

Summary
The structures of glycoproteins that mediate enveloped virus entry into cells have revealed dramatic structural changes that accompany membrane fusion and provided mechanistic insights into this process. The group of class I viral fusion proteins includes the influenza hemagglutinin, paramyxovirus F, HIV env and other mechanistically related fusogens, but these proteins are unrelated in sequence and exhibit clearly distinct structural features. Recently determined crystal structures of the paramyxovirus F protein in two conformations, representing prefusion and postfusion states, reveal a novel protein architecture that undergoes large-scale, irreversible refolding during membrane fusion, extending our understanding of this diverse group of membrane fusion machines.
INTRODUCTION
Enveloped virus entry into cells
Viruses have evolved a variety of architectures that are designed to protect and transmit their nucleic acid genomes, ensuring their survival despite fundamental dependencies on their hosts for replication and spread [1]. For all viruses, recognition of and subsequent penetration into host cells are key steps in the viral life cycle and many adaptations in structures, mechanisms and entry pathways have evolved to overcome the fundamental barriers in this process, such as crossing the topological barrier presented by the cellular lipid bilayer [1,2]. Many viruses themselves, so-called enveloped viruses, are surrounded by a lipid bilayer that is acquired during prior budding from infected cells. In contrast to those viruses coated by a protein shell, enveloped viruses are presented with the problem of uniting two lipid bilayers during entry into cells, one membrane from the target cell and one from the virus. As more has been learned about the structural aspects of enveloped virus entry mechanisms, many common mechanistic features have been identified that appear to hold for proteins of widely different structures.
Classes of viral fusion proteins
Pioneering studies of the influenza virus hemagglutinin (HA) carried out by the Wiley and Skehel laboratories, beginning in the 1970s and extending for over two decades, provided the first insights into the membrane fusion machinery of enveloped viruses [3–7]. The activation of membrane fusion by HA is triggered by low pH, after virus trafficking into endosomal compartments of the cell [2]. Structures of the HA in its pre- and post-fusion conformations revealed a dramatic refolding of the C-terminal portion of the protein into a hairpin-like conformation that suggested how HA might lower the barriers to lipid bilayer fusion [4–7]. At two ends of the “hairpin” are two membrane-interacting segments within the fusion protein sequence – one of these being a fusion peptide initially sequestered in the core of the trimeric HA and the other being the C-terminal transmembrane anchor regions. Importantly, the observation of this hairpin and associated refolding changes within previously identified heptad repeat segments of HA, lead to the proposal of a “spring-loaded” mechanism for HA-mediated membrane fusion, in which the hydrophobic fusion peptide would be projected towards the target cell bilayer by a coil-helix transition in HA, followed by a folding back of the C-terminal end of the protein, resulting in the close juxtaposition of the fusion peptides and transmembrane domains of the HA trimer [4,5,8]. Mutations in HA that affect the optimal pH of this refolding are located throughout the structure, suggesting that HA might function as a “global” pH sensor [4], although it is possible that ionization of specific residues may play an important initiating role in the refolding process.
These structural insights into the HA fusion mechanism also suggested that aspects of this process might be extendable to other viral fusion proteins. In particular, it was recognized that the fusion proteins from unrelated viruses, such as Ebola, HIV, paramyxoviruses and others, contained identifiable sequence motifs that appeared to be related to those in HA. These motifs include an internal, often furin-like, cleavage site near or adjacent to a stretch of 20–25 hydrophobic amino acids (a fusion peptide), followed by 1 or 2 heptad repeat regions (Figure 1). Subsequent structural, biochemical and functional studies of the heptad repeat regions from these viral fusion proteins (Figure 2), demonstrated their assembly into trimeric hairpin-like structures similar to that of the low-pH induced HA, indicative of commonalities in the mechanism of membrane fusion [9–16]. In all structures, three hydrophobic fusion peptides are located near the N-termini of helices forming a centralized coiled coil, while an antiparallel structure, often helical and deriving from a second heptad repeat in the sequence, positions the transmembrane anchors at the same end of a rod-like structure (Figure 2).
Figure 1. Schematic of three prototypical class I viral fusion proteins.

Schematics of the paramyxovirus F, influenza virus HA and HIV env glycoproteins are shown with important sequence features annotated. All three proteins are produced as intact polypeptide chains, which form trimers that are subsequently cleaved (gap) during biosynthesis. Paramyxovirus F is cleaved into two subunits, F1 and F2, HA into HA1 and HA2 and HIV env into gp120 and gp41. The site of cleavage is adjacent to a hydrophobic stretch of 20–25 amino acids known as the fusion peptide (FP; blue) followed by an N-terminal heptad repeat sequence (HRA; green). In paramyxovirus F and HIV env, a separate C-terminal heptad repeat sequence is easily identifiable (HRB; magenta). In the influenza HA sequence, we have used HRB to indicate the C-terminal portion of the primary heptad repeat that is observed to reverse its orientation during the low pH conformational change. Transmembrane anchors (TM) are also indicated and these are proximal to HRB. In paramyxovirus F, 250 amino acids separate HRA and HRB, in contrast to their much closer positioning in HA and env sequences.
Figure 2. Core structures of class I viral fusion proteins.

Representative core structures of class I viral fusion proteins, corresponding to the HRA and HRB regions in Figure 1, are shown in their presumed postfusion conformations. The paramyxovirus F (PIV5), influenza HA2 (bacterially expressed), SARS spike glycoprotein (S), Ebola GP2 and HIV gp41 form similar hairpin-like arrangements, though with clear differences in the details of their structures. The chains are colored from blue (N-terminus) to red (C-terminus), with the blue tips of the coiled coils corresponding to the positions of the fusion peptides and the red C-termini corresponding to the positions of the TM domains. In all proteins, these membrane interacting segments are localized to the same end of the structures.
