

Abstract
Mice heterozygous for the ApcMin (Min) mutation develop adenomas throughout the intestinal tract. Apc is believed to be involved in cell migration, adhesion, and polarity. Adenoma multiplicity and growth rate are modulated by an unlinked modifier locus, Mom1. The secretory phospholipase Pla2g2a is a candidate for Mom1. Here, we investigate the range of action of Apc and Mom1. Analysis of chimeric Min mice indicates that the actions of both Apc and Mom1 are localized within the cell lineage that gives rise to intestinal tumors.
Humans carrying a germline mutation in the APC (adenomatous polyposis coli) gene develop multitudes of colonic adenomas (1, 2). Somatic mutation of APC occurs frequently in both familial and sporadic colonic adenomas (3, 4). Similarly, mice carrying Min (multiple intestinal neoplasia), a nonsense mutation at codon 850 in the mouse Apc gene, develop adenomas throughout the intestinal tract (5, 6). The adenomas in Min/+ mice also show loss of the wild-type allele of Apc (7, 8). The APC polypeptide has been detected in a number of intracellular locations (9–12). These results support the idea that Apc is a cell-autonomous tumor suppressor gene.
APC is believed to play a role in maintaining epithelial organization and integrity by indirectly modulating the activity of E-cadherin in cell adhesion, migration, and cell polarity (13). Thus, despite its intracellular localization, APC may have intercellular effects. Loss of APC in a single cell may influence junctions with adjacent cells and the integrity of the entire crypt epithelium.
The study of Min has been enhanced by the identification of loci that modify the intestinal tumor phenotype of Min mice. An understanding of the molecular nature, function, and mechanism of action of these modifier loci will provide new molecular targets for chemoprevention and chemotherapy. Genetic variability between inbred strains of mice such as AKR/J (AKR) and C57BL/6J (B6) has permitted the identification of one such modifier locus, Modifier of Min-1 (Mom1) (14, 15). B6 Mom1B6/B6 Min/+ mice develop on average 26.1 tumors at 120 days of age (16). Tumor multiplicity drops to 7.8 in age-matched B6 Mom1AKR/AKR Min/+ mice (16). Mom1 acts semidominantly, affecting the net growth rate, size, and number of Min-induced adenomas (16). Intestinal tumors from Mom1AKR/B6 Min/+ mice show no evidence of loss of heterozygosity at markers flanking Mom1 (17, 18). This observation indicates that Mom1 may not be a classical, cell-autonomous tumor suppressor gene.
Pla2g2a, encoding a secretory phospholipase, has been proposed as a candidate for Mom1 (19). Strains such as AKR, which carry a resistance allele at Mom1, carry a wild-type allele at Pla2g2a, whereas strains such as B6, which carry a sensitivity allele at Mom1, carry a mutant allele at Pla2g2a (18, 19). This phospholipase, primarily known for promoting inflammation through arachidonic acid metabolites, is secreted by mast cells, neutrophils, fibroblasts, and Paneth cells (20, 21).
A gene may influence tumorigenesis either by acting within or between tumor cells, or by acting either locally or systemically from outside the tumor. A priori, one can hypothesize either cell-autonomous or non-cell-autonomous action for Apc and for Mom1. Using intestinal isografts, we have shown that neither Apc nor Mom1 acts systemically to influence tumor development (22). Here, we use aggregation chimeras and a histochemical marker to investigate the range of action of Apc and Mom1 in intestinal neoplasia.
MATERIALS AND METHODS
Mice.
Mice were bred and housed at the McArdle Laboratory for Cancer Research. The B6 mice used were derived from B6 mice obtained from The Jackson Laboratory. B6 Min/+ mice were generated by crossing B6 females to congenic B6 Min/+ males. B6 Min/+ mice and B6.Mom1AKR/AKR Min/+ were identified by a PCR-based assay described previously from DNA isolated from blood (15).
A B6 R26/+ line was generated from (129 × B6) hybrid ROSA26 (R26) mice by crossing B6 females to R26/+ males from each backcross generation. The B6 R26/+ mice used in these experiments were generated by crossing B6 females to B6 R26/+ males. R26/+ mice were identified by mixing 50 μl of blood with 50 μl of 5-bromo-4-chloro-3-indolyl β-d-galactopyranoside (X-Gal) stain (1.6 μg/ml X-Gal/5 mM potassium ferricyanide/5.75 mM potassium ferrocyanide/2 mM MgCl2 in PBS with 2.5% DMSO). Samples were incubated at 37°C for at least 2 hr and then centrifuged for 1 min at 650 × g to pellet the erythrocytes. The serum from mice carrying R26 stains blue, whereas that of non-R26 mice is yellow.
