

Abstract
Retinoic acid is one of the most promising drugs for chemotherapy and chemoprevention of cancer. Either blocking activator protein-1 (AP-1) activity or activating retinoic acid response element (RARE) have been proposed to be responsible for its antitumor activity. However, evidence for this hypothesis is lacking in vivo studies. To address this issue, we used an AP-1-luciferase transgenic mouse as a carcinogenesis model and new synthetic retinoids that are either selective inhibitors of AP-1 activation or selective activators of the RARE. The results showed that the SR11302, an AP-1 inhibition-specific retinoid, and other AP-1 inhibitors such as trans-retinoic acid and fluocinolone acetonide, markedly inhibit both 12-O-tetradecanoylphorbol-13-acetate-induced papilloma formation and AP-1 activation in 7,12-dimethyl benz(a)anthracene-initiated mouse skin (P < 0.05). In contrast, repeated applications of SR11235, a retinoid with RARE transactivating activity, but devoid of AP-1 inhibiting effect, did not cause significant inhibition of papilloma formation and AP-1 activation (P > 0.05). These results provide the first in vivo evidence that the antitumor effect of retinoids is mediated by blocking AP-1 activity, but not by activation of RARE.
The transcription factor activator protein-1 (AP-1) regulates the transcription of various genes with the consensus DNA recognition sequence TGA(C/G)TCA, designated as 12-O-tetradecanoylphorbol-13-acetate (TPA)-responsive element (TRE) in their promoter region (1). AP-1 is consisted of a family of Jun/Fos dimers that include different Jun proteins (c-Jun, JunB, and JunD) and Fos proteins (c-Fos, FosB, Fra-1, Fra-2, and FosB2) (2–8). Each of these proteins consists of a “leucine zipper,” which permits Jun proteins to form homodimers or heterodimers among themselves or with Fos proteins (2), but Fos proteins cannot dimerize and are not able to bind DNA on their own (2). AP-1 and its regulated gene expression has been shown to play an important role in the preneoplastic-to-neoplastic progression in cell culture models (9–18). Because the binding of the AP-1 protein to DNA does not always result in an induction of transcription (19), AP-1 DNA binding activity measured by gel-shift assay may not correlate with AP-1 transcriptional activity (19). Thus, the ideal in vivo model to study the relevance of AP-1 activation to tumor promotion is to use AP-1-luciferase reporter transgenic mice. The transgenic mouse, which expressed a 2X TRE luciferase in all the cells of mouse, developed by Rincón and Flavell (20), made it possible to study the role of AP-1 activity in tumor promotion and the mechanism of some chemopreventional drugs in animal models.
Retinoids can inhibit tumor cell growth and induce the differentiation and reversion of certain malignant cells to normal phenotype (21, 22). Retinoic acid has been proven to be effective in inhibiting papilloma formation in a mouse model and tumor promoter-induced transformation in JB6 cells (21, 23–26). Clinical studies indicated that retinoic acid is effective for treatment of certain types of leukemia (27, 28) and a chemopreventive agent against the occurrence of secondary head and neck cancers (29). However, the clinical usefulness of retinoic acid is limited by its side effects, such as lipostrichia, bleeding, hyperostosis, and teratogenicity (30). The biological activities of retinoids are believed to be mediated by transcriptional activation of retinoic acid response element (RARE) and inhibition of AP-1 activity, acting through distinct nuclear receptors, namely the retinoic acid receptors (RARs) and the retinoid X receptors (RXRs) (31–33). To distinguish these two different effects of retinoic acid, Fanjul and coworkers (34, 35) screened the transcriptional activities of 50 synthetic retinoids. They found that some retinoids, such as SR11302 (Fig. 1), inhibit AP-1 activity without activating the transcription of RARE. In contrast, SR11235 (Fig. 1) selectively activates transcriptional activity of the RARE without inhibiting AP-1 activity (35). By using these retinoids, Li et al. (21) showed that cell transformation was blocked by SR11302, but not by SR11235, in the JB6 cell system (21). This poses the intriguing question of whether inhibition of AP-1 or activation of RARE is responsible for the antitumor promotion effect of retinoic acid. Here we addressed this question by using AP-1-luciferase transgenic mice and these synthesized retinoids displaying selective biologic activity (35).
