

Abstract
Homopurine (AG) and homopyrimidine (CT) oligodeoxyribonucleotides predicted to form triple-helical (triplex) structures have been shown to specifically suppress gene expression when supplied to cultured cells. Here we present evidence that homopurine RNA (effector) sequences designed to form a triplex with a homopurine⋅homopyrimidine sequence 3′ to the termination codon of the insulin-like growth factor type I receptor (IGF-IR) structural gene can efficiently suppress IGF-IR gene transcription. Transfection vectors were constructed to drive transcription of either AG or CT variant triplex-forming strands. To increase the probability of obtaining stable transfectants with adequate expression of effector sequences, these were designed to be transcribed together with cDNA sequences conferring neomycin resistance as a fusion transcript. Rat C6 glioblastoma cells transfected with the AG variant showed dramatic reduction of IGF-IR transcripts compared with untransfected cells. The AG transfectants also exhibited marked down-regulation of the IGF-I, and an enhanced accumulation of serine protease inhibitor nexin-I mRNA. Similar changes in gene expression were observed following transfection of C6 cells with constructs transcribing antisense RNA to IGF-IR transcripts, but were not observed in C6 cells transfected with either the CT triplex variant or with vector lacking triplex-forming sequences. Moreover, C6 cells transfected with AG triplex variant displayed a dramatic inhibition of tumor growth when injected into nude mice. The results suggest that a triple-helix strategy can be used to inhibit transcription elongation of the IGF-IR gene, and emphasize the efficacy of triplex-mediated gene inhibition in an animal model.
Keywords: homopurine, triplex, nexin-I, tumorigenesis, gene therapy
The type I insulin-like growth factor receptor (IGF-IR) plays an important role in the maintenance of the malignant phenotype (1). A large number of cancers and cancer-derived cell lines overexpress the IGF-IR (2). Antisense expression vectors directed against the IGF-IR have proven effective in suppressing tumor growth of C6 rat glioblastoma (3, 4), hamster mesothelioma (5), and rat prostate cancer (6). Antisense oligonucleotides (7) and the α-IR3 antibody for the IGF-IR (8–10) have also been shown to inhibit cellular proliferation in a number of cancer cell lines. Because the IGF-I receptor plays a critical role in cell proliferation and transformation, it is important to develop additional and more efficient strategies to inhibit its function.
Sequence-specific, stable triple-helical structures can be formed by hydrogen bonding of polypurine or polypyrimidine-rich oligodeoxyribonucleotides (ODNs) to polypurine tracts of double-stranded DNA in vitro (11–13). Thus, ODN-mediated triplex formation offers a potentially effective method for experimental or therapeutic modification of gene expression (14). Compared with antisense strategies, triplex formation targets fewer sequence copies (two versus multiple and regenerative mRNA transcripts) while maintaining high sequence specificity and stability (15, 16). ODN-mediated triplex formation can disrupt the regulation of gene expression at several points. Triplex formation was demonstrated to interfere with sequence-specific binding of transcription factors both in vitro and in cultured cells (see ref. 17 for review) and in addition was shown to inhibit DNA replication when directed against DNA polymerase binding sites (18–21). Reports which demonstrate that triplex formation can disrupt gene expression through inhibition of transcription elongation (22–24) suggest that it is possible to target any suitable region within the entire transcribed portion of a given gene.
The effects of exogenously supplied unmodified ODNs used to inhibit gene expression are transient since they are susceptible to extra- and intracellular degradation, thereby limiting their potential therapeutic use (25). To overcome this problem, we developed an approach in which a third strand for a potential triple helix is continuously supplied intracellularly. A recent report from our group showed that a plasmid-encoded purine oligoribonucleotide contributing a potential third strand (effector strand) for triplex formation could inhibit insulin-like growth factor type I (IGF-I) expression in stably transfected rat C6 glioblastoma cells and reduce the tumorigenicity of these cells in an animal model (26). As a target for the effector strand, we used a homopurine sequence in the promoter region of the IGF-I gene. In addition to IGF-I inhibition, a dramatic up-regulation of the serine protease inhibitor nexin-I mRNA was observed in these transfected cells.