The B6.Mom1AKR/AKR Min/+ and +/+ mice were obtained from our B6.Mom1AKR line segregating for Min. This line carries a 35-cM region of mouse chromosome 4, between the markers D4Mit9 and D4Mit180, from the AKR strain (16). Mom1 is the only modifier in this line (16). The mice used were generated by crossing B6.Mom1AKR/AKR +/+ mice with B6.Mom1AKR/AKR Min/+ mice in each parental orientation.
Generation of Chimeras.
Chimeras were generated by morula aggregation as described by Hogan et al. (23). The composition of each chimera is described by indicating the genotypes of the two embryos that produced it separated by a double arrow: genotype 1 ↔ genotype 2. For production of Min/+ ↔ Apc+/+ R26/+ chimeras, single embryos produced in a B6 × B6 Min/+ cross were aggregated with single embryos produced in a B6 × B6 R26/+ cross. Mice recovered from such aggregations that carried both Min and R26 were necessarily Min/+ ↔ Apc+/+ R26/+ chimeras.
To produce Min/+ R26/+ ↔ Apc+/+ +/+ chimeras, individual embryos from a Min/+ × B6 R26/+ cross were aggregated with B6 +/+ embryos. Mice recovered from these aggregations that were chimeric for Min and R26 on the basis of allelic ratios were Min/+ R26/+ ↔ Apc+/+ +/+ chimeras.
The Mom1B6/B6 Min/+ ↔ Mom1AKR/AKR +/+ and Mom1AKR/AKR Min/+ ↔ Mom1B6/B6 +/+ chimeras were produced by aggregating individual embryos from a Min/+ ×B6 R26/+ cross with single embryos from a Mom1AKR/AKR Min/+ × Mom1AKR/AKR +/+ cross. Mice were genotyped for Min, R26, and Mom1 to identify chimeras of interest.
DNA was isolated from blood as previously described (15). Samples were amplified in duplicate. For each assay, the PCR products were separated on a 7.5% denaturing polyacrylamide gel. With a PhosphorImager (Molecular Dynamics), the products from each allele were quantitated. By comparing the ratio of alleles at a particular locus for each sample with the average allelic ratio for the control samples, we could determine whether a given animal was chimeric.
To identify mice that were chimeric for Min, we amplified DNA from each animal, using a quantitative PCR-based assay described previously (8). The controls used in this assay were DNA samples isolated from blood of Min/+ and +/+ animals.
To identify mice that were chimeric for R26, we typed each animal at D6Mit36. For this marker, the reverse primer was 32P end-labeled as described previously by Luongo et al. (8) except that reactions contained 1.0 μM primer and 0.17 μM [γ-32P]ATP (6,000 Ci/mmol; 1 Ci = 37 GBq; Dupont).
Each sample, 4 μl of DNA prepared from blood, was amplified in a 20-μl reaction containing 0.05 μM unlabeled reverse primer; 0.1 μM forward primer; 0.28 μl end-labeling reaction (0.001 μM labeled reverse primer); 240 μM concentrations (each) of dATP, dCTP, dGTP, and dTTP; 1.85 mM MgCl2; 10 mM Tris⋅HCl (pH 9.0 at 25°C); 50 M KCl; 0.1% Triton X-100; and 1.6 units of Taq polymerase (Promega). Samples were amplified for 30 cycles as described (8, 24). The controls used in this assay were DNA samples isolated from blood of R26/+ (129/B6) and +/+ (B6/B6) animals.
Animals potentially chimeric with respect to Mom1 were typed at D4Mit13, which is tightly linked to Mom1. For this marker, the DNA samples were amplified as described for D6Mit36 above, except that 1.55 mM MgCl2 was used. The controls used in this assay were DNA samples isolated from blood of Mom1AKR/AKR, Mom1AKR/B6, and Mom1B6/B6 animals.
Isolation of DNA from Fixed and Stained Intestinal Samples.
Sections of normal intestinal tissue were excised after staining with X-Gal. Excision was performed under the dissecting microscope (×30) to ensure that the sections removed did not contain adenomas, as this would skew the results of the subsequent determination of the Min/Apc+ allelic ratio. One proximal and one distal section were excised from each of the four segments of the intestine that are scored for tumors (5). DNA was isolated from each excised section as described (22).
Determining the Percentage of Min/+ Cells in Chimeric Intestines.
DNA isolated from the excised pieces of intestines (2 μl) was used in the quantitative assay for genotyping at the Apc locus. DNAs isolated from intestines of Min/+ and +/+ mice were used as controls. The allelic ratios of the two DNA samples from each segment were averaged to obtain a single allelic ratio for each segment. If the allelic ratios of the two samples varied by more than 10%, the amplification was repeated. The average allelic ratios of the four scored segments were averaged to obtain a single average allelic ratio for the entire scored area.
Scoring of Tumors.