These similarities have been recognized by a nomenclature that places viral fusion proteins with these sequence and structural features into the so-called class I viral fusion protein group [17–19]. It is generally thought that the class I viral fusion proteins fold to a prefusion, metastable conformation, which is then activated to undergo a large conformational rearrangement to a lower energy state, thereby providing the energy needed to accomplish membrane fusion. The process is irreversible and independent of the use of ATP. A class II group, that includes flavivirus and alphavirus membrane fusion proteins, has a different, primarily beta-sheet architecture, and different mechanistic details involving changes in oligomerization state and domain repositioning that are distinct from the refolding transitions observed in HA and paramyxovirus F proteins, and that are presumed to occur in other class I viral fusion proteins. Nonetheless, class II fusion proteins are also thought to assemble into a hairpin-like conformation during membrane fusion [17,20]. Recent structural studies of the VSV G protein in two conformations, and its surprising structural homology to the herpesvirus gB protein, suggest that these two proteins may define a third class of fusion protein [21–23].
Despite this classification scheme, it has been clear that class I fusion proteins from different viruses do not exhibit any significant sequence homologies, despite the presence of some similar architectural motifs, and that these class I proteins probably represent distinct structural subfamilies –much more different than the alphavirus and flavivirus fusion proteins. For example, the distribution of heptad repeats, cleavage sites and fusion peptides in HA and paramyxovirus F are very different: in F, over 250 amino acids separate the two heptad repeats that assemble into a 6 helix bundle (6HB), whereas in HA very few residues separate the heptad repeat regions that form the base of the hairpin structure (Figures 1 & 2). Despite overall analogous hairpin arrangements, the core region structures from different class I proteins are also quite different in many of their structural details (Figure 2). Finally, the mechanisms for activation of different viral fusion proteins are distinct. Some, like HA, are activated by low pH, others, like HIV env and paramyxovirus F are activated by receptor-binding events, either by direct interactions (HIV env) or indirectly through a viral attachment protein (paramyxovirus F). Ebola GP-mediated fusion requires its trafficking through the endosomal pathway, but this likely reflects a need for further GP processing by lysosomal enzymes, such as cathepsins, to trigger virus entry, rather than simply low pH itself [24]. Thus many questions regarding the structures, folding and activation of class I viral fusion proteins remain to be addressed. Recent structural results on the paramyxovirus F protein have provided new insights into just how different these class I viral fusion proteins may be and suggest what structural parallels may apply across this class of membrane fusogens.
Introduction to paramyxovirus entry
The paramyxoviruses are enveloped, negative-strand RNA viruses that cause both respiratory and systemic disease. The paramyxovirus family includes, among others, mumps virus, measles virus, Sendai virus, Newcastle disease virus (NDV), human respiratory syncytial virus (hRSV), parainfluenza virus 5 (PIV5; formerly known as SV5), human parainfluenza viruses 1–4 (hPIV) [25–27] and the deadly Nipah and Hendra viruses [28,29]. Members of this viral family are among the most significant human and animal pathogens, being directly responsible for many human deaths and hospitalizations each year, and for infections of farm animals that have major economic consequences. For example, measles virus is still a major cause of death in children in developing countries and hRSV is the primary cause of infant hospitalization for respiratory infection in the US, accounting for ~70% of viral bronchiolitis cases [30].
Two viral glycoproteins are involved in the infection of cells – an attachment protein, called HN, H or G, depending on the virus, and the fusion (F) protein. In all paramyxoviruses, the F protein catalyzes membrane fusion, after the attachment protein mediates binding of the virus to the cell surface [31,32]. Although the F protein sequences can vary substantially between viruses, the majority of F cysteine residues involved in disulfide bonds are conserved. Given this and their similar biological activities, it is likely that representative F structures provide insight into the shared F function of membrane fusion, but there are likely to be important virus-specific sequence and structural differences. For many of the paramyxoviruses (NDV, hPIV1–4, PIV5 and others), the attachment protein is a hemagglutinin/neuraminidase (HN) protein, which binds to and can also cleave sialic acid structures. The morbilliviruses, such as measles virus, express a hemagglutinin (H) protein in place of HN, while the pneumoviruses (RSV) and henipaviruses (Nipah and Hendra) express a distinct attachment glycoprotein (G) [32,33]. In contrast to HN, H from measles interacts with CD46 or CDw150/SLAM [34,35], and RSV G has been shown to interact with heparin sulfate [32,36]. In many of the paramyxoviruses, it is the attachment protein interaction with receptors that is thought to initiate conformational changes in F, thereby activating membrane fusion at the right time and right place. Thus in contrast to influenza virus, paramyxoviruses carry out membrane fusion and entry at the cell surface and at neutral pH.
REVIEW
Crystal structures of paramyxovirus F proteins
Crystal structures of fragments of F proteins from different paramyxoviruses provided the first insights into the F structure and revealed a conserved 6 helix bundle (6HB) arrangement formed by two heptad repeat regions (HRA and HRB; Figure 1, 2), whose assembly is tightly coupled to membrane fusion [11,12]. Peptides spanning HRA and HRB regions are inhibitors of F-mediated membrane fusion and mechanistic studies identified two distinct F refolding intermediates during the process [31,37]. One intermediate can be trapped at low temperatures and is susceptible to inhibition by exogenously added HRA peptide [37]. At higher temperatures, a second intermediate in refolding becomes susceptible to inhibition by both HRA and HRB peptides. These results suggest that endogenous heptad repeat segments of the F protein become sequentially exposed along the refolding pathway. In the low-temperature trapped intermediate, selective exposure/unfolding of the HRB region would enable interactions with exogenously added HRA peptide, while at higher temperatures the unfolding of F and exposure of both endogenous HRA and HRB regions would explain susceptibility to inhibition by the exogenous peptides. These studies suggested that F undergoes a sequential, stepwise unfolding and collapse or refolding, different from influenza HA, yielding a final trimer of F hairpins coincident with 6HB formation. Importantly, this stepwise exposure of endogenous HRB and HRA regions at different stages of the fusion pathway provided key evidence that the two segments could not form a 6HB in the prefusion conformation of F, even if it was of reduced stability.