Figure 1.
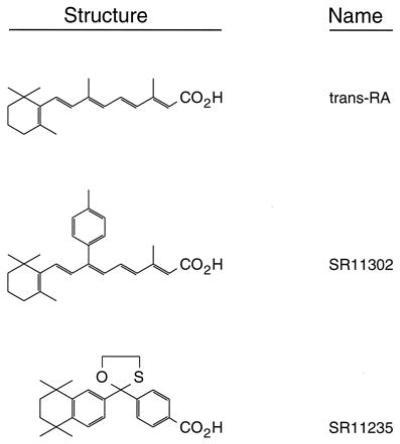
The structure of retinoids.
MATERIALS AND METHODS
Animals and Reagents.
TPA was from Calbiochem, and dimethyl sulfoxide was from Pierce. 7,12-Dimethylbenz(a)anthracene (DMBA), fluocinolone acetonide (FA), and trans-retinoic acid (RA) were from Sigma. The synthetic retinoids, SR11302 and SR11235, were synthesized as previously reported (35). 2X TRE-luciferase reporter transgenic mice were originally established by Rincón and Flavell (20). A C57BL/6 male mouse carrying the 2X TRE-luciferase transgene was crossed with DBA2 females. The F1 offspring were screened by testing both the basal level and TPA-induced level of luciferase activity for the presence of the AP-1 luciferase reporter gene. Males and females were housed separately in solid bottom polycarbonate cages on ventilated animal racks (4–5 mice per cage, individualized by incisions in the ears) under temperature, humidity, and yellow light controlled conditions. Food and water were available ad libitum and the dorsal skin of the mice was shaved every week during the experiment period.
Tumor Induction and Prevention.
Both the basal level and TPA-induced level of luciferase activity were determined in the mice 2 weeks before DMBA treatment. The AP-1-luciferase reporter-bearing male and female mice (6–9 weeks old) were randomly divided into six groups. There were 16–19 mice in each group. DMBA (51.2 μg dissolved in 300 μl of acetone for each mouse) was used as a tumor initiator and applied to mouse dorsal skin. Fourteen days following initiation, the mice were promoted twice a week (on Monday and Thursday) with 17 nmol TPA dissolved in 300 μl of acetone for the next 18 weeks. For the chemoprevention groups, mice were treated with 34 nmol of various retinoids or 1 nmol FA dissolved in 300 μl of acetone 1 hr prior to each promotion with TPA. Negative control mice were treated with acetone alone. The number of papillomas in each mouse were counted weekly.
Assay of AP-1 Activity in Vivo.
The AP-1-luciferase transgenic mice were identified, grouped, housed, and initiated with DMBA as described in tumor induction and prevention. Two weeks after initiation with DMBA, both the basal levels and TPA-induced levels of luciferase activity were measured by dorsal skin punch biopsy using biopsy punch (1.5 mm, Acuderm, Ft. Lauderdale, FL). Two weeks after the last punch biopsy, the mice were treated topically four times over 7 days with 34 nmol various retinoids or 1 nmol FA dissolved in 300 μl acetone prior to promotion with TPA. The last of the four topical doses of retinoids or FA was given 1 hr prior to 17 nmol TPA treatment on the dorsal skin of mouse. Treatment of DMBA-initiated mouse skin results in rapid induction of AP-1 activity, which reached the peak at 12 hr post-TPA treatment (data not shown). Thus, 12 hr after TPA treatment, the mice were punch biopsied to determine the effect of retinoids or FA on TPA-induced AP-1 activity in the epidermis. One-hundred microliters of lysis buffer was added to each skin biopsy and kept at 4°C for 12 hr. The luciferase activity of punch biopsied epidermis in supernatant was measured by a luminometer (Monolight 2010, Analytical Luminescence Laboratory, San Diego) 10 sec after mixing the extract and luciferase assay reagent as described (14, 20). The relative AP-1 activity was presented as relative to the basal level of luciferase activity of each mouse.
Statistical Analysis.
The significance of the difference in the tumor multiplicity data and AP-1 activity was determined with the Student’s t test.