All mice were killed by CO2 asphyxiation. The intestinal tract was removed, prepared, and scored for tumors as described (5). In this method, three 4-cm sections of the small intestine and the entire large intestine were examined for tumors.
Whole-Mount Staining of Intestines with X-Gal.
After the tumors were counted, intestines from chimeras were fixed flat in 0.2% glutaraldehyde for 30 min. Intestines were then incubated in wash buffer and stained overnight in an X-Gal solution at 37°C as described by Sanes et al. (25).
Statistics.
All statistical comparisons were performed with the Wilcoxon rank sum test. One-sided P values are given.
RESULTS AND DISCUSSION
ROSA26 as a Cellular Lineage Marker.
We used the lacZ transgenic line ROSA26 (R26) as a cell lineage marker in chimeric mice (26). R26/+ mice show high levels of β-galactosidase activity in all cells of all adult tissues examined thus far, including the epithelium of the small and large intestine (Fig. 1A and data not shown; ref. 27). To determine whether lacZ is expressed in Min-induced intestinal adenomas, we crossed R26/+ females with Min/+ males. Tumors in R26/+ Min/+ progeny showed high levels of β-galactosidase activity (Fig. 1B). In tumors greater than 1 mm in diameter, the central region failed to stain in whole mounts, presumably due to insufficient penetration of the X-Gal. Therefore, we limited subsequent analyses to tumors less than 1 mm in diameter.
Figure 1.
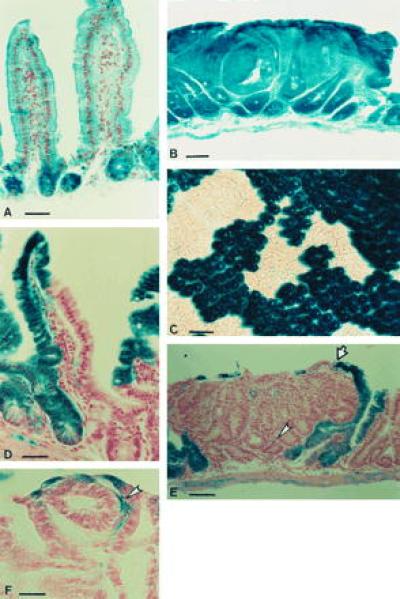
X-Gal staining in ROSA26 and chimeric mice. After X-Gal staining, samples were post-fixed in 10% neutral buffered formalin, cleared in 70% ethanol, processed, and serially sectioned at 10 μm. Sections were counterstained with Nuclear Fast Red (A, B, and D–F). In R26 mice, staining is observed in all cells of the small intestinal epithelium (A). The β-galactosidase activity is maintained in intestinal adenomas from R26/+ Min/+ mice (B). Crypts in the colon of R26/+ ↔ +/+ mice are monoclonal (C). Villi in the small intestine of R26/+ ↔ +/+ mice are polyclonal (D). Epithelial cells within tumors in R26/+ ↔ Min/+ mice are derived from the Min/+ lineage and are therefore unstained with X-Gal. The normal epithelial layer encapsulating the tumors is polyclonal (indicated by arrows). Scattered X-Gal-positive cells (indicated by arrowheads) in these tumors are lymphocytes and stromal cells (E, F). [Bars = 40 μm (A, B, and E), 80 μm (C), 20 μm (D), and 15 μm (F).]
Next, we confirmed that the R26 line did not contain modifiers of the Min phenotype. This step is essential because R26 was generated in the 129/Sv strain, which carries an allele of at least one unmapped modifier of Min (18). R26/+ females from the third to fifth backcross generation to B6 were mated to Min/+ males. The average tumor multiplicity in resulting Min/+ mice was 26.9. This value did not differ significantly from the average of 27.2 tumors in age-matched B6 Min/+ mice (P = 0.48). Furthermore, the average tumor multiplicity of 25.3 in R26/+ Min/+ mice did not differ significantly from the average of 27.7 tumors in their non-R26 Min/+ siblings (P = 0.34). Thus, after several backcross generations, the R26 line did not contain any dominant modifier alleles from 129/Sv either linked or unlinked to R26. Nevertheless, we backcrossed the R26 strain to the N9 generation before generating chimeras.
To genotype chimeric mice, we needed a DNA marker to identify carriers of R26. Therefore, we used mice from the early backcross generations to B6 to map the insertion site of R26. Evidence for linkage was detected with several markers on chromosome 6 (data not shown). The marker D6Mit36 is tightly linked to the insertion site and was, therefore, used for identification of mice chimeric for R26.
Crypt Clonality and Patch Size in Chimeras.