In order to understand the folding and rearrangements of F that accompany membrane fusion, a number of groups have pursued the structures of F proteins using crystallographic and electron microscopy methods [38–40]. A partial structure of the Newcastle Disease virus (NDV) F protein was solved by X-ray crystallographic methods, providing a model for extensive regions of the F ectodomain, including residues linking HRA and HRB [40,41]. However, key regions of the F structure could not be built, including the N-terminal regions of HRA, the entire HRB region and the fusion peptide and neighboring cleavage site. Partial proteolysis of the F protein had occurred during crystallization [39], and although models for the F fusion mechanism were proposed, this partial structure raised many questions and could not explain available functional data on F-mediated membrane fusion.
Subsequently, we determined the crystal structure of a related F protein from human parainfluenza virus 3 (hPIV3) [42]. The hPIV3 F protein was generated using recombinant baculovirus technology and secreted from insect cells. Crystals of the hPIV3 F protein were obtained with no evident proteolytic cleavage and the structure was solved using a model based on the previously determined NDV F protein structure (Figure 3). The most revealing observation in the hPIV3 F structure was the presence of the HRA/HRB 6HB, providing a key structural insight that was lacking in the previous NDV F structure. The hPIV3 F 6HB marked this structure as likely representing a post-fusion form of the protein. Furthermore, when mutations known to either stabilize or destabilize the metastable form of F were mapped onto this structure, this did not provide any insight into how these residues could influence the activation of F mediated membrane fusion [42–44]. Despite the fact that the hPIV3 F protein had been engineered to prevent its proteolytic cleavage, with the goal of stabilizing the prefusion form, the secreted protein had clearly adopted the post-fusion conformation. Interestingly, cryo-EM reconstructions of intact Sendai virus F protein solubilized in detergents, appeared generally similar to both F crystal structures, raising further questions about the potential similarities and differences between the pre- and post-fusion conformations [38].
Figure 3. Structures of paramyxovirus F and influenza HA trimers in two conformations.
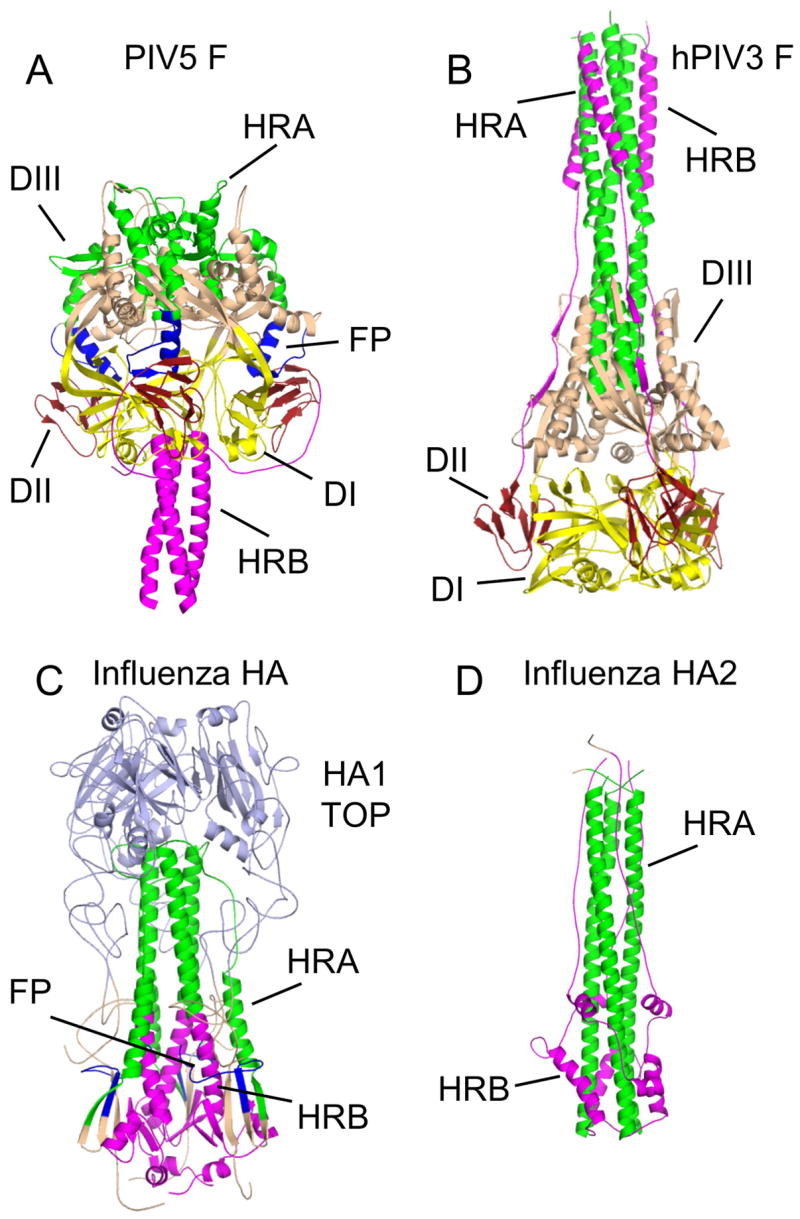
(A) The prefusion conformation of the PIV5 F trimer. HRA is colored green, HRB and its N-terminal linker region are colored magenta and the fusion peptides are colored blue. Domains I (DI), II (DII) and III (DIII) are indicated. The DI domains, which remain relatively invariant during the conformational change, are shown in yellow and DII domains are in dark red. (B) The postfusion conformation of the hPIV3 F trimer. Coloring of the protein is as for the PIV5 F trimer in (A). (C) The prefusion conformation of the influenza HA trimer. The coloring of HRA, HRB (and its C-terminal linker to the TM domain), and the fusion peptides follow those used for the PIV5 trimer in (A). The region of HA1 that forms a globular receptor-binding domain (HA1 TOP), is colored light blue. (D) The postfusion conformation of the influenza HA trimer, as revealed by the crystal structure of the C-terminal HA2 subunit. Coloring of the protein is as in (C). The HA1 subunit, including HA1 TOP, is thought to dissociate from HA2, becoming linked by disordered and protease-sensitive tethers during exposure to low pH.