We show here that IGF-IR transcription can be suppressed by a similar strategy. However, in this case we sought to interfere with transcription elongation by targeting potential triplex-forming oligoribonucleotides against a region of DNA encoding sequences downstream of the termination codon of the IGF-IR mRNA. To accomplish this, we designed vectors that direct transcription of the triplex-forming effector sequence of either the polypurine or polypyrimidine motif and the mRNA of a selection marker (neomycin resistance) from one promotor element as a single transcript. C6 cells transfected with the homopurine effector sequence (but not the homopyrimidine effector sequence) exhibited suppression of IGF-IR transcripts accompanied by up-regulation of nexin-I mRNA. We also observed marked down-regulation of IGF-I transcripts in these C6 cell clones. Moreover, a dramatic suppression of tumor growth in nude mice was observed, which demonstrates that the triplex strategy can be applied as a gene therapy approach to a biological model.
MATERIALS AND METHODS
Construction of Plasmids pTH-AG-IGFIR, pTH-CT-IGFIR, and pAS-IGFIR.
For assembly of plasmids appropriate for forming a triple helix with sequences in the 3′ untranslated region of the rat IGF-IR gene (accession no. L29232L29232; from nucleotide positions 4504 to 4525), oligonucleotides were synthesized on an automated oligonucleotide synthesizer (Applied Biosystems).
The oligonucleotides IGFIR-AG (5′-GGGGTACCTCTAGAGGAAGGGAGAGAGAGGAGAGGGAATTCC-3′) and IGFIR-CT (3′-CCCCATGGAGATCTCCTTCCCTCTCTCTCCTCTCCCTTAAGG-5′) contain restriction sites for the enzymes EcoRI, XbaI, and KpnI. After annealing and digestion the oligonucleotide duplexes were cloned in both orientations into a vector designated pTH-CMV (Fig. 1). This vector was derived from the eukaryotic expression vector pRc-CMV (Invitrogen), with the sequences spanning the region 995-2100 deleted by restriction digestion with the enzymes ApaI and SmaI. The resulting ApaI overhang was endfilled with T4 DNA Polymerase (GIBCO/BRL) according to the supplier’s instructions, and the vector was religated. The digested oligonucleotides were inserted into EcoRI and KpnI sites of vector pTH-CMV yielding the vector designated pTH-AG-IGFIR or into XbaI and EcoRI sites yielding the vector designated as pTH-CT-IGFIR. This cloning strategy positioned inserted sequences into the 5′ untranslated region of the neomycin resistance gene. Transcription of this fusion transcript is driven by the constitutive cytomegalovirus (CMV) promotor.
Figure 1.

Schematic representation of plasmid construction. Transcription vectors pTH-AG-IGFIR and pTH-CT-IGFIR code for the polypurine and polypyrimidine variants of triple-helix third strand, respectively. Py, polypyrimidine; Pu, polypurine; CMV, cytomegalovirus; MCS, multiple cloning site; BGH, bovine growth hormone polyadenylylation signal; M13, origin for the rescue of single strand; SV40, origin of replication; Neo, neomycin resistance gene; polyA, SV40 polyadenylylation sequence.
To prepare the antisense IGF-IR expression construct (pAS-IGFIR) a 696-bp human IGF-IR cDNA fragment (from nucleotide position 42 in exon 1 to nucleotide position 738 in exon 3) was inserted in antisense orientation into the episome-based vector pMT/EP containing the ZnSO4-inducible mouse metallothionein-1 (MT-1) promotor as described previously (6).
Cell Culture.
The rat glioblastoma cell line C6 (27) was obtained from the American Type Culture Collection (Rockville, MD). To reduce intrinsic heterogeneity, we used the clone C6(t1), which was derived from parental C6 cells for experiments in this study. Cells for routine culture were maintained in Dulbecco’s modified Eagle medium (BioWhittaker) supplemented with 10% fetal bovine serum (GIBCO/BRL).
Transfection.
C6(t1) cells were transfected with plasmids of the vectors pTH-AG-IGFIR, pTH-CT-IGFIR, pTH-CMV (containing no insert), or pAS-IGFIR using Lipofectin (GIBCO/BRL) according to the manufacturer’s instructions. Following transfection, the cells were cultured in nonselective OPTI-MEM I reduced serum medium (GIBCO/BRL) for 24 hr. Selection of pTH-AG-IGFIR, pTH-CT-IGFIR, and pTH-CMV transfectants was carried out in the presence of 0.5 mg/ml G418 (GIBCO/BRL) in the culture medium, and pAS-IGFIR transfectants were selected in medium containing 0.5 mg/ml hygromycin (Calbiochem). Clonal rings (Nalgene) were used for the isolation of single transfectants. A minimum of six single cell clones of each of the transfectants was expanded under continued selection pressure.
RNA Isolation and Hybridization.