RESULTS
Inhibition of Tumor Promotion by Retinoid SR11302, But Not by SR11235, in AP-1-Luciferase Transgenic Mice.
Previous in vitro studies by us and others suggest that AP-1 plays a crucial role in tumor promoter-induced cell transformation (1–7). To test whether inhibition of tumor promotion by RA occurs through blocking AP-1 activation but not through RARE activation, we used transgenic mice with AP-1 luciferase reporter and the well-characterized DMBA-TPA 2-stage skin carcinogenesis model. Each mouse was initiated with 0.2 nmol (51.2 μg) DMBA dissolved in 300 μl acetone. After 14 days following initiation, the mice were grouped and promoted twice a week (on Monday and Thursday) with 17 nmol of TPA for 18 weeks. The mice of the experimental groups were treated with 34 nmol of various retinoids 1 hr prior to each promotion with TPA. RA and FA were used as positive controls for tumor inhibition. The results are shown in Fig. 2. The repeated TPA treatment alone resulted in 27.1 papillomas per mouse at week 18 of TPA promotion (n = 16), whereas no papillomas were observed in the acetone negative control group (n = 19). Pretreatment with FA (n = 17) or RA (n = 17) 1 hr prior to each TPA promotion effectively inhibited the papilloma formation induced by TPA (P < 0.05). Application of retinoid SR11302 (n = 18) at 1 hr prior to each twice-weekly TPA promotion inhibited papilloma formation by 67.9%, as determined by the number of papillomas per mouse (P < 0.05) (Fig. 2). In contrast, no significant inhibition of papilloma induction was observed in the group pretreated with the SR11235 (n = 17) (P > 0.05) (Fig. 2). The dynamic papilloma formation was consistent with the results described above (Fig. 3). These results demonstrate that FA, RA, and SR11302, but not SR11235, have significant antitumor promotion activity and further suggests that activation of RARE transcription activity by the retinoids is not required for their antitumor promotion activity.
Figure 2.
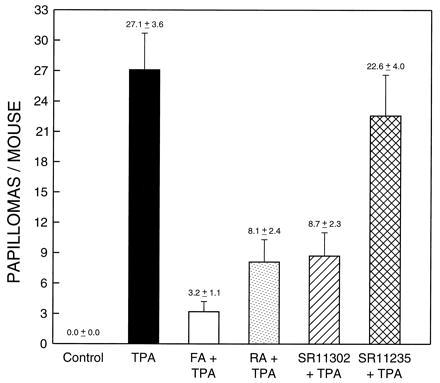
Inhibition of tumor promotion by retinoid SR11302, but not by SR11235, in AP-1-luciferase transgenic mice. Transgenic mice that expressed a 2X TRE luciferase reporter gene were grouped and initiated with DMBA as well as treated twice a week with TPA with or without different tumor prevention drugs such as FA, RA, and SR11302 or SR11235, as described. SR11302 and SR11235 are new synthetic retinoids with selective inhibiting AP-1 activation and activating RARE activity, respectively. The results are shown as the average number of papillomas per mouse with the mean ± SE at week 18 after tumor promotion.
Figure 3.
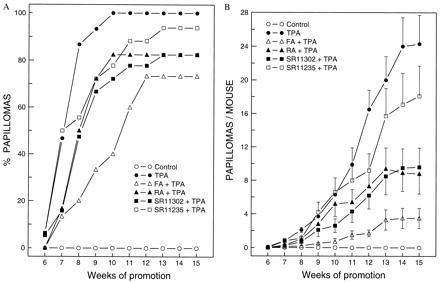
Inhibition of papilloma formation by retinoid SR11302 during the course of the tumor promotion. The mice were prepared and initiated with DMBA as well as treated with TPA with or without different tumor prevention drugs (such as FA, RA, SR11302, or SR11235) twice a week, as described. The incidence of papillomas was observed weekly. The results were presented as (A) percentage of mice bearing papillomas at different promotion points and (B) average number of papillomas per mouse with the mean ± SE.
Retinoid SR11302, But Not SR11235, Blocks TPA-induced AP-1 Transactivation in AP-1 Luciferase Transgenic Mice.