In the R26 ↔ non-R26 chimeras, all cells of each intestinal crypt either stained blue (R26/+) or were unstained (non-R26) after incubation with X-Gal (Fig. 1C). These data are consistent with the idea that intestinal crypts are monoclonal (28). The X-Gal staining pattern revealed that in the intestines of chimeric mice, patches of R26 and non-R26 crypts were small, often containing one crypt or a few crypts, and highly intermixed (Fig. 1C). Small intestinal villi, supplied with cells by 6–10 surrounding crypts, are polyclonal (Fig. 1D).
Analysis of Gene Action in the Intestinal Epithelium and Neoplasm.
The interpretation of the experiments presented here must take into account the biology of the intestinal epithelium. The villi of the small intestine and surface epithelial cuffs of the large intestine are supplied with differentiated cells that emerge from the crypts (29). Near the base of each crypt reside 4–16 multipotential stem cells, which give rise to cells that proliferate, migrate, and differentiate to provide cells for the surface epithelium (30, 31). Cells also migrate to the base of the crypt after commitment to Paneth cell differentiation. Because the cohort of 4–16 multipotential stem cells in each crypt is derived from a single progenitor, each intestinal crypt is monoclonal (28, 32). Thus, each crypt gives rise to a crypt lineage, a contiguous somatic clone of cells extending from the base of the crypt, through the crypt, and in relatively straight lines up to the tips of 6–10 surrounding villi (33).
Because of this clonal architecture, it is difficult to determine whether a gene acts between cells derived from a single crypt or whether it acts in a strictly cell-autonomous fashion. Using chimeric mice, one can resolve the issue of autonomy only to the level of the crypt lineage (crypt lineage autonomous).
One important unresolved issue is whether intestinal tumors, including those in Min mice, are monoclonal or polyclonal in origin (34–36). However, this uncertainty does not impact the analysis of the action of Apc and Mom1.
Action of Apc Is Localized to the Crypt Lineage.
To investigate the interaction between Min/+ and +/+ cells, we generated 17 Min/+ ↔ +/+ chimeras. In 12 chimeras, R26 was present either as a cellular marker for the +/+ population (seven Min/+ ↔ +/+ R26/+) or else for the Min/+ population (five Min/+ R26/+ ↔ +/+). Five Min/+ ↔ +/+ mice did not contain R26.
To determine whether the +/+ cells affected the ability of the adjacent Min/+ intestinal cells to form tumors, we examined the relationship between tumor multiplicity and the percentage of Min/+ cells within the intestines of these chimeric mice. If the +/+ cells inhibit tumor formation, then the tumor multiplicity in the chimeric mice would be smaller than the fraction of Min/+ cells would predict. Because there is no detectable systemic effect of either the wild type or the Min allele of Apc on tumor multiplicity (22), only the percentages of Min/+ and +/+ cells within the intestine need to be considered for this analysis. These percentages and the raw tumor multiplicity data are shown in Table 1. To compare the tumor multiplicity data from the chimeras with those of control animals, we normalized these data to calculate an expected tumor multiplicity for a composition of 100% Min/+ cells. The distribution of normalized tumor multiplicities is shown in Fig. 2. Because tumor multiplicity in B6 Min/+ mice does not change between 80 and 120 days of age (16), the data from these two groups were pooled. The average tumor multiplicity in these 13 chimeras is 27.7. This value does not differ significantly from the average tumor multiplicity of 25.3 observed in a control population of non-chimeric B6 Min/+ mice (P = 0.23) The +/+ cells do not detectably influence the ability of Min/+ cells to form tumors.
Table 1.
Tumor multiplicity in 13 Min/+ ↔ +/+ chimeras
| % Min/+ intestinal cells | Raw tumor multiplicity |
|---|---|
| 100* | 16 |
| 99 | 8 |
| 96 | 21 |
| 92 | 38 |
| 87 | 25 |
| 86 | 3 |
| 84 | 25 |
| 62 | 26 |
| 62 | 25 |
| 57 | 31 |
| 55 | 24 |
| 40 | 6 |
| 34 | 1 |
This animal showed chimerism in its blood DNA (85% Min/+).
Figure 2.
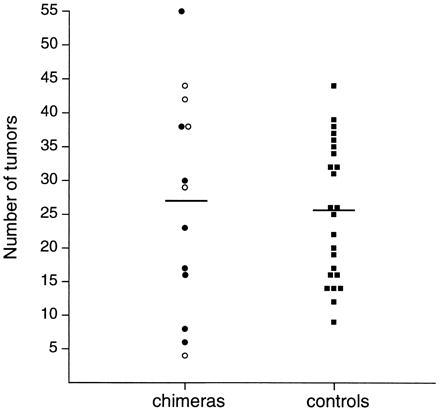
Normalized tumor multiplicity in Min/+ ↔ +/+ chimeras. Each circle or square indicates the tumor multiplicity in a single animal. The chimeras were killed at 80 days of age (•) or 120 days (○). B6 Min/+ controls were killed at 80 days of age (▪). The horizontal bars indicate the positions of the means.