Stabilization of the prefusion F structure
Another important insight that came from the hPIV3 F crystal structure was the realization that the prefusion F conformation might be dependent upon its anchoring in the membrane. Secreting the F protein, by truncating the polypeptide chain before the TM domain, might affect the F structure in two different ways. The folding landscape of F could be perturbed, either interfering with the folding to its metastable form, or by lowering the energy barrier between the metastable and post-fusion conformations. In either case, the hPIV3 structure suggested that the prefusion form might only be crystallized by mimicking the effects of the missing TM domains and cytoplasmic tails in the secreted protein.
We chose to use a soluble, 3-helix bundle, derived from studies of Harbury and Kim on the sequence determinants of GCN4 oligomerization, as a TM domain mimic [45,46]. The trimeric GCN4 derivative (GCNt) was placed in register with C-terminal HRB of the PIV5 F protein [47]. The recombinant PIV5 F protein was expressed and crystallized, but the structure could not be solved using models derived from the NDV or hPIV3 F structures, because of substantial refolding and rearrangements of the polypeptide chain.
Refolding to drive membrane fusion
A comparison of the conformational states of the PIV5 and hPIV3 F proteins is shown in Figures 3 & 4. In both forms, a more globular head region sits at one end of a helical stalk, although the direction of the stalk is inverted relative to the beta-sheet domains comprising this head region. In the two structures, individual domains (domain I, domain II and core elements of domain III) retain similar overall folds and the arrangement of domain I around the trimer axis remains relatively constant during the refolding transition, perhaps acting as an anchor of the oligomer during the structural transition. In the hPIV3 F structure, segments of the ectodomain corresponding to the hydrophobic fusion peptide and upstream cleavage site were not observed and are exposed on the outside of the helical stalk region. In the PIV5 crystal structure, these regions are well ordered and the fusion peptide is observed nestled in between two subunits of the trimer in the midsection of the globular head. Removal of the fusion peptide from this interface results in a movement of domain II towards the central trimer axis and an overall compaction of the head as F progresses from pre- to post-fusion conformations.
Figure 4. Structures of paramyxovirus F and influenza HA subunits in two conformations.
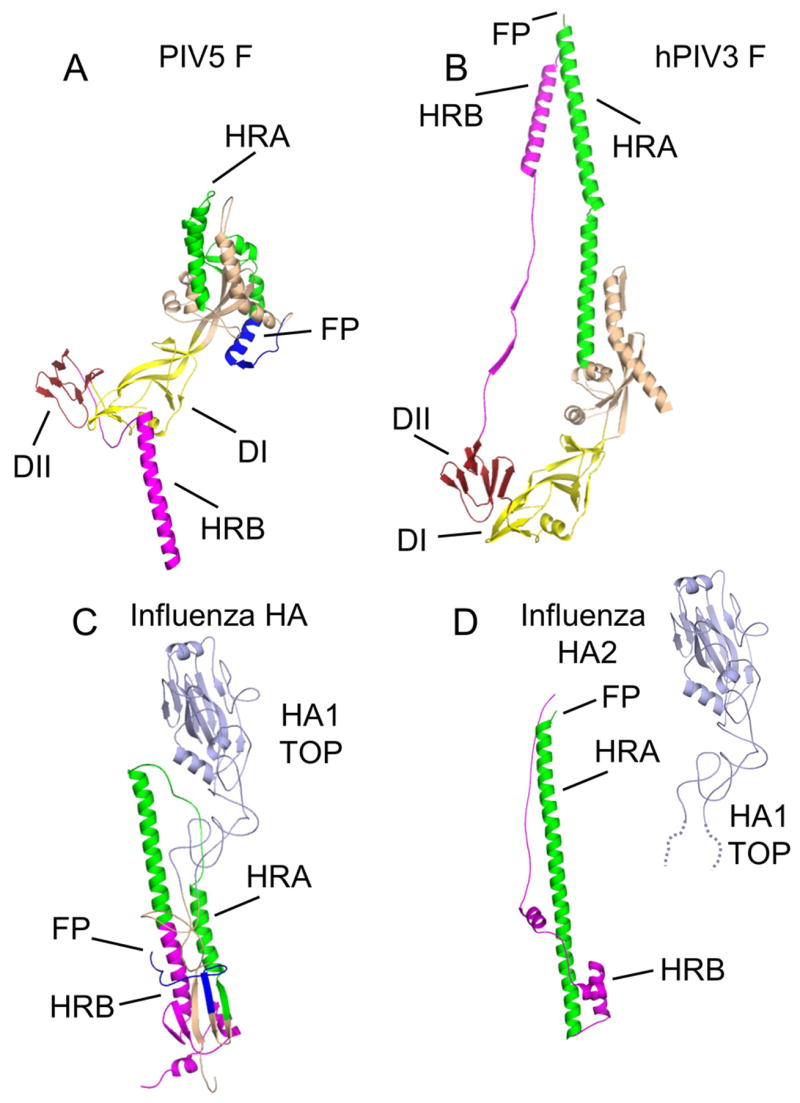
(A) The prefusion conformation of a single PIV5 F subunit. (B) The postfusion conformation of a single hPIV3 F subunit. (C) The prefusion conformation of a single influenza HA subunit. (D) The postfusion conformation of a single subunit of the influenza HA2 protein. The flexible linkage of HA2 to HA1 TOP is indicated by dotted lines representing amino acids present in HA1, that extend from the globular HA1 TOP domain. The HA1–HA2 disulfide bond is omitted for clarity. Coloring of the subunits follows those used in Figure 3.