To determine whether the inhibition of papilloma formation by FA, RA, SR11302, or SR11235 correlated with the inhibition of AP-1 activity, we investigated the influence of these agents on TPA-induced AP-1 activity in AP-1-luciferase reporter transgenic mice. Since inhibition of skin papilloma formation requires repeated applications of these agents to the DMBA-initiated skin, the inhibition of AP-1 activity induced by TPA was determined following multiple applications of retinoids to mouse skin initiated with DMBA. The results are shown in Fig. 4. Treatment of mouse skin with TPA induced a 550-fold induction of AP-1 activity (n = 10). TPA-induced AP-1 activity was markedly inhibited by either FA (n = 10), RA (n = 10), or SR11302 (n = 10) (P < 0.05), whereas SR11235 did not show significant inhibition of TPA-induced AP-1 activity in mouse skin (n = 10) (P > 0.05) (Fig. 4). Taken together with our previous findings, we conclude that inhibition of tumor promotion by retinoids is mediated by blocking the tumor promoter-induced AP-1 activity.
Figure 4.
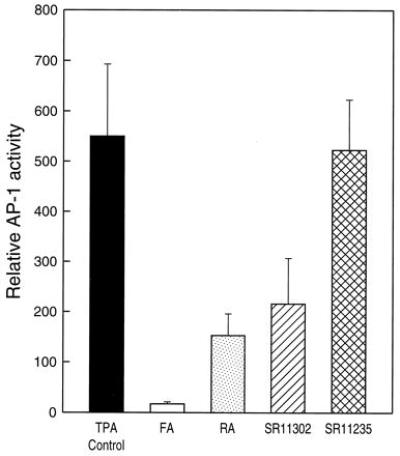
Retinoid SR11302, but not SR11235, blocks TPA-induced AP-1 transactivation in AP-1-luciferase transgenic mice. The AP-1-luciferase reporter transgenic mice were first initiated with 0.2 nmol (51.2 μg) of DMBA in 300 μl acetone. Fourteen days after initiation, the mice were treated with TPA with or without different tumor prevention drugs and punch biopsied (1.5 mm) on the dorsal skin of each mouse to determine the luciferase activity as described. The results were shown with the mean and standard error of the relative AP-1 activity from mice in each group.
DISCUSSION
Our results indicate that the retinoids RA and SR11302 inhibit both TPA-induced AP-1 activity and papilloma formation in AP-1 reporter transgenic mice that were initiated with DMBA. In contrast, no significant inhibition of papilloma formation and AP-1 activation was observed in the SR11235-treated group. These data provide the first in vivo evidence that AP-1 plays a crucial role in tumor promotion. The inhibition of tumor promotion by RA occurs through blocking AP-1 activation, but not through activation of RARE.
Previous studies using different cell models have suggested the important role of AP-1 activation in cell transformation (10–15). The activation of AP-1 appears to be required for the preneoplastic-to-neoplastic progression of JB6 cells. Overexpression of c-Jun in P+ cells caused neoplastic transformation and an introduced dominant negative mutant of c-Jun was found to block tumor promoter-induced transformation (11). Many antitumor promoters, such as FA and RA, were shown to be effective inhibitors of AP-1 activation in cell culture systems (36, 37) and our present data provide the first in vivo evidence for the role of AP-1 activation in tumor promotion. The results show that TPA induces both papilloma induction and high levels of AP-1 activation in AP-1-luciferase transgenic mouse skin initiated with DMBA. The glucocorticoid FA, a well known inhibitor of tumor promotion, inhibits TPA-induced papilloma induction and AP-1 activity. The extent of inhibition of AP-1 activity by FA is similar to that observed for inhibition of papilloma formation at the same dosage. Thus, AP-1 activation plays a crucial role in tumor promotion and the inhibitory effect of FA on tumor promotion occurs through blocking AP-1 activation induced by the tumor promoter. Many of the TPA-responsive genes include several protooncogenes, such as c-fos, c-jun, and matrix degrading metalloproteinases contain the AP-1 binding sequence or TRE (1–3, 26). The downstream genes with a regulatory element AP-1 may involve the process of tumor promotion. Recently, Wilson et al. (18) reported that an absence of metalloproteinase matrilysin resulted in a reduction in mean tumor multiplicity of ≈60% and metalloproteinase matrilysin contribute to very early stage in tumor development.