To determine whether +/+ epithelial cells can be recruited into the tumor during Min-induced neoplasia, we examined tumors from chimeric mice in which R26 marked either the Min/+ or the +/+ population. Only tumors at borders between patches of Min/+ and +/+ cells were examined. Scoring serial sections of 40 tumors revealed no evidence that +/+ epithelial cells could contribute to or be recruited into Min-induced tumors. This finding also suggests that normal, differentiated +/+ epithelial cells do not often become trapped within the tumor, implying that the differentiated epithelial cells found in the adenomas of Min mice (14) are derived from the tumor lineage and that Min-induced tumors are derived from a multipotential stem cell population. In these chimeras, asymmetric with respect to the Apc genotype, tumors, whether monoclonal or polyclonal, are derived from a single lineage (Min/+).
We observed that stromal cells, lymphoid cells, and vascular endothelial cells within the tumors as well as cells in the normal epithelial monolayer encapsulating the tumors were often derived from the +/+ lineage. The external monolayer is polyclonal, consisting of cells derived from normal crypts adjacent to the tumor (Fig. 1E).
To explore the possibility that Min/+ ↔ +/+ chimeras may have an extended lifespan relative to B6 Min/+ mice, four chimeras were killed only when moribund. One animal, with 93% Min/+ intestinal cells, had 15 tumors when killed at 170 days of age. The lifespan and tumor multiplicity of this chimera is within the normal distribution of B6 Min/+ mice (5, 16). Two mice with only 2–3% Min/+ cells in the intestine were killed at 408 days of age. One mouse had one tumor and the other was tumor-free. Chimerism in these mice was confirmed by genotypic analysis of DNA from blood. The fourth long-lived chimera, having 39% Min/+ cells within the intestine, was sacrificed at 561 days of age and had two tumors. The low tumor multiplicities in these three chimeras may be due to the fact that a small percentage of cells carry Min and thus have tumorigenic potential. Another possible interpretation of these observations is that tumors in Min mice are polyclonal in origin (36). This hypothesis would predict a nonlinear relationship between the percentage of Min/+ cells and tumor multiplicity Min/+ ↔ +/+ chimeras, especially those with a low percentage of Min/+ cells. Analysis of tumor multiplicities in the 80- and 120-day-old chimeras, in which 11 of 12 mice have greater than 40% Min/+ cells in the intestine, suggests that when at least a certain percentage of Min/+ cells are present, the relationship between the percentage of Min/+ cells and tumor multiplicity is monotonic.
Analysis of Min/+ ↔ +/+ chimeric mice indicates that the wild type and the Min allele of Apc each act autonomously within the crypt lineage. The subcellular localization and tumor-specific allelic loss of APC suggests that APC may be a classical cell-autonomous tumor suppressor (3, 4, 7–12). Further experiments are required to determine the range of action of Apc within the crypt lineage.
Localized Action of Mom1.
To investigate the action of Mom1, we generated mice that were chimeric with respect to Mom1 genotype. Three Mom1AKR/AKR Min/+ ↔ Mom1B6/B6 +/+ chimeras were analyzed. The percentage of Mom1AKR/AKR Min/+ intestinal cells and the number of tumors in these chimeras were determined. Although the percentage of Min/+ cells in the intestines of each of these chimeras was high (67%, 80%, and 95%), the chimeras developed few tumors (four, one, and two, respectively). Normalizing these to 100% Min/+ cells, we obtained a normalized average tumor multiplicity of 3.1. This value is not significantly different from the average tumor multiplicity of 4.4 in age-matched Mom1AKR/AKR Min/+ nonchimeric control mice (P = 0.22; Fig. 3). By contrast, the normalized average of 3.1 for the chimeras does differ significantly from the average tumor multiplicity of 25.3 in age-matched Mom1B6/B6 Min/+ nonchimeric controls (P = 2.7 × 10−3; Fig. 3). These comparisons indicate that the Mom1AKR allele within the Min/+ lineage results in a reduction in the number of Min-induced tumors in these mice.
Figure 3.
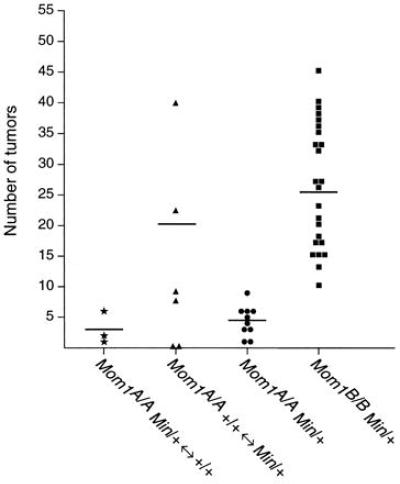
Normalized tumor multiplicity in Mom1 chimeras. Each symbol indicates the tumor number in a single animal. ★, Mom1B6/B6 +/+ ↔ Mom1AKR/AKR Min/+ mice; ▴, Mom1AKR/AKR +/+ ↔ Mom1B6/B6 Min/+ mice; •, a control population of age-matched Mom1AKR/AKR Min/+ mice; ▪, a control population of Mom1B6/B6 Min/+ mice. All mice were killed at 80 days of age. The horizontal bars indicate the positions of the means.
