

Abstract
Recombinants of amphotropic murine leukemia virus (A-MuLV) have found widespread use in retroviral vector systems due to their ability to efficiently and stably infect cells of several different species, including human. Previous work has shown that replication-competent recombinants containing the amphotropic env gene, encoding the major SU envelope glycoprotein that determines host tropism, induce lymphomas in vivo. We show here that these viruses also induce a spongiform encephalomyelopathy in mice inoculated perinatally. This fatal central nervous system disease is characterized by noninflammatory spongiform lesions of nerve and glial cells and their processes, and is associated with moderate astro- and microgliosis. The first clinical symptoms are ataxia, tremor, and spasticity, progressing to complete tetraparesis and incontinence, and finally death of the animal. Sequences within the amphotropic env gene are necessary for disease induction. Coinfection of A-MuLV recombinants with nonneuropathogenic ecotropic or polytropic MuLV drastically increases the incidence, degree, and distribution of the neurodegenerative disorder. The consequence of these results in view of the use of A-MuLV recombinants in the clinic is discussed.
Keywords: gene therapy, retrovirus, retrovirus pathogenicity, central nervous system, packaging cell lines
As early as 1982 murine leukemia virus (MuLV) genomes have been usurped to generate powerful gene transfer systems (1, 2). More recently, these retroviral vector systems have been the system of choice for several gene therapy approaches currently being used in the majority of approved clinical trials (3). The best characterized MuLV belong to the type C subgenus. These viruses can be further divided into several different host range or interference classes (4–6), determined by the env-coded major surface glycoprotein (SUgp70) expressed by the different viral isolates. Binding of the SUgp70 to its specific cell surface receptor triggers endocytosis and entry into the host cell. Amphotropic (A)-MuLV can efficiently infect a wide range of cell types from many different species including human (5, 7), and thus have been used as the basis of retroviral systems for clinical trials.
A-MuLV-based transfer systems are currently being used in over 70% of the approved gene marking and gene therapy protocols in the United States and Europe. These protocols primarily involve ex vivo retroviral-mediated transfer into normal or tumorigenic cells of patients using replication-incompetent packaging systems and reintroduction into the donor; however, several protocols have been approved and initiated in which the virus-producing packaging cells are directly injected in vivo in the diseased tissue (e.g., glioblastoma, ovarian, and breast cancers) (8). The aim of these latter protocols is the in situ infection of all or most of the tumorigenic tissue with vectors carrying “suicide” genes (e.g., herpes simplex-thymidine kinase) that render the cells susceptible to nontoxic pharmacologic agents (9). Strategies are also being developed that allow direct administration of high titer retroviral particles in vivo, eliminating the complexity and costs of ex vivo manipulations (10, 11). Significantly, although advanced packaging systems have been developed that limit the chance of replication-competent virus release due to recombination, these systems are not always tight (3, 12, 13).
Despite the already wide use of A-MuLV recombinants in both the research and clinical setting, only a few limited studies have examined the pathogenicity of either the wild-type A-MuLV or Moloney (Mo)-AmphoV. The latter is a recombinant carrying the amphotropic env gene in a Mo-MuLV background and is the basis of the majority of retroviral packaging cell lines used to date. The studies performed have shown that these viruses induce lymphatic leukemias at a 25 and 60% incidence, respectively, in both wild-type and inbred mice (NIH Swiss) after a relatively long latency period (7–15 months) (14–16). A high incidence of myeloid leukemia 2–6 months after infection of DBA/2 mice undergoing a chronic inflammatory response has also been observed (17). Inadvertent infection of immunosuppressed rhesus macaques with high titers of replication-competent Mo-AmphoV also resulted in lymphoma induction in 3 out of 10 macaques (13, 18), in contrast to earlier reports in which no disease induction was observed (19), presumably due to virus inactivation by the complement system.
In the course of assaying several MuLV recombinants for altered disease specificity, we observed a rather high incidence (20 to 30%) of a neurological disorder in mice, not previously described, 5 to 10 months after perinatal infection with Mo-AmphoV. The studies described here were performed to determine histopathological changes in the central nervous system (CNS) in infected mice and to define the viral determinants of disease induction.
MATERIALS AND METHODS
Mouse Strains and Infection Protocol.
With the exception of NIH (NIH/OlaHsd, inbred) and NMRI (HsdWin:NMRI, outbred), which were purchased from Harlan–Winkelmann (Borchen, Germany), all mice were bred in the Heinrich-Pette-Institut animal quarters. DBA/2J (Rmcfr), Balb/cJ, and 129/SvJ were originally obtained from Jackson Laboratories, and DDD (Fv-2s, inbred) were obtained from T. Odaka (University of Tokyo, Tokyo). Mice were infected intraperitoneally either within 24–48 hr or 8 days after birth with 50 μl virus-containing supernatant with a virus titer between 3 × 105 and 1 × 106/ml.