More dramatic refolding occurs in the HRA and HRB regions of the F polypeptide. In the PIV5 F structure, the ~46 Å long helical stalk is formed by a 3-stranded coiled coil of the C-terminal HRB region, which is further extended by the GCNt domain. In the hPIV3 F structure, the stalk region is formed by an ~107 Å long helical bundle, with the core 3 helix coiled coil formed by HRA segments, with the HRB segments packing along the outside. HRA is initially collapsed into a compacted set of structures located within the top half of the PIV5 head region, which, in the hPIV3 structure, erupt into long helices that would move the fusion peptide away from the viral membrane and towards the target cell. None of this HRA coiled coil is present in the prefusion structure and the structural mechanism by which this large conformational change is orchestrated remains to be better understood. Instead HRA adopts both helical and beta strand structures that are closely associated with domain III elements, the domain III core, which appear to remain relatively invariant during the conformational change. The domain III core is comprised of a 3-stranded beta sheet and associated helices. The observed HRA beta strands supplement this core beta sheet off of one edge, while the collapsed HRA helical segments pack on both sides of the domain III core, giving the impression of a well folded globular domain. However, structural database searches with the entire prefusion domain III do not show any related protein folds. The prefusion stalk region, formed by HRB, must detach from the head, dissociate the 3 helix coiled coil and flip around the head to form the post-fusion 6HB observed in the hPIV3 F structure. During this conformational rearrangement, the C-terminal residues in HRB move over 196 Å. It remains to be established how the paramyxovirus attachment proteins (HN, H or G) can activate these folding changes in F.
Direct conversion of the prefusion F to the postfusion F conformation
While the comparison of the hPIV3 F and PIV5 F structures undoubtedly provide good models for the pre- and postfusion structures of all paramyxovirus F proteins, changes in the amino acid sequences between the two proteins may also contribute to some of the smaller structural differences observed. For example, no residue-to-residue contacts between subunits are preserved in the two structures, despite the overall similar positioning of domain I. This might be a direct result of the conformational change or might also be due to sequence differences between the two proteins, resulting in slightly different packing interactions in domain I. While the crystal structures of the same F protein in the two conformations have not yet been determined, electron microscopy studies (Figure 5) have provided direct evidence that the PIV5 F can be converted from the prefusion to postfusion states, revealing single molecule structures (a postfusion golf-tee and prefusion ball-and-stem) reminiscent of the X-ray models [48]. In addition, biochemical studies of both F forms have been carried out demonstrating quantitative differences in membrane insertion and hydrophobic aggregation of the two proteins [48]. The PIV5 prefusion structure does not change shape or expose the fusion peptide after proteolytic cleavage until heated to 50 °C. The conversion of the PIV5 structure to the postfusion conformation by heat is consistent with the interpretation that it is stabilized in a metastable state that requires energy input to overcome the kinetic barrier to this change. There is no obvious need for disulfide bond rearrangements to occur in the conformational change, as has been suggested for NDV F [49]. Heating of PIV5 F to 50 °C followed by proteolytic cleavage lead to conversion of F to its postfusion form and the formation of F rosettes, indicating that the hydrophobic fusion peptides can translocate to the end of the postfusion stalk with little or no energy barrier after cleavage.
Figure 5. Electron microscopy of the PIV5 F protein and progression from the pre-fusion to the post-fusion conformation.
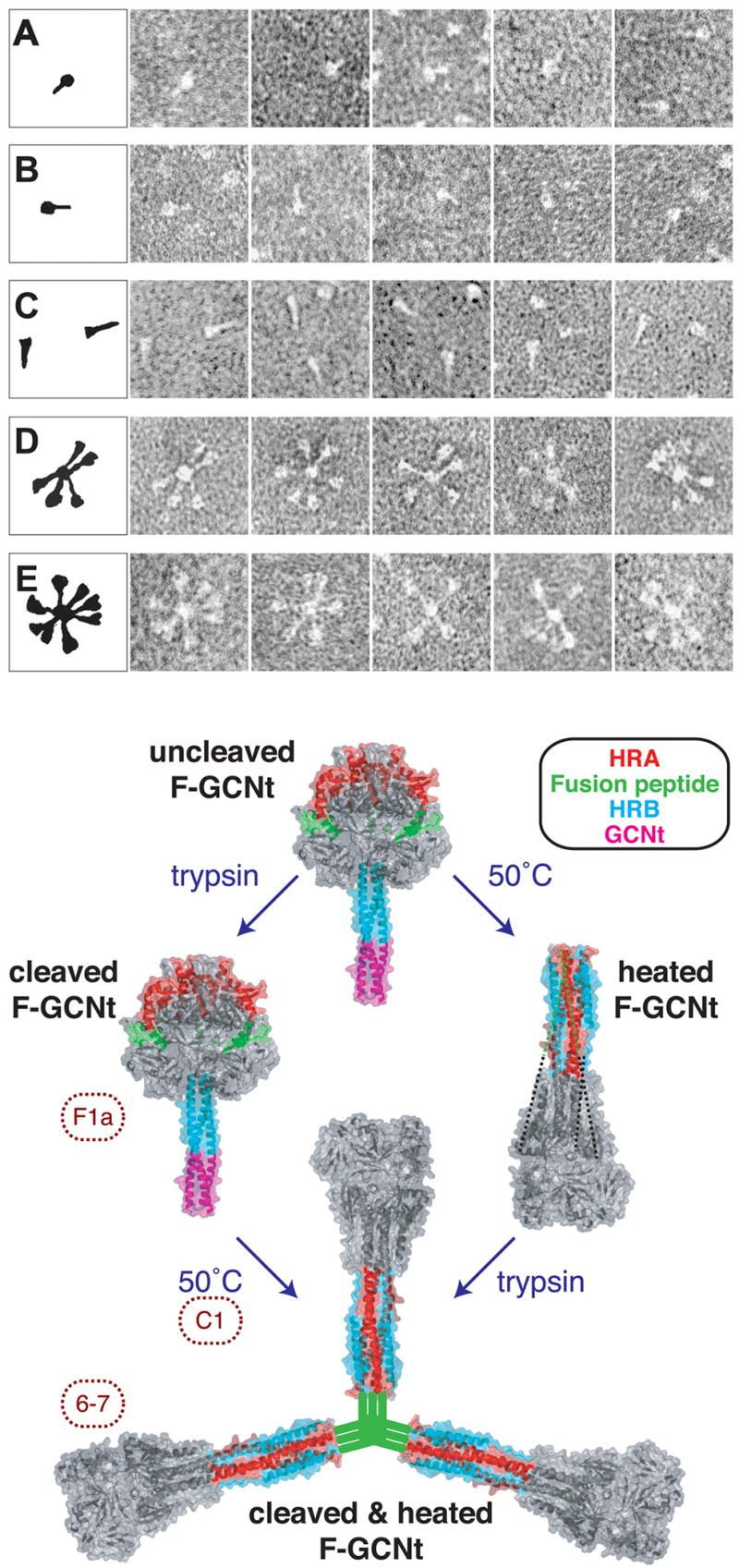
Top Panel. (A). F-GCNt, (B) F-GCNt trypsin digested, (C) F-GCNt heated to 50°C for 30 min, (D) F-GCNt digested with trypsin and then heated to 50°C for 30 min, (E) F-GCNt heated to 50°C for 30 min and then digested with trypsin. Bottom panel: The unheated and heated proteins are depicted by the PIV5 F-GCNt (Protein data bank [PDB] 2B9B) and hPIV3 solF0 (PDB 1ZTM) structures, respectively. The HRA (red), fusion peptide (green), HRB (blue), and GCNt (magenta) regions are colored. When F-GCNt is cleaved with trypsin (left side), the protein does not refold to its post-fusion conformation, but a gain of MAb F1a reactivity indicates a subtle change. When cleaved F-GCNt is heated, the protein converts to the post-fusion golf tee-like conformation, aggregates into rosettes through its fusion peptide, and gains MAb 6–7 reactivity. If C1 peptide (HRA) is added during the heating of cleaved F-GCNt, it can bind to the protein and most likely trap the pre-hairpin intermediate. When F-GCNt is heated without cleavage (right side), the protein refolds into its uncleaved post-fusion conformation. The 47 residues for which there is no interpretable density in the hPIV3 electron density map, including the residues encoding the cleavage site and fusion peptide, have been added as dotted lines. When heated F-GCNt is cleaved, the protein aggregates into rosettes. It is anticipated that the TM domain or GCNt domain would be adjacent to the fusion peptide in rosettes, but for clarity this has been omitted.