Six Mom1B6/B6 Min/+ ↔ Mom1AKR/AKR +/+ chimeras were analyzed to determine whether Mom1AKR can act at a distance. The percentage of Mom1B6/B6 Min/+ cells and the number of tumors in each of these chimeras were determined. In four of these chimeras, the percentage of Min/+ cells was relatively high (53%, 63%, 67%, and 81%). The observed tumor multiplicity of these mice was 4, 15, 6, and 28, respectively. As compared with the Mom1AKR/AKR Min/+ ↔ Mom1B6/B6 +/+ chimeras above, these four chimeras (Mom1B6/B6 Min/+ ↔ Mom1AKR/AKR +/+) have a lower average percentage of Min/+ cells (66% vs. 81%) but a higher average tumor multiplicity (13.3 vs. 2.3). Normalizing the data to 100% Min/+ composition, we calculate an average tumor multiplicity of 20.0. The observed normalized value of 20.0 is significantly different from the average tumor multiplicity of 4.4 in age-matched Mom1AKR/AKR Min/+ control (P = 5 × 10−3), but not from the average tumor multiplicity of 25.3 in a population of age-matched Mom1B6/B6 Min/+ controls (P = 0.22; Fig. 3). These comparisons demonstrate that Mom1AKR in the +/+ lineage does not reduce the number of tumors that develop in the juxtaposed Min/+ lineage (Mom1B6/B6). Because these two genotypic lineages are highly intermixed in the chimeras (see Fig. 1C), we conclude that Mom1 acts in a localized fashion to influence tumor multiplicity, not freely over substantial distances within the intestine. Models of partial Mom1AKR action over a range beyond a crypt lineage are not excluded by these experiments.
Two Mom1B6/B6 Min/+ ↔ Mom1AKR/AKR +/+ chimeras contained only a small proportion of Min/+ cells within the intestine (5% and 8%). These mice developed very few tumors (zero and one, respectively). As discussed above, there are several possible explanations for the lower-than-expected tumor multiplicity in these mice.
Analysis of tumor multiplicity in chimeric mice suggests that the action of Mom1 is largely, if not entirely, autonomous to the crypt lineage. Markers that flank the Mom1 locus maintain heterozygosity in Min-induced intestinal adenomas in Mom1AKR/B6 Min/+ mice (17, 18). One interpretation of these results is that Mom1 is not a classical tumor suppressor gene. This interpretation is consistent with the idea that Mom1 encodes a secreted factor not produced by the tumor and the prediction of a non-cell-autonomous mode of action for Mom1. However, it is instead possible that the observed maintenance of heterozygosity in the Mom1 region reflects the fact that somatic mutation of Mom1 occurs through small deletion or intragenic point mutation. Such events would not have been detected in the analysis of genetic markers that flank Mom1.
Mom1 Action and Pla2g2a.
Intriguing correlative and mapping data support the idea that Mom1 may encode Pla2g2a (18, 19). However, this hypothesis has not yet been rigorously tested (13, 18). Nonautonomous action for Mom1 is predicted by the hypothesis that Mom1 encodes a secreted phospholipase (18, 19). It is unclear how the level of Pla2g2a secreted locally by the Paneth cells within the crypt could influence Min-induced tumorigenesis. On the one hand, if tumors in Min mice arise from the single, ultimate stem cell within the crypt, the only local source of Pla2g2a would be the small number of differentiated Paneth cells within the tumor. In this scenario, one would expect selection for allelic loss at Pla2g2a. However, heterozygosity at Pla2g2a is maintained in adenomas from Min mice (18). On the other hand, if tumors in Min mice arise from just one of several multipotent precursor cells present within the crypt, a second local source of Pla2g2a would also be present; the normal multipotential precursor cells of the cohort within the crypt would generate Paneth cells that secrete Pla2g2a. In these non-neoplastic cells, there would be no selection for loss of the resistance allele of Mom1. At present, we cannot distinguish between these two possibilities. Each interpretation links together the current data regarding the mechanism of action of Mom1, the hypothesis that Mom1 encodes a secretory phospholipase, and the fact that the Mom1 region and Pla2g2a maintain heterozygosity in Min-induced adenomas.
Prospectus.