Construction of Recombinant Virus Genomes.
The basis of all proviral constructs was a plasmid containing the Mo-MuLV cl MLV-K (20). The plasmids pAM and pAMS (differing in flanking sequences) encoding Mo-AmphoV were generated by exchanging a SalI–ClaI fragment containing 3′ pol sequences and all except 35 bp of the env-coding region with an equivalent fragment from a molecular clone of A-MuLV cl 4070 and were kindly provided by A. D. Miller (Fred Hutchinson Cancer Research Center). To generate the plasmid R275 encoding Mo-AmphoV-MP, a ClaI fragment of plasmid pAMS containing the entire Mo-AmphoV provirus except the 3′ long terminal repeat (LTR) was inserted in a plasmid containing the 3′ LTR of the myeloproliferative virus (MPSV) (21). The plasmid encoding MES-AmphoV-MP (R320) was generated by substituting a KpnI–PstI fragment containing the U5 LTR and leader sequence of R275 with that from murine embryonic stem cell virus (MESV) (plasmid p5Gneo) (22).
Virus Production and Titration.
Recombinant viruses were generated by transfecting recombinant plasmid DNA of cloned proviral genomes into SC1 fibroblasts or MS-5 stroma cells (23). Virus spread was monitored by a reverse transcriptase assay (24). SC1 cells producing the Mirand strain of F-MuLV cloned by end-point dilution (cl 643/22N) (24) were provided by W. Ostertag (Heinrich-Pette-Institut). NIH 3T3 cells producing either mink cell focus-forming virus (F-MCFV) cl1 (25) or Mo-MuLV (26) were kindly provided by R. Friedrich (Universität Giessen, Giessen, Germany) and C. Löliger (Universität Krankenhaus Eppendorf, Hamburg, Germany), respectively.
Virus titers were measured by end-point dilution on SC1 cells. Eight weeks after infection, virus-positive cells were detected by PCR of genomic DNA or reverse transcriptase activity of the supernatant, and is expressed as infectious units (i.u.) per ml. The PCR was performed in a final volume of 25 μl in the buffer provided by the manufacturer with 200 ng genomic DNA, 1.0 μM oligonucleotide primers, 0.2 mM dNTP, 1.5 mM Mg2Cl2, and 0.625 units of Taq Polymerase (GIBCO/BRL). Thirty cycles were performed (1 min at 94°; 1 min at 65°; and 2 min at 72°). To detect amphotropic viruses the primer pairs 5′-CGTATGTCGGGTATGGCTGCA and 5′-GCTGTGGGACACTTGGACTTGTAG spanning positions 266 to 883 of the amphotropic env gene were used, whereas the primer pair 5′-CACCCTCTGTGGACTTGGTG and 5′-CAGCTTAAGCTGTTCCAGGC spanning 193 to 417 of the ecotropic env sequence was used to detect ecotropic virus.
Histopathology of Infected Mice.
Brains were fixed in situ in the dissected neurocrania by use of 4% paraformaldehyde solution containing 1% acetic acid. For better penetration of the fixative, the occipital bones of the skull were removed. After fixation, vertebral columns containing the spinal cords were decalcified in a saturated EDTA buffer solution (pH 7.2) prior to embedding. Embedding in paraffin and staining of sections (hematoxylin-eosin, cresyl violet, elastica-van Gieson) was according to standard procedures.
Screening for Viral Recombinants.
Cell-free brain homogenants of diseased mice were used to infect SC1 mouse cells. One week p.i. genomic DNA was extracted and subjected to Southern analysis. Genomic DNA was digested with HindIII, BamHI, XbaI, PvuII, EcoRV, and EcoRI restriction enzymes. Probes specific for ecotropic (nucleotides 194–949) or amphotropic (nucleotides 266–883 and an EcoRI–ClaI fragment) SUgp70 sequences were used for hybridization.
RESULTS
The Amphotropic Mo-AmphoV Recombinant Induces a Spongiform Encephalomyelopathy in Infected Mice.
To determine the pathogenic potential of Mo-AmphoV, mice were infected 24–48 hr after birth. Because the immune system is still poorly developed in neonates, infection during this time frame maximizes virus spread and thus detection of disease. The Mo-AmphoV recombinant used in these studies is depicted in Fig. 1. As expected, the majority of infected mice succumbed to a lymphatic leukemia within a mean latency of 6 months, confirming the results of others (15, 16). However, unexpectedly, in 22% of the infected mice, we observed overt signs of a complex neurological syndrome, including tremor, ataxia, hindleg paralysis, and spasticity. The clinical symptoms progressed to complete tetraparesis and incontinence, and finally death of the animals at a mean age of 8 months.