Total RNA was extracted from cells by the acid guanidine thiocyanate method (28). Poly(A)+ RNA was isolated by oligothymidylated cellulose using the “messagemaker” kit (GIBCO/BRL) according to the supplier’s instructions. RNA was separated on a denaturing agarose gel and transferred to a Hybond-N+ nylon membrane (Amersham). The cDNA probes were labeled with [32P]dCTP (NEN) by the random hexanucleotide primer method (29) and hybridized to Northern blots in 5× standard saline citrate (SSC), 5× Denhardt’s solution, 0.1% (wt/vol) SDS and 100 μg/ml denatured salmon sperm DNA at 65°C for 18 h. The filters were washed at room temperature for 15 min in 2× SSC followed by 5–15 min in 0.5× SSC, 0.5% (wt/vol) SDS at 65°C and exposed to x-ray film for 24 h. Northern blot hybridization was carried out with a human IGF-IR cDNA insert (see above), a 160-bp portion of nexin-I cDNA (26), a 500-bp rat IGF-I cDNA fragment (30), and chicken β-actin cDNA (31) as probes.
Nude Mouse Experiments.
Transfected C6(t1) cells were detached using Versene reagent (GIBCO/BRL) and washed in serum-free medium prior to injection into nude mice (HSD nu/nu, Case Western Reserve University Animal Resource Center, Cleveland). Cells (1.5 × 106 in 0.1 ml PBS) were injected subcutaneously over the right scapula of 6-week-old athymic nude mice with a 22-gauge needle. Thirteen, 12, and 10 animals were used for tumor growth assays of pTH-AG-IGFIR-, pTH-CT-IGFIR-, and pTH-CMV-transfected cells, respectively. Animals were sacrificed after 15 days and the tumors excised and weighed. Data are presented as mean ± SE.
RESULTS
A polypurine effector sequence was constructed to suppress transcription of the IGF-IR gene. This sequence was theoretically appropriate for triplex formation with a 24-base homopurine target sequence in the 3′ untranslated region of the IGF-IR gene (Fig. 2). The sequence was incorporated into the 5′ untranslated region of the neomycin resistance gene of vector pTH-CMV to maximize the probability that transfected G418-resistant cell clones would express high levels of potential triplex effector sequences (Fig. 1). C6(t1) glioblastoma cells were transfected with the resulting construct, producing several clones. Fig. 3A depicts a Northern blot of poly(A)+ RNA from a clone (TH-AG2) transfected with vector pTH-AG-IGFIR and from untransfected cells probed with IGF-IR cDNA. A marked reduction of IGF-IR mRNA is apparent in the pTH-AG-IGFIR-transfected clone (lane 2) compared with untransfected cells (lane 1). The same blot was reprobed with chicken β-actin sequences to confirm comparable RNA loading (Fig. 3B).
Figure 2.

Homology between effector strands targeted against the IGF-I gene and the IGF-IR gene. Two nucleotides (X) differ between IGF-IR and IGF-I effector strand (boldface type) sequences, which may form Hoogsteen bonds (∗) with target DNA double-strand sequences. Identical nucleotide sequences are designated by |.
Figure 3.
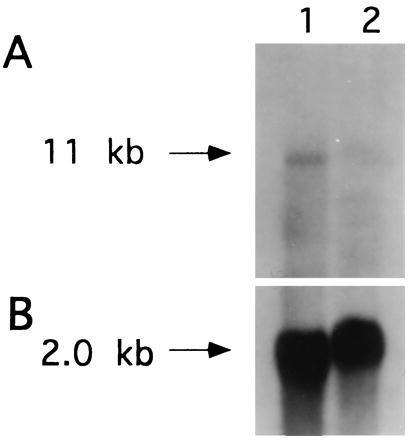
Suppression of IGF-IR transcripts in C6(t1) cells transfected with pTH-AG-IGFIR construct. (A) Poly(A)+ RNA (5 μg per lane) derived from untransfected C6(t1) cells (lane 1) and a C6(t1) cell clone (TH-AG2) transfected with the pTH-AG-IGFIR construct (lane 2) were analyzed by Northern blot. An IGF-IR cDNA was used as a probe. (B) Rehybridization of the same blot with chicken β-actin cDNA. Probes were labeled with 32P-dCTP.