Comparison to influenza HA and other class I fusion proteins
The influenza HA and paramyxovirus F proteins bear no discernable structural relationship and appear to represent independent, convergent solutions to developing a membrane fusion machine. The fusogenic C-terminal fragment of HA, HA2, undergoes a refolding that is less involved than that observed for F. HA trimerization is maintained by a coiled coil region in HA2 that is present in both pre- and postfusion structures [5]. Refolding of an extended chain segment into a helix adds to this invariant coiled coil, projecting the fusion peptide towards the target cell membrane. That refolding in HA requires the dissociation of the HA1 head region, which becomes flexibly tethered to HA2. The HA hairpin is achieved by melting out a portion of the C-terminal prefusion coiled coil to form a turn that allows C-terminal residues to engage the newly formed coiled coiled and assemble an N-cap structure important for driving membrane fusion [5,7].
Despite the clear differences in details of their structural transitions, the comparison of HA and F allows some potentially general parallels to be drawn. In both structures, the complete folding of the postfusion coiled coil is prevented in the prefusion form and its assembly during the transition is important for the translocation of the hydrophobic fusion peptide in the direction of the target membrane. The fusion peptides are in both cases shielded from solvent by their burial at intersubunit interfaces in the prefusion form, and these interfaces change during refolding, forming new intersubunit packing interactions in F and defining the site of the reversal of direction of the C-terminal residues in HA2. Finally, other structural elements participate in prefusion interactions with both HR regions, presumably regulating the conversion to the final post-fusion hairpin.
Although it remains to be established how other class I viral fusion proteins function, it seems likely that there will be significant parallels, despite differences in the details of these structures. For example, if a general feature of class I fusion proteins is the regulation of the N-terminal coiled coil folding, enabling the projection of the fusion peptide towards the cell membrane, then this may be accomplished by different folding/refolding strategies in different fusion proteins. In the case of SIV/HIV env, structures of the N-terminal gp120 domain reveal a two-domain arrangement, with a relatively constant “outer” domain and a more flexible “inner” domain, which is adjacent to the C-terminal, fusogenic gp41 region in sequence [50–52]. Perhaps this inner domain could influence the folding of the N-terminal heptad repeat in gp41 in a manner analogous to that observed for domain III in F. Structures of env and other class I fusion proteins will undoubtedly provide greater insights into the common features of this strategy for coupling metastable protein folding to membrane fusion.
Acknowledgments
We thank past and present members of the Jardetzky and Lamb Laboratories. This research was supported in part by NIH research grants to T.S.J. and R.A.L. R.A.L. is an investigator of the Howard Hughes Medical Institute.