Figure 1.
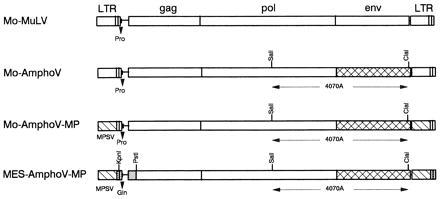
Structure of the proviral genome of MuLV recombinants assayed in newborn mice. Mo-AmphoV differs from Mo-MuLV by a SalI–ClaI fragment encoding SUgp70. Recombinant Mo-AmphoV-MP is identical to Mo-AmphoV, except that the U3 region of the Mo-MuLV LTR has been replaced with that of MPSV (21). MES-AmphoV-MP differs from Mo-Ampho-MP in 354 bp of “leader” sequences immediately downstream of the 5′ LTR but upstream of the p65gag start codon derived from MESV (22).
Histological examination confirmed a CNS disorder, revealing marked, noninflammatory spongiform degeneration of predominantly white matter tracts in the spinal cord and cerebellum, whereas in the medulla oblongata and more rostrally white and gray matter structures were equally affected (Fig. 2 A and B). No signs of inflammation or disturbances of the blood–brain barrier could be detected. Furthermore, the vacuolar changes of the glial and neuronal cells were associated with moderate astro- and microgliosis (Fig. 2C).
Figure 2.
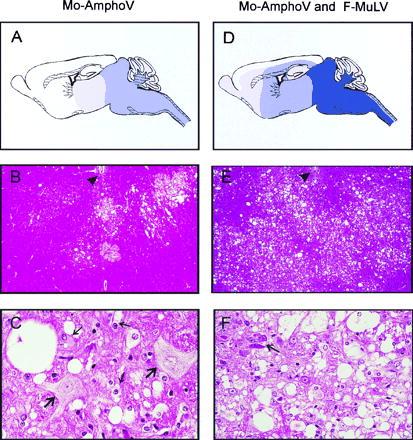
Neuropathology of DDD mice infected with Mo-AmphoV alone (A–C), or coinfected with Mo-AmphoV and F-MuLV (D–F). (A) Schematic presentation of distribution and severity of spongiform alterations along the neuraxis and hemispheres. Dark-blue shading denotes extreme; medium-blue shading, severe; and light-blue shading, moderate to mild tissue damage. (B) Overview of frontal sections of the medulla oblongata at the level of the foramina of Luschka demonstrating circumscribed foci of spongiform tissue lesions. Ependymum of the fourth ventricle floor is indicated by an arrowhead (×40). (C) Higher magnification (×245) reveals isolated single, large vacuoles of the neuropil, perikaryal swelling, and vacuolization of oligodendrocytes and astrocytes (thin arrows) and two hypertropic, swollen neurons (thick arrows) with pale nuclei and loss of Nissl substance in the cytoplasm. (D–F) Same as A–C, respectively, but mice were coinfected with Mo-AmphoV and F-MuLV. (D) Note lesions aggravate, extend more rostrally, and also involve the hemispheres. (E) In contrast to mice infected with Mo-AmphoV alone, widespread lesioning is observed. (F) Lesions in mice coinfected with F-MuLV are characterized by a higher degree of spongiform degeneration with confluent vacuolization and loss of neurons; the arrows point to shrunken, degenerated neurons.
Significantly, both the incidence and latency of the neurological disease was drastically altered when newborn mice were coinfected with nonneuropathogenic ecotropic Friend (F)-MuLV (cl 643/22N) (24). Between 25 and 107 days after infection, greater than 90% of the infected mice had succumbed to a spongiform encephalomyelopathy when coinfected with Mo-AmphoV and F-MuLV (Fig. 3). Two other A-MuLV recombinants incorporating sequences from MPSV or MESV (Fig. 1), Mo-MuLV derivatives that exhibit increase transcription efficiency in both hematopoietic and embryonic stem cells (27–29), were also tested. These two recombinants produced similar results as Mo-AmphoV when coinfected with F-MuLV (Fig. 3), indicating that the CNS lesioning was not a particularity of the Mo-AmphoV construct. None of the 19 control mice infected with F-MuLV alone exhibited neurological symptoms over an observation period of 125 days.
Figure 3.