Analysis of chimeric mice will permit the dissection of the interactions within or between tumor cells as well as the interaction of these cells with locally or systemically acting factors. These analyses are facilitated by the use of a cell lineage marker that is both ubiquitously expressed and phenotypically neutral. When such experiments are performed on defined genetic backgrounds, the effects of single genetic differences can be observed.
Acknowledgments
This paper is dedicated to the memory of Dr. Elizabeth C. Miller, on the 10th anniversary of her premature death, and to Dr. Roswell K. Boutwell, on the occasion of his 80th birthday. These two founding colleagues created the environment for the genetic analysis of cancer with the laboratory mouse at the McArdle Laboratory. We thank Dr. Camille Connelly for technical advice; Charles Rolfsmeyer for excellent animal husbandry; Glenn Friedrich and Dr. Philippe Soriano for the R26 mice; Dr. Amy R. Moser and Alex R. Shoemaker for helpful discussions and critical evaluation of the manuscript; and Bob Cormier and Drs. Andrea Bilger, Norman Drinkwater, Richard Halberg, Anita Merritt, and Ilse Riegel for critical evaluation of the manuscript. This is publication No. 3469 from the Laboratory of Genetics. This work was supported by Core Grant CA-07175, Training Grant CA-09135, and Research Grant R01CA-63677 from the National Cancer Institute.
ABBREVIATIONS
- R26
ROSA26 mouse strain
- AKR
AKR/J mouse strain
- B6
C57BL/6J mouse strain
- X-Gal
5-bromo-4-chloro-3-indolyl β-d-galactopyranoside
References
- 1.Groden J, Thliveris A, Samowitz W, Carlson M, Gelbert L, et al. Cell. 1991;66:589–600. doi: 10.1016/0092-8674(81)90021-0. [DOI] [PubMed] [Google Scholar]
- 2.Kinzler K W, Nilbert M C, Su L-K, Vogelstein B, Bryan T M, et al. Science. 1991;253:661–665. doi: 10.1126/science.1651562. [DOI] [PubMed] [Google Scholar]
- 3.Nishisho I, Nakamura Y, Miyoshi Y, Miki Y, Ando H, et al. Science. 1991;253:665–669. doi: 10.1126/science.1651563. [DOI] [PubMed] [Google Scholar]
- 4.Miyoshi Y, Nagase H, Ando H, Horii A, Ichii S, Nakatsuru S, Aoki T, Miki Y, Mori T, Nakamura Y. Hum Mol Genet. 1992;1:229–233. doi: 10.1093/hmg/1.4.229. [DOI] [PubMed] [Google Scholar]
- 5.Moser A R, Pitot H C, Dove W F. Science. 1990;247:322–324. doi: 10.1126/science.2296722. [DOI] [PubMed] [Google Scholar]
- 6.Su L-K, Kinzler K W, Vogelstein B, Preisinger A C, Moser A R, Luongo C, Gould K A, Dove W F. Science. 1992;256:668–670. doi: 10.1126/science.1350108. [DOI] [PubMed] [Google Scholar]
- 7.Levy D B, Smith K J, Beazer-Barclay Y, Hamilton S R, Vogelstein B, Kinzler K W. Cancer Res. 1994;54:5953–5958. [PubMed] [Google Scholar]
- 8.Luongo C, Moser A R, Gledhill S, Dove W F. Cancer Res. 1994;54:5947–5952. [PubMed] [Google Scholar]
- 9.Smith K J, Johnson K A, Bryan T M, Hill D E, Markowitz S, Wilson J K V, Pareskeva C, Petersen G M, Hamilton S R, Vogelstein B, Kinzler K W. Proc Natl Acad Sci USA. 1993;90:2846–2850. doi: 10.1073/pnas.90.7.2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miyashiro I, Senda T, Matsumine A, Baeg G-H, Kuroda T, Shimano T, Miura S, Noda T, Kobayashi S, Monden M, Toyoshima K, Akiyama T. Oncogene. 1995;11:89–96. [PubMed] [Google Scholar]
- 11.Matsumine A, Ogai A, Senda T, Okumura N, Satoh K, Baeg G-H, Kawahara T, Kobayashi S, Okada M, Toyoshima K, Akiyama T. Science. 1996;272:1020–1023. doi: 10.1126/science.272.5264.1020. [DOI] [PubMed] [Google Scholar]
- 12.Näthke I, Adams C L, Polakis P, Sellin J H, Nelson W J. J Cell Biol. 1996;134:165–167. doi: 10.1083/jcb.134.1.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shoemaker A R, Gould K A, Luongo C, Moser A R, Dove W F. Biochim Biophys Acta. 1997;1332:F25–F48. doi: 10.1016/s0304-419x(96)00041-8. [DOI] [PubMed] [Google Scholar]