We determined levels of IGF-I transcripts (Fig. 4A) in pTH-AG-IGFIR-transfected cell clones as well as in two types of control cells: cells transfected with vector sequence (pTH-CMV) and cells transfected with the polypyrimidine variant of the effector sequence (pTH-CT-IGFIR; Fig. 1). As shown in Fig. 4A, lanes 6 and 7, IGF-I transcripts were dramatically reduced in C6(t1) cell clones TH-AG2 and TH-AG3 that were transfected with pTH-AG-IGFIR. In contrast, two C6(t1) cell clones transfected with pTH-CT-IGFIR (lanes 3 and 4) or vector (pTH-CMV; lane 2) contain IGF-I transcript levels comparable to untransfected cells (lane 1). Fig. 4B demonstrates integrity of loaded RNAs using chicken β-actin cDNA sequences for hybridization. These data indicate that inhibition of IGF-IR transcription by the homopurine effector sequence is accompanied by suppression of IGF-I.
Figure 4.
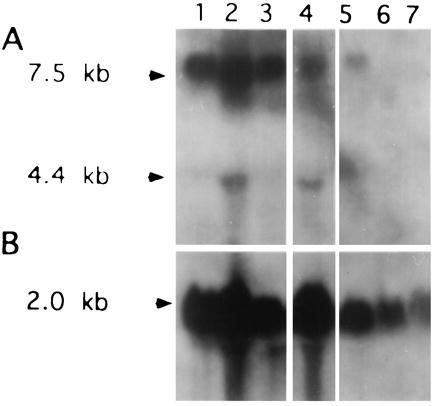
Suppression of IGF-I detected by Northern blot analysis. (A) An IGF-I cDNA was used as a hybridization probe to analyze total RNA (20 μg per lane) from C6(t1) untransfected cells (lane 1), vector transfected cells (lane 2), cell clones TH-CT1 (lane 3), and TH-CT-2 (lane 4) transfected with pTH-CT-IGFIR, or pTH-AG-IGFIR-transfected cell clones TH-AG1 (lane 5), TH-AG2 (lane 6), and TH-AG3 (lane 7). (B) Rehybridization of the same filter was performed with a cDNA probe for chicken β-actin. Blots were exposed to x-ray film for 3 days.
To verify whether C6(t1) cells transfected with the homopurine effector sequence displayed alterations in nexin-I levels, a Northern blot of the same RNAs shown in Fig. 4A was carried out using nexin-I cDNA as a probe (Fig. 5A). RNA from two cell clones (TH-AG2 and TH-AG3) transfected with pTH-AG-IGFIR in which IGF-I transcripts were reduced showed a dramatic increase in nexin-I transcripts (lanes 9 and 10), compared with RNA from cell clones transfected with either pTH-CT-IGFIR (lanes 3–7), vector without insert pTH-CMV (lane 2), or untransfected cells (lane 1). In contrast, elevated nexin-I transcript levels were not detected in RNA from the clone TH-AG1 (Fig. 5A, lane 8). The IGF-I transcript level seen in clone TH-AG1 (Fig. 4A, lane 5) is similar to control levels and correlates with the lack of nexin-I up-regulation. These data indicate that enhanced expression of nexin-I in C6(t1) cells can act as a marker for suppression of IGF-IR as well as IGF-I. Comparable quantities of RNAs were confirmed by reprobing the same blot with chicken β-actin cDNA sequences (Fig. 5B).
Figure 5.
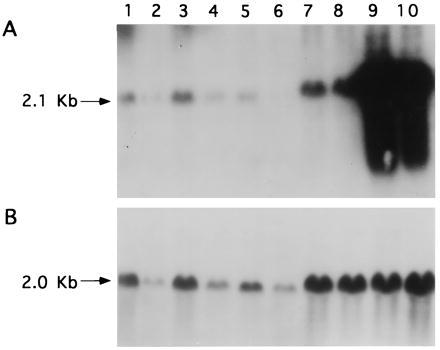
Up-regulation of nexin-I transcripts detected by Northern blot analysis. (A) A nexin-I cDNA was used as a hybridization probe to analyze total RNA (20 μg per lane) derived from untransfected C6(t1) cells (lane 1), C6(t1) cells transfected with vector sequences only (lane 2), C6(t1) cell clones TH-CT1 through TH-CT5 (lanes 3–7) transfected with pTH-CT-IGFIR, or C6(t1) cell clones TH-AG1 (lane 8), TH-AG2 (lane 9), or TH-AG3 (lane 10) transfected with pTH-AG-IGFIR. (B) Rehybridization of the same filter using chicken β-actin cDNA probe.