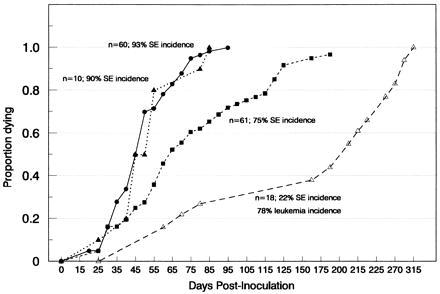
Cumulative total mortality of DDD neonates infected with three different amphotropic recombinants together with F-MuLV. The mortality curve for mice infected with Mo-AmphoV alone (▵), Mo-AmphoV and F-MuLV (▴), Mo-AmphoV-MP and F-MuLV (▪), and Mes-AmphoV-MP and F-MuLV (•) are shown. The percentage of mice dying with spongiform encephalomyelopathy (SE) is given.
In addition to decreasing the latency and increasing the incidence of the spongiform encephalomyelopathy in mice coinfected with F-MuLV, the topographical distribution of the neurological lesions was much more widespread (Fig. 2 D and E). Gray and white matter of the spinal cord, cerebellum, and brain stem exhibited extensive spongiform lesions, and the cerebral cortex and white matter structures also were involved. Moribund animals exhibited a nearly complete loss of neurons in affected areas. Destruction of nerve cells apparently occurred due to extensive cellular swelling and vacuolization or a shrinking process similar to ischemic nerve cell alteration (Fig. 2F).
Amphotropic-Derived Sequence Are Responsible for Disease Induction.
To confirm that sequences derived from A-MuLV and not other components of the Mo-AmphoV recombinant are responsible for the induction of spongiform encephalomyelopathy, newborn mice were infected with supernatants containing wild-type Mo-MuLV. Mo-AmphoV differs from Mo-MuLV in sequences encoding the SUgp70 (Fig. 1). No signs of neurological disease were observed in a total of 20 infected mice over a 4-month observation period, even when coinfected with F-MuLV. Instead, within this period, all mice succumbed to lymphoid leukemias typical of these two viral strains (Table 1). Taken together with the fact that Mo-AmphoV alone was sufficient to induce neurodegeneration in infected mice, these results clearly assign a causal role to the amphotropic SUgp70 sequences.
Table 1.
Infection of newborn DDD mice with Mo-AmphoV alone or together with F-MuLV, Mo-MuLV, or F-MCFV
| Virus and coninfectant* | No. of mice | Spongiform degeneration†
|
Leukemia‡
|
||
|---|---|---|---|---|---|
| Incidence, % | Latency (mean) | Incidence, % | Latency (mean) | ||
| Mo-AmphoV | |||||
| Alone | 16 | 22 | 250 | 78 | 188 |
| F-MuLV | 10 | 90 | 53 | 10 | 44 |
| Mo-MuLV | 16 | 100 | 64 | 0 | — |
| F-MCFV | 10 | 100 | 41 | 0 | — |
| F-MuLV | |||||
| Alone | 19 | 0 | — | 100 | 110 |
| Mo-MuLV | 16 | 0 | — | 100 | 95 |
| F-MCFV | 10 | 0 | — | 100 | 96 |
DDD mice were infected i.p. between 24 and 48 hr after birth with 50 μl of virus supernatant with titers between 3 × 105 and 1 × 106 units/ml.
Onset of spongiform encephalomyelopathy was determined by penetrance of hindleg paralysis, and, in most cases, the disease was confirmed by histopathology.
Leukemias were morphologically classified as lymphatic leukemias (cells were Thy1+).
To further collaborate these results, experiments were performed to confirm the presence of Mo-AmphoV in the diseased tissue. Staining using monospecific antibodies that recognize the gag-encoded CAp30 and the env-encoded SUgp70 demonstrated a striking correlation between affected tissue and viral protein expression, with microglial and capilliary endothelial cells as the most prominent positive cell type (J.L., unpublished results). However, as no antibodies are available that are specific for Mo-AmphoV, the detected antigens may be attributed to activated endogenous retrovirus. PCR analysis using primers specific for amphotropic env sequences was thus used to confirm the presence of amphotropic SUgp70 sequences in the brain. In all diseased mice infected with either Mo-AmphoV alone or Mo-AmphoV plus F-MuLV, but not in F-MuLV-infected control mice, amphotropic sequences were readily detected in the brain (data not shown).
Genetic Background and Age Influence Mice Susceptibility to the Virus-Induced Neurodegenerative Disorder.
To determine whether susceptibility to Mo-AmphoV-induced neurodegeneration is lost with age, DDD mice were infected 8 days after birth with one of the A-MuLV recombinants together with F-MuLV. In a total of 46 infected mice, no symptoms of the CNS disorder were observed; rather, all mice became leukemic after a mean latency of 139 days. Similar results have been reported with the ecotropic CAS-BrE MuLV and its recombinants, as well as two other ecotropic MuLV variants that induce CNS degeneration (30–32).