Footnotes
Ethics and conflicts of interest
The authors declare that this manuscript represents their original work and that they have no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Harrison SC. Principles of Virus Structure. In: Knipe DM, Howley PM, editors. Fields Virology. 5. Vol. 1 Wolters Kluwer: Lippincott WIlliams & Wilkins; 2007. pp. 59–98. [Google Scholar]
- 2.Helenius A. Virus Entry and Uncoating. In: Knipe DM, Howley PM, editors. Fields Virology. Vol. 1 Wolters Kluwer: Lippincott WIlliams & Wilkins; 2007. pp. 99–118. [Google Scholar]
- 3.Wilson IA, Skehel JJ, Wiley DC. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 Å resolution. Nature. 1981;289:366–375. doi: 10.1038/289366a0. [DOI] [PubMed] [Google Scholar]
- 4.Bullough PA, Hughson FM, Skehel JJ, Wiley DC. Structure of influenza haemagglutinin at the pH of membrane fusion. Nature. 1994;371:37–43. doi: 10.1038/371037a0. [DOI] [PubMed] [Google Scholar]
- 5.Skehel JJ, Wiley DC. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu Rev Biochem. 2000;69:531–569. doi: 10.1146/annurev.biochem.69.1.531. [DOI] [PubMed] [Google Scholar]
- 6.Chen J, Wharton SA, Weissenhorn W, Calder LJ, Hughson FM, Skehel JJ, Wiley DC. A soluble domain of the membrane-anchoring chain of influenza virus hemagglutinin (HA2) folds in Escherichia coli into the low-pH-induced conformation. Proc Natl Acad Sci USA. 1995;92:12205–12209. doi: 10.1073/pnas.92.26.12205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen J, Skehel JJ, Wiley DC. N- and C-terminal residues combine in the fusion-pH influenza hemagglutinin HA(2) subunit to form an N cap that terminates the triple-stranded coiled coil. Proc Natl Acad Sci U S A. 1999;96:8967–8972. doi: 10.1073/pnas.96.16.8967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carr CM, Kim PS. A spring-loaded mechanism for the conformational change of influenza hemagglutinin. Cell. 1993;73:823–832. doi: 10.1016/0092-8674(93)90260-w. [DOI] [PubMed] [Google Scholar]
- 9.Weissenhorn W, Carfi A, Lee KH, Skehel JJ, Wiley DC. Crystal structure of the Ebola virus membrane fusion subunit, GP2, from the envelope glycoprotein ectodomain. Mol Cell. 1998;2:605–616. doi: 10.1016/s1097-2765(00)80159-8. [DOI] [PubMed] [Google Scholar]
- 10.Malashkevich VN, Schneider BJ, McNally ML, Milhollen MA, Pang JX, Kim PS. Core structure of the envelope glycoprotein GP2 from Ebola virus at 1.9 Å resolution. Proc Natl Acad Sci U S A. 1999;96:2662–2667. doi: 10.1073/pnas.96.6.2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baker K, Dutch RE, Lamb RA, Jardetzky TS. Structural basis for paramyxovirus-mediated membrane fusion. Molecular Cell. 1999;3:309–319. doi: 10.1016/s1097-2765(00)80458-x. [DOI] [PubMed] [Google Scholar]
- 12.Zhao X, Singh M, Malashkevich VN, Kim PS. Structural characterization of the human respiratory syncytial virus fusion protein core. Proc Natl Acad Sci U S A. 2000;97:14172–14177. doi: 10.1073/pnas.260499197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chan DC, Fass D, Berger JM, Kim PS. Core structure of gp41 from the HIV envelope glycoprotein. Cell. 1997;89:263–273. doi: 10.1016/s0092-8674(00)80205-6. [DOI] [PubMed] [Google Scholar]
- 14.Weissenhorn W, Dessen A, Harrison SC, Skehel JJ, Wiley DC. Atomic structure of the ectodomain from HIV-1 gp41. Nature. 1997;387:426–430. doi: 10.1038/387426a0. [DOI] [PubMed] [Google Scholar]
- 15.Caffrey M, Cai M, Kaufman J, Stahl SJ, Gronenborn AM, Clore GM. Three-dimensional solution structure of the 44 kDa ectodomain of SIV gp41. EMBO J. 1998;17:4572–4584. doi: 10.1093/emboj/17.16.4572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Russell CJ, Luque LE. The structural basis of paramyxovirus invasion. Trends Microbiol. 2006;14:243–246. doi: 10.1016/j.tim.2006.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kielian M, Rey FA. Virus membrane-fusion proteins: more than one way to make a hairpin. Nat Rev Microbiol. 2006;4:67–76. doi: 10.1038/nrmicro1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kielian M. Class II virus membrane fusion proteins. Virology. 2006;344:38–47. doi: 10.1016/j.virol.2005.09.036. [DOI] [PubMed] [Google Scholar]
- 19.Earp LJ, Delos SE, Park HE, White JM. The many mechanisms of viral membrane fusion proteins. Curr Top Microbiol Immunol. 2005;285:25–66. doi: 10.1007/3-540-26764-6_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jardetzky TS, Lamb RA. Virology: a class act. Nature. 2004;427:307–308. doi: 10.1038/427307a. [DOI] [PubMed] [Google Scholar]
- 21.Heldwein EE, Lou H, Bender FC, Cohen GH, Eisenberg RJ, Harrison SC. Crystal structure of glycoprotein B from herpes simplex virus 1. Science. 2006;313:217–220. doi: 10.1126/science.1126548. [DOI] [PubMed] [Google Scholar]
- 22.Roche S, Bressanelli S, Rey FA, Gaudin Y. Crystal structure of the low-pH form of the vesicular stomatitis virus glycoprotein G. Science. 2006;313:187–191. doi: 10.1126/science.1127683. [DOI] [PubMed] [Google Scholar]
- 23.Roche S, Rey FA, Gaudin Y, Bressanelli S. Structure of the prefusion form of the vesicular stomatitis virus glycoprotein G. Science. 2007;315:843–848. doi: 10.1126/science.1135710. [DOI] [PubMed] [Google Scholar]
- 24.Chandran K, Sullivan NJ, Felbor U, Whelan SP, Cunningham JM. Endosomal proteolysis of the Ebola virus glycoprotein is necessary for infection. Science. 2005;308:1643–1645. doi: 10.1126/science.1110656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lamb RA, Parks GD. Paramyxoviridae: The viruses and their replication. In: Knipe DM, Howley PM, editors. Fields Virology. 5. Vol. 1 Wolters Kluwer: Lippincott WIlliams & Wilkins; 2007. pp. 1449–1496. [Google Scholar]
- 26.Karron RA, Collins PL. Parainfluenza Viruses. In: Knipe DM, Howley PM, editors. Fields Virology. 5. Vol. 1 Wolters Kluwer: Lippincott WIlliams & Wilkins; 2007. pp. 1497–1526. [Google Scholar]