To determine whether inhibition of IGF-IR by the polypurine triplex effector sequence affects tumor growth, nude mice were injected with transfected C6(t1) cell clones. Tumor growth was significantly decreased in all mice injected with cells transfected with pTH-AG-IGFIR (clone TH-AG2; Fig. 6C). Mice injected with C6(t1) cells transfected with vector pTH-CMV (Fig. 6A) or with C6(t1) cells transfected with pTH-CT-IGFIR (Fig. 6B) bore large tumors. Fig. 7 illustrates mean weights of the tumors recovered from animals that were injected with C6(t1) cells transfected with pTH-AG-IGFIR (solid bar), pTH-CT-IGFIR (striped bar), or pTH-CMV (open bar). Tumor weights were more than 80% lower in animals injected with pTH-AG-IGFIR transfectants as compared with the other groups.
Figure 6.
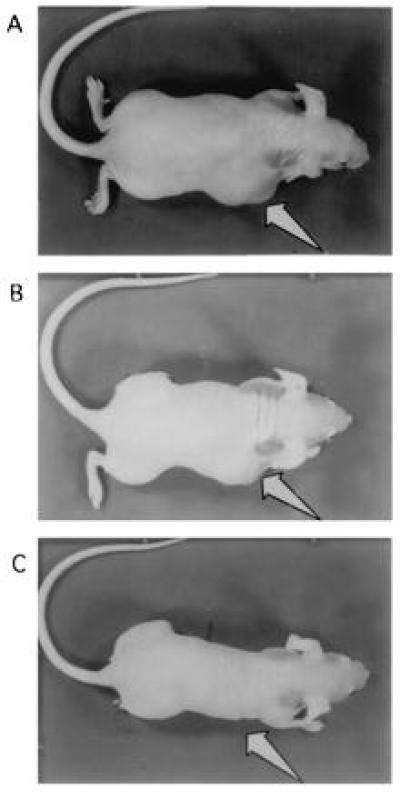
Tumor growth in nude mice. Photographs of mice bearing tumors (arrows) derived from C6(t1) cells transfected with vector sequences (A), and from cells transfected with potential triplex-forming sequences of the pyrimidine motif (pTH-CT-IGFIR) (B). C is a photograph of a mouse injected with cells from the pTH-AG-IGFIR-transfected cell clone TH-AG2. Arrows point to regions of tumor development.
Figure 7.
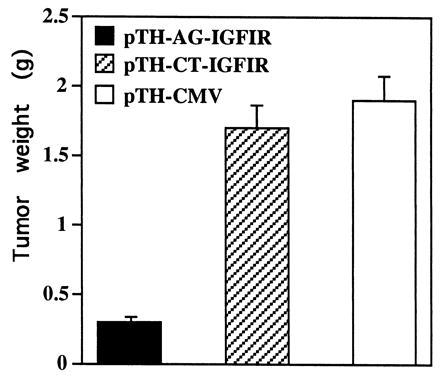
Suppression of tumorigenesis by pTH-AG-IGFIR-transfected C6(t1) cells in nude mice. A total of 13, 12, and 10 nude mice were injected with 1.5 × 106 cells transfected with pTH-AG-IGFIR (solid bar), pTH-CT-IGFIR (striped bar), or vector sequences only (pTH-CMV) (open bar), respectively. Mice were sacrificed 15 days postinjection, tumors were excised, and the tumor weights were determined. Data are presented as mean ± SE.
The homopurine target sequence in the IGF-IR gene contains a stretch of 19 nucleotides of which only 2 deviate from the sequence previously shown to be an effective target for inhibition of IGF-I ligand expression in C6(t1) cells (ref. 26; Fig. 2).
Therefore, the decreased levels of IGF-I transcripts observed in pTH-AG-IGFIR-transfected cells might be accounted for by triplex formation with the target sequence in the IGF-I gene. The possibility of crossreactivity of the IGF-IR effector sequence with the IGF-I gene was examined by Northern blot analysis. C6(t1) cells that had been rendered IGF-IR deficient by the independent method of antisense inhibition showed decreased levels of IGF-I transcripts in three different C6(t1) cell clones transfected with the antisense vector pAS-IGFIR (Fig. 8, lanes 3–5) compared with cells transfected with vector (Fig. 8, lane 2) or untransfected cells (Fig. 8, lane 1). Comparable RNA quantities were demonstrated by hybridization of the same blot with chicken β-actin sequences (Fig. 8B).
Figure 8.