To determine whether the induction of spongiform encephalomyelopathy by Mo-AmphoV was due to a unique genetic background of DDD mice, five other mice strains were tested. To decrease the latency period, newborn mice were infected with both Mo-AmphoV and F-MuLV. All Balb/cJ and NMRI mice (n = 31 and n = 19, respectively) succumbed to leukemia within a relatively short (2- to 4-month) latency period without any clinical signs of neurological disorders. Similarly, NIH mice (n = 16) also developed leukemias (latency 2–3 months, unpublished results); however, histological analysis of three diseased animals showed clear signs of concurrent spongiform lesions in the CNS, representative of early stages of the disease. Of the strains tested, neuropathology was most prevalent in DBA/2J and 129/SvJ mice. Three to four months after infection, 68% and 100% of the moribund DBA/2J (n = 25) and 129/SvJ (n = 7) mice, respectively, had clear clinical symptoms (tremor, ataxia, and spasticity) of the CNS disorder. No significant differences in the histopathology or topographical distribution of spongiform degeneration was observed in the different mouse strains. These results demonstrate that induction of spongiform encephalomyelopathy by Mo-AmphoV is not unique to DDD mice; however, the genetic background does contribute to the latency and degree of disease. Significantly, the disease is often masked by severe leukemias in some mice strains.
Coinfection of Mo-AmphoV with Either Polytropic or Ecotropic Retrovirus Increases Incidence and Degree of Disease.
It can be envisaged that the enhanced degree and incidence of spongiform encephalomyelopathy caused by coinfection with F-MuLV could be due to either the synergistic action of proteins expressed by both viruses or, alternatively, pseudotyping of the A-MuLV recombinant genome by F-MuLV viral capsids, increasing the efficiency of viral spread into regions of the CNS. To determine which component of the F-MuLV contributed to the increased neuropathogenicity, newborn mice were coinfected with Mo-AmphoV and either the ecotropic Mo-MuLV or a polytropic F-MCFV (Table 1). Somewhat surprisingly, both viruses increased the incidence and degree of spongiform degeneration to similar levels observed when Mo-AmphoV was coinfected with F-MuLV. Again, neither virus alone or together with F-MuLV induced neuropathological alterations in infected mice (Table 1).
A Recombination Event Between Mo-AmphoV and Coinfectant Is Unlikely to Contribute to Altered Pathogenicity.
To rule out the possibility that recombinants between the coinfectants were generated that contributed to the disease induction, proviral DNA from virus released from diseased brain from mice coinfected with Mo-AmphoV and Mo-MuLV was examined by Southern blot analysis. Recombinants were observed in the brain of two independent mice; however, the recombination junction in both instances was within pol. Because these sequences share 97% amino acid identity (33) (C.S. and P. Lührs, unpublished results), these recombinant viruses are practically identical to the original inoculants and are thus unlikely to contribute to the increased neurodegeneration. Furthermore, the prerequisite to generate polytropic MCF recombinants for CNS disease induction can be ruled out because DBA/2J mice, which carry the Rmcfr locus inhibiting the spread of MCFV (34), succumb to the Mo-AmphoV-induced spongiform encephalomyelopathy.
DISCUSSION
The results presented here clearly demonstrate that A-MuLV recombinants expressing the amphotropic env-encoded SUgp70 induce a fatal degenerative CNS disorder in mice. Although four independent ecotropic variants have also been shown to induce a similar pathological process (30–32), none of these viruses induces a CNS disturbance with such a wide topographical distribution, even when the same viral transcriptional machinery is used to express the env gene.
Substitution of a SalI–ClaI fragment, containing 3′ pol and env sequences of A-MuLV 4070, was sufficient to confer the altered pathogenicity to Mo-MuLV. Although we cannot entirely exclude that the subtle differences in the pol-encoded RT and IN proteins contribute to the pathogenicity, the env-encoded SUgp70 is the most probable pathogenic determinant. Previous analysis of ecotropic retroviruses that induce neurodegeneration have also confirmed a causal role of env-derived sequences, although the mechanism is still unclear (30, 35, 36). Because the SUgp70 glycoprotein is a surface protein, it could be envisaged that its interaction with cellular receptors may trigger the neurodegenerative process. A sodium phosphate transporter ubiquitously expressed on the cell membrane has been shown to bind the amphotropic SUgp70 and enable cell entry. Interestingly, this receptor is expressed at high levels in the brain and is well conserved between human and rodents (37–39). Alternatively, there may exist other surface proteins that bind with the amphotropic SUgp70 but have not yet been identified. Although the neurodegeneration may be due to a direct interaction of the env product with neurons, evidence is accumulating in studies of other viral-induced spongiform disorders that supporting cells in the CNS (e.g., microglial or endothelial capillary cells) may be the target cell of the virus (31, 36). Interference with their function or release of noxious factors may indirectly trigger neuron killing.