Retinoids have long been known to modulate the differentiation and proliferation of cells (38). It is also well known that action of retinoic acid is mediated by at least two distinct classes of nuclear retinoid receptors, including RARs and RXRs (31–33). Both types are coded for by three genes (α, β, and γ) from which three subtypes of both RARs (α, β, and γ) and RXRs (α, β, and γ) can be generated by differential splicing and use alternative promoters (39–42). RARs bind and are activated by both RA and 9-cis-retinoic acid, whereas RXRs only interact with 9-cis-retinoic acid (43, 44). Although both these receptors can directly activate the RARE or RXRE, and indirectly inhibit the activation of AP-1 activity, several lines of evidence suggest that some synthetic retinoids clearly exhibit distinct receptor selectivities in transcription of RARE and anti-AP-1 activity. For instance, SR11235 is an RXRα-selective transcriptional activator with <20% of anti-AP-1 activity. In contrast, SR11302 show strong anti-AP-1 activity with selective binding with RARα and RARγ, but not with RARβ and RXRα (35).
In several studies, the importance of retinoids in antitumor promotion has been demonstrated (21, 23–25). However, the use of RA for treatment of solid malignancies and for prevention of cancer has been hindered because the prolonged use of RA has significant side effects (30). Thus, elucidating the molecular mechanisms that underlie the diverse effects of retinoids is one of the most challenging projects in the cancer research field. The clarity of this issue will generate useful information to help develop more effective retinoids with fewer side effects for prevention and treatment of human cancer. Based on this view, progress has been made in the synthesis of selective ligands for retinoid receptors (34, 35). These agents may help to determine which mechanisms are responsible for therapeutic effects and which ones produce side effects.
In summary, by using an AP-1-luciferase reporter transgenic mouse and synthesized retinoids displaying selective inhibition of AP-1 activity or activation of RARE, we demonstrated that AP-1 activation is required for tumor promotion. Inhibition of tumor promotion of RA is mediated by blocking AP-1 activation. This study can lead to development of better retinoids for cancer prevention and therapy.
Acknowledgments
We thank Dr. Harald H. O. Schmid for critical reading, Ms. Ruth Morton for helping with the animal care, and Ms. Jeanne A. Ruble and Ms. Carmen Perleberg for editorial and secretarial assistance. This work was supported by the Hormel Foundation. R.A.F. is an investigator at the Howard Hughes Medical Institute.
ABBREVIATIONS
- AP-1
activator protein-1
- RARE
retinoic acid response element
- RAR
retinoic acid receptor
- RA
trans-retinoic acid
- DMBA
7,12-dimethyl benz(a)anthracene
- FA
fluocinolone acetonide
- TPA
12-O-tetradecanoylphorbol-13-acetate
- TRE
TPA responsive element
- RXR
retinoid X receptor
References
- 1.Angel P, Imagawa M, Chiu R, Stein B, Imbra R J, Rahmsdorf H J, Jonat C, Herrlich P, Karin M. Cell. 1987;49:729–739. doi: 10.1016/0092-8674(87)90611-8. [DOI] [PubMed] [Google Scholar]
- 2.Angel P, Karin M. Biochim Biophys Acta. 1991;1072:129–157. doi: 10.1016/0304-419x(91)90011-9. [DOI] [PubMed] [Google Scholar]
