI] [PubMed] [Google Scholar]
- 18.Sikorski R S, Heiter P. Genetics. 1989;12:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guthrie C, Fink G R. Guide to Yeast Genetics and Molecular Biology. San Diego: Academic; 1991. [Google Scholar]
- 20.Kamb A, Skolnick M H, Becher H, Chang-Claude J. Important Adv Oncol. 1996;13:23–35. [PubMed] [Google Scholar]
- 21.Friedman L S, Ostermeyer E A, Szabo C I, Dowd P, Lynch E D, Rowell S E, King M-C. Nat Genet. 1994;8:399–404. doi: 10.1038/ng1294-399. [DOI] [PubMed] [Google Scholar]
- 22.Frebourg T, Kassel J, Lam K T, Gryka M A, Barbier N, Andersen T I, Børresen A-L, Friend S H. Proc Natl Acad Sci USA. 1992;89:6413–6417. doi: 10.1073/pnas.89.14.6413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Futreal P A, Liu Q, Shattuck-Eidens D, Cochran C, Harshman K, et al. Science. 1994;266:120–122. doi: 10.1126/science.7939630. [DOI] [PubMed] [Google Scholar]
- 24.Gayther S A, Harrington P, Russell P, Kharkevich G, Garkavtseva R F, Ponder B A. Am J Hum Genet. 1996;58:451–456. [PMC free article] [PubMed] [Google Scholar]
- 25.Durocher F, Shattuck-Eidens D, McClure M, Labrie F, Skolnick M H, Goldgar D E, Simard J. Hum Mol Genet. 1996;5:835–842. doi: 10.1093/hmg/5.6.835. [DOI] [PubMed] [Google Scholar]
- 26.Struewing J P, Brody L C, Erdos M R, Kase R G, Giambarresi T R, Smith S A, Collins F S, Tucker M A. Am J Hum Genet. 1995;57:1–7. [PMC free article] [PubMed] [Google Scholar]
- 27.Lancaster J M, Cochran C J, Brownlee H A, Evans A C, Berchuck A, Futreal P A, Wiseman R W, Lancaster J M, Wiseman R W, Berchuck A. J Natl Cancer Inst. 1996;88:552–554. doi: 10.1093/jnci/88.8.552. [DOI] [PubMed] [Google Scholar]
- 28.Collins F S. N Engl J Med. 1996;334:186–188. doi: 10.1056/NEJM199601183340311. [DOI] [PubMed] [Google Scholar]
- 29.Ishioka C, Ballester R, Engelstein M, Vidal M, Kassel J, The I, Bernards A, Gusella J F, Friend S H. Oncogene. 1995;10:841–847. [PubMed] [Google Scholar]
- 30.Scully R, Chen J, Plug A, Xiao Y, Weaver D, Feunteun J, Ashley T, Livingston D M. Cell. 1997;88:265–275. doi: 10.1016/s0092-8674(00)81847-4. [DOI] [PubMed] [Google Scholar]
- 31.Beaudet A L, Tsui L C. Hum Mutat. 1993;2:245–248. doi: 10.1002/humu.1380020402. [DOI] [PubMed] [Google Scholar]