Although A-MuLV recombinants have been widely used, no group has previously demonstrated an association between amphotropic SUgp70 expression and CNS degeneration. This can most likely be attributed to several factors. First, neurological disease symptoms are only visible after a rather long latency (5 to 10 months) and at a low incidence (20 to 30%) and thus are probably often masked by the coincident or more quickly appearing symptoms of lymphoid leukemias. Second, mouse strains vary in their susceptibility to the neurodegenerating property of A-MuLV. Finally, there exists a rather limited susceptibility window to the virus-induced spongiform encephalomyelopathy.
Remarkably, coinfection of A-MuLV recombinants with either ecotropic F-MuLV or Mo-MuLV or polytropic F-MCFV drastically altered the incidence, latency, and distribution of the disease. Particularly revealing is the synergistic effect observed with ecotropic Mo-MuLV, as it can be concluded that the enhancing effect must be due to SUgp70 sequences. This was unexpected, because it has been previously shown that sequences in the LTR and upstream gag sequences of F-MuLV can greatly increase the neuropathogenic capacity of the ecotropic CAS-BrE retrovirus, which also induces a spongiform encephalomyelopathy in newborn mice (40–42). Because polytropic and ecotropic SUgp70 proteins share only 40% similarity in the N terminus (the C terminus being highly conserved between all three types of virus) (43), we find it unlikely that these proteins directly synergize with the amphotropic SUgp70 protein to induce the neurological lesions. Similarly, as recombinants between A-MuLV and the coinfectant did not result in the generation of chimeric env genes or altered transcription controls, we find it unlikely that generation of a unique recombinant virus is necessary for disease induction. Rather, the coinfectants most likely act to target the amphotropic env sequences to the appropriate cells during early stages of infection through the generation of viral pseudotypes. A similar mechanism has also been attributed to polytropic MCF recombinants in the induction of leukemias by ecotropic MuLV. Conceivably, virus spread is more efficient when two types of receptors are available (e.g., ecotropic and amphotropic). Thus, the availability of a second virus receptor would allow more efficient spread of A-MuLV; amphotropic spread to the CNS has been previously reported to be quite inefficient compared with ecotropic variants (44).
The importance of targeting SUgp70 expression in the CNS for induction of spongiform encephalomyelopathy is also reflected in the age-dependent restriction observed here and previously reported for neuropathogenic ecotropic virus. It has been shown that this restriction is due to the inability of the virus to mount a productive infection of the CNS. Although this is most likely mediated by the immune system, other studies have implicated the importance of the blood–brain barrier and/or a developmental window in which specific cells are replicating and thus are still permissive for viral infection (45–47). Thus, conditions that increase the levels of A-MuLV in the CNS and thereby enhance the expression of amphotropic SUgp70, will likely increase the incidence and severity of spongiform neurodegeneration.
Significantly, the Mo-AmphoV recombinant used in these studies is the basis of all approved packaging cell lines used in gene therapy trials (3). In light of the results presented here, the wide use of amphotropic recombinants both in the laboratory as well as in clinical settings needs to be examined. Although several studies have clearly demonstrated the effectiveness of the human complement system in eliminating MuLV infection, this protection is not 100% when virus is released from human cells or when patients are immunodeficient (11, 13). Similarly, direct injection of amphotropic virus-producing cells or pseudotypes carrying amphotropic sequences within the CNS, as proposed in several gene therapy protocols (9, 48), would occur in an environment in which plasma complement factors and other elements of the defense system are normally excluded by the blood–brain barrier. Perhaps more important, spontaneous or induced immunosuppression in patients will also abrogate the immunological restriction of virus replication and spreading. In addition, it should be kept in mind that disease processes are frequently associated with re- or de novo expression of cellular determinants or functions that are normally developmentally regulated—such changes may enable viral entry into cells normally restrictive to infection in adults. Significantly, it has been demonstrated in the mouse system that direct introduction of cell-associated, neuropathogenic retrovirus in the adult CNS is sufficient to induce spongiform neurodegeneration (46).
Acknowledgments
We are grateful to W. Ostertag for stimulating discussions and to A. D. Miller for providing the pAM and pAMS plasmids used in these studies. We also thank K. Heigl and G. Arman-Kalcek for expert technical assistance. This paper is based on a doctoral study by C.M. in the faculty of Biology, University of Hamburg. Support by the Bundesministerium für Forschung und Technologie is acknowledged. The Heinrich-Pette-Institut is supported by the Freie und Hansestadt Hamburg and the Bundesministerium für Gesundheit.