- 3.Cohen D R, Curran T. Mol Cell Biol. 1988;8:2063–2069. doi: 10.1128/mcb.8.5.2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Halazonetis T D, Georgopoulos K, Greenberg M E, Leder P. Cell. 1988;55:917–924. doi: 10.1016/0092-8674(88)90147-x. [DOI] [PubMed] [Google Scholar]
- 5.Hirai S I, Ryseck R P, Mechta F, Bravo R, Yaniv M. EMBO J. 1989;8:1433–1439. doi: 10.1002/j.1460-2075.1989.tb03525.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nakabeppu Y, Ryder K, Nathans D. Cell. 1988;55:907–915. doi: 10.1016/0092-8674(88)90146-8. [DOI] [PubMed] [Google Scholar]
- 7.Ryder K, Lanahan A, Perez-Albuerne E, Nathans D. Proc Natl Acad Sci USA. 1989;86:1500–1503. doi: 10.1073/pnas.86.5.1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zerial M, Toschi L, Ryseck R P, Schuermann M, Muller R, Bravo R. EMBO J. 1989;8:805–813. doi: 10.1002/j.1460-2075.1989.tb03441.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alani R, Brown P, Binetruy B, Dosaker H, Rosenberg R K, Angel P, Karin M, Birrer M J. Mol Cell Biol. 1991;11:6286–6295. doi: 10.1128/mcb.11.12.6286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Domann F E, Levy J P, Birrer M J, Bowden G T. Cell Growth Differ. 1994;5:9–16. [PubMed] [Google Scholar]
- 11.Dong Z G, Birrer M J, Wats R G, Matrisian L M, Colburn N H. Proc Natl Acad Sci USA. 1994;91:609–613. doi: 10.1073/pnas.91.2.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dong Z G, Lavrovsky V, Colburn N H. Carcinogenesis. 1995;16:749–756. doi: 10.1093/carcin/16.4.749. [DOI] [PubMed] [Google Scholar]
- 13.Dong Z G, Watts R G, Sun Y, Zhan S N, Colburn N H. Int J Oncol. 1995;7:359–364. doi: 10.3892/ijo.7.2.359. [DOI] [PubMed] [Google Scholar]
- 14.Huang C, Ma W-Y, Dong Z. Mol Cell Biol. 1996;16:6427–6435. doi: 10.1128/mcb.16.11.6427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang C S, Ma W-Y, Dong Z G. Int J Oncol. 1996;8:389–393. doi: 10.3892/ijo.8.2.389. [DOI] [PubMed] [Google Scholar]
- 16.Domann F E, Levy J P, Finch J S, Bowden G T. Mol Carcinog. 1994;9:61–66. doi: 10.1002/mc.2940090202. [DOI] [PubMed] [Google Scholar]
- 17.Holladay K, Fujiki H, Bowden G T. Mol Carcinog. 1992;5:16–24. doi: 10.1002/mc.2940050106. [DOI] [PubMed] [Google Scholar]
- 18.Wilson C L, Heppner K J, Labosky P A, Hogan B L M, Matrisian L M. Proc Natl Acad Sci USA. 1997;94:1402–1407. doi: 10.1073/pnas.94.4.1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Herr I, Van Darn H, Angel P. Carcinogenesis. 1994;15:1105–1113. doi: 10.1093/carcin/15.6.1105. [DOI] [PubMed] [Google Scholar]
- 20.Rincón M, Flavell R A. EMBO J. 1994;13:4370–4381. doi: 10.1002/j.1460-2075.1994.tb06757.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li J-J, Dong Z, Dawson M I, Colburn N H. Cancer Res. 1996;56:483–489. [PubMed] [Google Scholar]
- 22.Gudas L J. Cell Growth Differ. 1992;3:655–662. [PubMed] [Google Scholar]
- 23.Verma A K, Garcia C T, Ashendel C L, Boutwell R K. Cancer Res. 1983;43:3045–3049. [PubMed] [Google Scholar]
- 24.Verma A K, Shapas B G, Rice H M, Boutwell R K. Cancer Res. 1979;39:419–425. [PubMed] [Google Scholar]
- 25.Athar M, Agarwal R, Wang Z Y, Lloyd J R, Bickers D R, Mukhtar H. Carcinogenesis. 1991;12:2325–2329. doi: 10.1093/carcin/12.12.2325. [DOI] [PubMed] [Google Scholar]