- 14.Moser A R, Dove W F, Roth K A, Gordon J I. J Cell Biol. 1992;116:1517–1526. doi: 10.1083/jcb.116.6.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dietrich W F, Lander E S, Smith J S, Moser A R, Gould K A, Luongo C, Borenstein N, Dove W F. Cell. 1993;75:631–639. doi: 10.1016/0092-8674(93)90484-8. [DOI] [PubMed] [Google Scholar]
- 16.Gould K A, Dietrich W F, Borenstein N, Lander E S, Dove W F. Genetics. 1996;144:1769–1776. doi: 10.1093/genetics/144.4.1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dove W F, Luongo C, Connelly C S, Gould K A, Shoemaker A R, Moser A R, Gardner R L. Cold Spring Harbor Symp Quant Biol. 1994;59:501–508. doi: 10.1101/sqb.1994.059.01.055. [DOI] [PubMed] [Google Scholar]
- 18.Gould K A, Luongo C, Moser A R, McNeley M K, Borenstein N, Shedlovsky A, Dove W F, Hong K, Dietrich W F, Lander E S. Genetics. 1996;144:1777–1785. doi: 10.1093/genetics/144.4.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.MacPhee M, Chepenik K P, Liddell R A, Nelson K K, Siracusa L D, Buchberg A M. Cell. 1995;81:957–966. doi: 10.1016/0092-8674(95)90015-2. [DOI] [PubMed] [Google Scholar]
- 20.Pruzanski W, Vadas P. Immunol Today. 1991;12:143–146. doi: 10.1016/S0167-5699(05)80042-8. [DOI] [PubMed] [Google Scholar]
- 21.Kudo I, Murakami M, Hara S, Inoue K. Biochim Biophys Acta. 1993;117:217–231. doi: 10.1016/0005-2760(93)90003-r. [DOI] [PubMed] [Google Scholar]
- 22.Gould K A, Dove W F. Cell Growth Differ. 1996;7:1361–1368. [PubMed] [Google Scholar]
- 23.Hogan B, Beddington R, Constantini F, Lacy E. Manipulating the Mouse Embryo: A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1994. [Google Scholar]
- 24.Dietrich W F, Katz H, Lincoln S E, Shin H-S, Friedman J, Dracopoli N, Lander E S. Genetics. 1992;131:423–447. doi: 10.1093/genetics/131.2.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sanes J R, Rubenstein J L R, Nicolas J-F. EMBO J. 1986;5:3133–3142. doi: 10.1002/j.1460-2075.1986.tb04620.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Friedrich G, Soriano P. Genes Dev. 1991;5:1513–1523. doi: 10.1101/gad.5.9.1513. [DOI] [PubMed] [Google Scholar]
- 27.Wong M H, Hermiston M L, Snyder A J, Gordon J I. Proc Natl Acad Sci USA. 1996;93:9588–9593. doi: 10.1073/pnas.93.18.9588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ponder B A J, Schmidt G H, Wilkinson M M, Wood M J, Monk M, Reid A. Nature (London) 1985;313:689–691. doi: 10.1038/313689a0. [DOI] [PubMed] [Google Scholar]
- 29.Wright N A, Irwin G. Cell Tissue Kinet. 1982;15:595–609. doi: 10.1111/j.1365-2184.1982.tb01066.x. [DOI] [PubMed] [Google Scholar]
- 30.Cheng H, Leblond C P. Am J Anat. 1974;141:537–562. doi: 10.1002/aja.1001410407. [DOI] [PubMed] [Google Scholar]
- 31.Potten C S, Loeffler M. Development (Cambridge, UK) 1990;110:1001–1020. doi: 10.1242/dev.110.4.1001. [DOI] [PubMed] [Google Scholar]
- 32.Winton D J, Ponder B A J. Proc R Soc London B. 1990;241:13–18. doi: 10.1098/rspb.1990.0059. [DOI] [PubMed] [Google Scholar]
- 33.Schmidt G H, Wilkinson M M, Ponder B A J. Cell. 1985;40:425–429. doi: 10.1016/0092-8674(85)90156-4. [DOI] [PubMed] [Google Scholar]
- 34.Hsu S H, Luk G D, Krush A J, Hamilton S R, Hoover H H., Jr Science. 1983;221:951–953. doi: 10.1126/science.6879192. [DOI] [PubMed] [Google Scholar]
- 35.Fearon E R, Hamilton S R, Vogelstein B. Science. 1987;238:193–196. doi: 10.1126/science.2889267. [DOI] [PubMed] [Google Scholar]
- 36.Novelli M R, Williamson J A, Tomlinson I P M, Elia G, Hodgson S V, Talbot I C, Bodmer W F, Wright N A. Science. 1996;272:1187–1190. doi: 10.1126/science.272.5265.1187. [DOI] [PubMed] [Google Scholar]