ABBREVIATIONS
- MuLV
murine leukemia virus(es)
- MCFV
mink cell focus-forming virus
- A-MuLV
amphotropic MuLV
- Mo-MuLV
Moloney-MuLV
- F-MuLV
Friend-MuLV
- Mo-AmphoV
amphotropic recombinant between Mo-MuLV and A-MuLV
- CNS
central nervous system
- LTR
long terminal repeat
References
- 1.Tabin C J, Hoffman H W, Goff S P, Weinberg R A. Mol Cell Biol. 1982;2:426–435. doi: 10.1128/mcb.2.4.426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mann R, Mulligan R C, Baltimore D. Cell. 1983;33:153–159. doi: 10.1016/0092-8674(83)90344-6. [DOI] [PubMed] [Google Scholar]
- 3.Miller A D. Hum Gene Ther. 1990;1:5–14. doi: 10.1089/hum.1990.1.1-5. [DOI] [PubMed] [Google Scholar]
- 4.Levy J A. Nature (London) 1975;253:140–142. doi: 10.1038/253140a0. [DOI] [PubMed] [Google Scholar]
- 5.Rasheed S, Gardner M B, Chen E. J Virol. 1976;19:13–25. doi: 10.1128/jvi.19.1.13-18.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rein A. Virology. 1982;120:251–257. doi: 10.1016/0042-6822(82)90024-1. [DOI] [PubMed] [Google Scholar]
- 7.Hartley J W, Rowe W P. J Virol. 1976;19:19–25. doi: 10.1128/jvi.19.1.19-25.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ross G, Erickson R, Knorr D, Motulsky A G, Parkman R, Samulski J, Strauss S E, Smith B R. Hum Gene Ther. 1996;7:1781–1790. doi: 10.1089/hum.1996.7.14-1781. [DOI] [PubMed] [Google Scholar]
- 9.Culver K W, Ram Z, Walbridge S, Ishii H, Oldfield E H, Blaese R M. Science. 1992;256:1550–1552. doi: 10.1126/science.1317968. [DOI] [PubMed] [Google Scholar]
- 10.Irwin M J, Laube L S, Lee V, Austin M, Chada S, Anderson C-G, Townsend K, Jolly D J, Warner J F. J Virol. 1994;68:5036–5044. doi: 10.1128/jvi.68.8.5036-5044.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Takeuchi Y, Cosset F-L, Lachmann P J, Okada H, Weiss R A, Collins M K L. J Virol. 1994;68:8001–8007. doi: 10.1128/jvi.68.12.8001-8007.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chong H, Vile R G. Gene Ther. 1996;3:624–629. [PubMed] [Google Scholar]
- 13.Donahue R E, Kessler S W, Bodine D, McDonaugh K, Dunbar C, Goodman S, Agricola B, Byrne E, Raffeld M, Moen R, Bacher J, Zsebo K M, Nienhuis A W. J Exp Med. 1992;176:1125–1135. doi: 10.1084/jem.176.4.1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gardner M. Curr Top Microbiol Immunol. 1978;79:215–259. doi: 10.1007/978-3-642-66853-1_5. [DOI] [PubMed] [Google Scholar]
- 15.DesGrosseillers L, Jolicoeur P. J Virol. 1984;52:448–456. doi: 10.1128/jvi.52.2.448-456.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ott D E, Keller J, Sill K, Rein A. J Virol. 1992;66:6107–6116. doi: 10.1128/jvi.66.10.6107-6116.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wolff L, Koller R, Davidson W. J Virol. 1991;65:3607–3616. doi: 10.1128/jvi.65.7.3607-3616.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vanin E F, Kaloss M, Broscius C M, Nienhuis A W. J Virol. 1994;68:4241–4250. doi: 10.1128/jvi.68.7.4241-4250.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cornetta K, Morgan R A, Gillio A, Sturm S, Baltrucki L, O’Reilly R, Anderson W F. Hum Gene Ther. 1991;2:215–219. doi: 10.1089/hum.1991.2.3-215. [DOI] [PubMed] [Google Scholar]
- 20.Miller A D, Verma I M. J Virol. 1984;49:214–222. doi: 10.1128/jvi.49.1.214-222.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stocking C, Kollek R, Bergholz U, Ostertag W. Proc Natl Acad Sci USA. 1985;82:4098–4102. doi: 10.1073/pnas.82.17.5746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grez M, Akgün E, Hilberg F, Ostertag W. Proc Natl Acad Sci USA. 1990;87:9202–9206. doi: 10.1073/pnas.87.23.9202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Itoh K, Tezuka H, Sakoda H, Konno M, Uchiyama T, Uchino H, Mori K J. Exp Hematol. 1989;17:145–153. [PubMed] [Google Scholar]
- 24.Pragnell I B, McNab A, Harrison P R, Ostertag W. Nature (London) 1978;272:456–458. doi: 10.1038/272456a0. [DOI] [PubMed] [Google Scholar]