- 26.Dong Z, Colburn N H. In: Early Detection of Cancer Molecular Markers. Srivastava S, Lippman S M, Hong W K, Mulshine J L, editors. Armonk, NY: Futura; 1994. pp. 121–128. [Google Scholar]
- 27.Smith M A, Parkinson D R, Cheson B D, Friedman M A. J Clin Oncol. 1992;10:839–864. doi: 10.1200/JCO.1992.10.5.839. [DOI] [PubMed] [Google Scholar]
- 28.Huang M, Ye H C, Chen S R, Chai J R, Lu J X, Zhoa L, Gu L J, Wang Z. Blood. 1988;72:567–572. [PubMed] [Google Scholar]
- 29.Hong W K, Lippman S M, Itri L M, Karp D D, Lee J S, Byers R M, Schantz S P, Kramer A M, Lotan R, Peters L J, Dimery I W, Brown B W, Goepert H. N Engl J Med. 1990;323:795–801. doi: 10.1056/NEJM199009203231205. [DOI] [PubMed] [Google Scholar]
- 30.Bollag W, Holdener E E. Ann Oncol. 1992;35:13–26. doi: 10.1093/oxfordjournals.annonc.a058252. [DOI] [PubMed] [Google Scholar]
- 31.Giguere V, Ong E S, Segui P, Evans R M. Nature (London) 1987;330:624–629. doi: 10.1038/330624a0. [DOI] [PubMed] [Google Scholar]
- 32.Benbrook D, Lernhardt E, Pfahl M. Nature (London) 1988;333:669–672. doi: 10.1038/333669a0. [DOI] [PubMed] [Google Scholar]
- 33.Yang N, Schule R, Mangelsdorf D J, Evans R M. Proc Natl Acad Sci USA. 1991;88:3559–3563. doi: 10.1073/pnas.88.9.3559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lehmann J M, Jong L, Fanjul A, Cameron J F, Lu X P, Haefner P, Dawson M I, Pfahl M. Science. 1992;258:1944–1946. doi: 10.1126/science.1335166. [DOI] [PubMed] [Google Scholar]
- 35.Fanjul A, Dawson M I, Hobbs P D, Jong L, Cameron J F, Harlev E, Graupner G, Lu X-P, Pfahl M. Nature (London) 1994;372:107–111. doi: 10.1038/372107a0. [DOI] [PubMed] [Google Scholar]
- 36.Jonat C, Rahmsdorf H J, Park K-K, Cato A C B, Gebel S, Ponta H, Herrlich P. Cell. 1990;62:1189–1204. doi: 10.1016/0092-8674(90)90395-u. [DOI] [PubMed] [Google Scholar]
- 37.Schüle R, Rangarajan P, Yang N, Kliewer St, Ransone L J, Bolado J, Verma I M, Evans R M. Proc Natl Acad Sci USA. 1991;88:6092–6096. doi: 10.1073/pnas.88.14.6092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hong W K, Itri L M. In: The Retinoids: Biology, Chemistry, and Medicine. Sporn M B, Roberts A B, Goodman D S, editors. New York: Raven; 1994. pp. 597–630. [Google Scholar]
- 39.Chambon P. Semin Cell Biol. 1994;5:115–125. doi: 10.1006/scel.1994.1015. [DOI] [PubMed] [Google Scholar]
- 40.Glass C K, DiRenzo J, Kurokawa R, Han Z. Cell Biol. 1991;10:623–638. doi: 10.1089/dna.1991.10.623. [DOI] [PubMed] [Google Scholar]
- 41.Mangelsdorf D J, Umesono K, Evans R M. In: The Retinoids. Sporn M B, Roberts A B, Goodman D S, editors. Orlando, FL: Academic; 1994. pp. 319–349. [Google Scholar]
- 42.Pfahl M, Apfel R, Bendik I, Fanjul A, Graupner G, Lee M-O, La Vista N, Lu X-P, Piedratita J, Ortiz M A, Salbert G, Zhang X-K. Vitam Horm (Leipzig) 1994;49:327–382. doi: 10.1016/s0083-6729(08)61150-4. [DOI] [PubMed] [Google Scholar]
- 43.Heyman R A, Mangelsdorf D J, Dyck J A, Stein R B, Eichele G, Evans R M, Thaller C. Cell. 1992;68:397–406. doi: 10.1016/0092-8674(92)90479-v. [DOI] [PubMed] [Google Scholar]
- 44.Levin A A, Sturzenbecker L J, Kazmer S, Bosakowski T, Huselton C, Allenby G, Speck J, Kratzeisen C, Rosenberger M, Lovey A, Grippo J F. Nature (London) 1992;355:359–361. doi: 10.1038/355359a0. [DOI] [PubMed] [Google Scholar]