- 27.Carbone KM, Rubin SA. Mumps Virus. In: Knipe DM, Howley PM, editors. Fields Virology. 5. Vol. 1 Wolters Kluwer: Lippincott WIlliams & Wilkins; 2007. pp. 1527–1550. [Google Scholar]
- 28.Mackenzie JS, Field HE. Emerging encephalitogenic viruses: lyssaviruses and henipaviruses transmitted by frugivorous bats. Arch Virol Suppl. 2004:97–111. doi: 10.1007/978-3-7091-0572-6_8. [DOI] [PubMed] [Google Scholar]
- 29.Field H, Young P, Yob JM, Mills J, Hall L, Mackenzie J. The natural history of Hendra and Nipah viruses. Microbes Infect. 2001;3:307–314. doi: 10.1016/s1286-4579(01)01384-3. [DOI] [PubMed] [Google Scholar]
- 30.Openshaw PJ. Potential therapeutic implications of new insights into respiratory syncytial virus disease. Respir Res. 2002;3:S15–S20. doi: 10.1186/rr184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dutch RE, Jardetzky TS, Lamb RA. Virus membrane fusion proteins: biological machines that undergo a metamorphosis. Biosci Rep. 2000;20:597–612. doi: 10.1023/a:1010467106305. [DOI] [PubMed] [Google Scholar]
- 32.Morrison TG. Structure and function of a paramyxovirus fusion protein. Biochim Biophys Acta. 2003;1614:73–84. doi: 10.1016/s0005-2736(03)00164-0. [DOI] [PubMed] [Google Scholar]
- 33.Morrison TG. The three faces of paramyxovirus attachment proteins. Trends Microbiol. 2001;9:103–105. doi: 10.1016/s0966-842x(01)01959-x. [DOI] [PubMed] [Google Scholar]
- 34.Oldstone MB, Homann D, Lewicki H, Stevenson D. One, two, or three step: measles virus receptor dance. Virology. 2002;299:162–163. doi: 10.1006/viro.2002.1507. [DOI] [PubMed] [Google Scholar]
- 35.Yanagi Y, Ono N, Tatsuo H, Hashimoto K, Minagawa H. Measles virus receptor SLAM (CD150) Virology. 2002;299:155–161. doi: 10.1006/viro.2002.1471. [DOI] [PubMed] [Google Scholar]
- 36.Feldman SA, Hendry RM, Beeler JA. Identification of a linear heparin binding domain for human respiratory syncytial virus attachment glycoprotein G. J Virol. 1999;73:6610–6617. doi: 10.1128/jvi.73.8.6610-6617.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Russell CJ, Jardetzky TS, Lamb RA. Membrane fusion machines of paramyxoviruses: capture of intermediates of fusion. Embo J. 2001;20:4024–4034. doi: 10.1093/emboj/20.15.4024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ludwig K, Baljinnyam B, Herrmann A, Bottcher C. The 3D structure of the fusion primed Sendai F-protein determined by electron cryomicroscopy. Embo J. 2003;22:3761–3771. doi: 10.1093/emboj/cdg385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen L, Colman PM, Cosgrove LJ, Lawrence MC, Lawrence LJ, Tulloch PA, Gorman JJ. Cloning, expression, and crystallization of the fusion protein of Newcastle disease virus. Virology. 2001;290:290–299. doi: 10.1006/viro.2001.1172. [DOI] [PubMed] [Google Scholar]
- 40.Chen L, Gorman JJ, McKimm-Breschkin J, Lawrence LJ, Tulloch PA, Smith BJ, Colman PM, Lawrence MC. The structure of the fusion glycoprotein of Newcastle disease virus suggests a novel paradigm for the molecular mechanism of membrane fusion. Structure (Camb) 2001;9:255–266. doi: 10.1016/s0969-2126(01)00581-0. [DOI] [PubMed] [Google Scholar]
- 41.Colman PM, Lawrence MC. The structural biology of type I viral membrane fusion. Nat Rev Mol Cell Biol. 2003;4:309–319. doi: 10.1038/nrm1076. [DOI] [PubMed] [Google Scholar]
- 42.Yin HS, Paterson RG, Wen X, Lamb RA, Jardetzky TS. Structure of the uncleaved ectodomain of the paramyxovirus (hPIV3) fusion protein. Proc Natl Acad Sci U S A. 2005;102:9288–9293. doi: 10.1073/pnas.0503989102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Russell CJ, Jardetzky TS, Lamb RA. Conserved glycine residues in the fusion peptide of the paramyxovirus fusion protein regulate activation of the native state. J Virol. 2004;78:13727–13742. doi: 10.1128/JVI.78.24.13727-13742.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Russell CJ, Kantor KL, Jardetzky TS, Lamb RA. A dual-functional paramyxovirus F protein regulatory switch segment: activation and membrane fusion. J Cell Biol. 2003;163:363–374. doi: 10.1083/jcb.200305130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Harbury PB, Zhang T, Kim PS, Alber T. A switch between two-, three-, and four-stranded coiled coils in GCN4 leucine zipper mutants. Science. 1993;262:1401–1407. doi: 10.1126/science.8248779. [DOI] [PubMed] [Google Scholar]
- 46.Harbury PB, Kim PS, Alber T. Crystal structure of an isoleucine-zipper trimer. Nature. 1994;371:80–83. doi: 10.1038/371080a0. [DOI] [PubMed] [Google Scholar]
- 47.Yin HS, Wen X, Paterson RG, Lamb RA, Jardetzky TS. Structure of the parainfluenza virus 5 F protein in its metastable, prefusion conformation. Nature. 2006;439:38–44. doi: 10.1038/nature04322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Connolly SA, Leser GP, Yin HS, Jardetzky TS, Lamb RA. Refolding of a paramyxovirus F protein from prefusion to postfusion conformations observed by liposome binding and electron microscopy. Proc Natl Acad Sci U S A. 2006;103:17903–17908. doi: 10.1073/pnas.0608678103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jain S, McGinnes LW, Morrison TG. Thiol/disulfide exchange is required for membrane fusion directed by the Newcastle disease virus fusion protein. J Virol. 2007;81:2328–2339. doi: 10.1128/JVI.01940-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kwong PD, Wyatt R, Robinson J, Sweet RW, Sodroski J, Hendrickson WA. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature. 1998;393:648–659. doi: 10.1038/31405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen B, Vogan EM, Gong H, Skehel JJ, Wiley DC, Harrison SC. Structure of an unliganded simian immunodeficiency virus gp120 core. Nature. 2005;433:834–841. doi: 10.1038/nature03327. [DOI] [PubMed] [Google Scholar]
- 52.Huang CC, Tang M, Zhang MY, Majeed S, Montabana E, Stanfield RL, Dimitrov DS, Korber B, Sodroski J, Wilson IA, et al. Structure of a V3-containing HIV-1 gp120 core. Science. 2005;310:1025–1028. doi: 10.1126/science.1118398. [DOI] [PMC free article] [PubMed] [Google Scholar]