- 25.Troxler D H, Yuan E, Linemeyer D, Ruscetti S, Scolnick E. J Exp Med. 1978;148:639–653. doi: 10.1084/jem.148.3.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harbers K, Schnieke A, Stuhlmann H, Jähner D, Jaenisch R. Proc Natl Acad Sci USA. 1981;78:7609–7613. doi: 10.1073/pnas.78.12.7609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stocking C, Grez M, Ostertag W. In: Virus Strategies. Doefler W, Boehm P, editors. Heidelberg: VCH; 1993. pp. 433–455. [Google Scholar]
- 28.Grez M, Zörnig M, Nowock J, Ziegler M. J Virol. 1991;65:4691–4698. doi: 10.1128/jvi.65.9.4691-4698.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baum C, Hegewisch-Becker S, Eckert H-G, Stocking C, Ostertag W. J Virol. 1995;69:7541–7547. doi: 10.1128/jvi.69.12.7541-7547.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Portis J L. Curr Top Microbiol Immunol. 1990;160:11–27. doi: 10.1007/978-3-642-75267-4_2. [DOI] [PubMed] [Google Scholar]
- 31.Masuda M, Remington M P, Hofman P M, Ruscetti S K. J Virol. 1992;66:2798–2806. doi: 10.1128/jvi.66.5.2798-2806.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wong P K. Curr Top Microbiol Immunol. 1990;160:29–60. doi: 10.1007/978-3-642-75267-4_3. [DOI] [PubMed] [Google Scholar]
- 33.Miller A D, Chen F. J Virol. 1996;70:5564–5571. doi: 10.1128/jvi.70.8.5564-5571.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ruscetti S, Matthai R, Potter M. J Exp Med. 1985;162:1579–1587. doi: 10.1084/jem.162.5.1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paquette Y, Hanna Z, Avard P, Brousseau R, Robitaille Y, Jolicoeur P. Proc Natl Acad Sci USA. 1989;86:3896–3900. doi: 10.1073/pnas.86.10.3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.González-Scarano F, Nathanson N, Wong P K Y. In: The Retroviridae. Levy J A, editor. Vol. 4. New York; London: Plenum; 1995. pp. 409–490. [Google Scholar]
- 37.Miller D G, Edwards R H, Miller A D. Proc Natl Acad Sci USA. 1994;91:78–82. doi: 10.1073/pnas.91.1.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van Zeul M V, Johann S V, Closs E, Cunningham J, Eddy R, Shows T, O’Hara B. Proc Natl Acad Sci USA. 1994;91:1168–1172. doi: 10.1073/pnas.91.3.1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kavanaugh M P, Miller D G, Zhang W, Law W, Kozak S L, Kabat D, Miller A D. Proc Natl Acad Sci USA. 1994;91:7071–7075. doi: 10.1073/pnas.91.15.7071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gardner M B, Henderson B E, Officier J E, Rongey R W, Parker J C, Oliver C, Ester J D, Huebner R J. J Natl Cancer Inst. 1973;51:1243–1254. doi: 10.1093/jnci/51.4.1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Portis J L, Czub S, Caron C F, McAtee F. J Virol. 1990;64:1648–1656. doi: 10.1128/jvi.64.4.1648-1656.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Portis J L, Spangrude G J, McAtee F. J Virol. 1994;68:3879–3887. doi: 10.1128/jvi.68.6.3879-3887.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stoye J P, Coffin J M. J Virol. 1987;61:2659–2669. doi: 10.1128/jvi.61.9.2659-2669.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Oldstone M B A, Jensen F, Dixon F J, Lampert F. Virology. 1980;107:180–193. doi: 10.1016/0042-6822(80)90283-4. [DOI] [PubMed] [Google Scholar]
- 45.Czub M, Czub S, McAtee F L, Portis J L. J Virol. 1991;65:2539–2544. doi: 10.1128/jvi.65.5.2539-2544.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lynch W P, Robertson S J, Portis J L. J Virol. 1995;69:1408–1419. doi: 10.1128/jvi.69.3.1408-1419.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Robbins D S, Remington M P, Sarzotti M, Louis D S, Hoffman P M. J Virol. 1995;69:6847–6951. doi: 10.1128/jvi.69.11.6847-6851.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wei M X, Tamiya T, R K, Hurford J, Boviatsis E J, Tepper R I, Chiocca E A. Hum Gene Ther. 1995;6:437–443. doi: 10.1089/hum.1995.6.4-437. [DOI] [PubMed] [Google Scholar]