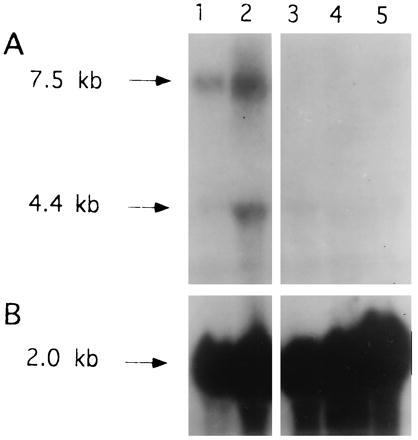
Suppression of IGF-I in IGF-IR antisense-transfected C6(t1) cells. (A) Total RNA (20 μg per lane) derived from untransfected C6(t1) glioblastoma cells (lane 1), vector-transfected cells (lane 2), and three pAS-IGFIR-transfected cell clones (lanes 3–5) was analyzed by Northern blot hybridization using IGF-I cDNA as a probe. (B) Rehybridization of the same filter was performed with a cDNA probe for chicken β-actin. Blots were exposed to x-ray film for 3 days.
Crossreactivity of the IGF-I triplex effector sequence pMT-AG-TH (Fig. 2) with sequences of the IGF-IR gene was evaluated by Northern blot hybridization of RNA from C6(t1) cells rendered IGF-I deficient (Fig. 9). Inhibition of IGF-I transcript accumulation was carried out by the triple-helix strategy recently reported (26) and by antisense strategy (32). Only C6(t1) cell clones transfected with AG-triplex effector sequences against the IGF-I gene (Fig. 9B, lanes 3–8) or C6(t1) cells transfected with antisense to IGF-I (Fig. 9B, lane 2) demonstrate suppression of IGF-I. Hybridization of the blot with cDNA sequences for IGF-IR shows that IGF-IR transcript levels in IGF-I-deficient C6(t1) cells (Fig. 9A, lanes 2–8) are similar to those detected in IGF-I-transcribing C6(t1) cells (lanes 1 and 9). Integrity of RNA was demonstrated by hybridization of the same blot with chicken β-actin sequences (Fig. 9C). The data presented in Figs. 8 and 9 indicate that triplex effector sequences directed against sequences of the IGF-IR or IGF-I genes do not crossreact.
Figure 9.
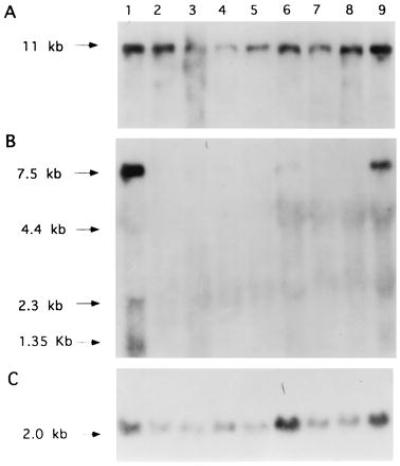
Maintenance of IGF-IR transcript levels in C6(t1) cells transfected with antisense or triplex effector sequences against IGF-I. Total RNA (15 μg per lane) from vector-transfected C6(t1) rat glioblastoma cells (lane 1), antisense to IGF-I-transfected cells (lane 2), six different cell clones transfected with polypurine triplex sequences against IGF-I (lanes 3–8), or a cell clone transfected with polypyrimidine triple-helix sequences against IGF-I (lane 9) was analyzed using an IGF-IR cDNA as a hybridization probe. Rehybridizations of the same filter were performed with cDNA probes for IGF-I (B) or chicken β-actin (C).
DISCUSSION
This study describes the application of a ribonucleotide sequence that can form a potential triple helix to suppress transcription of the IGF-IR gene in C6(t1) rat glioblastoma cells in culture as well as in an animal. The ability of ribonucleotide sequences to form triplexes is supported by several in vitro studies (33–35). Furthermore, our recent report demonstrated specific inhibition of IGF-I ligand expression by a plasmid-encoded ribonucleotide triplex effector sequence and showed a dramatic reduction in tumor growth rates in nude mice (26). In this study, it was also shown that inhibition of IGF-I expression by both plasmid-encoded antisense or potential triplex-forming ribonucleotide sequences resulted in up-regulation of nexin-I mRNA and cell surface expression of major histocompatibility complex I.
To avoid possible transcriptional repression of effector sequences due to integration site-specific inactivation (36), we employed a vector in which the triplex effector sequence and the neomycin resistance cassette were transcribed from a single promoter element. A consequence of utilizing such a fusion transcript is that the phenotypes of neomycin resistance and IGF-IR suppression require the transcript to be present in the nucleus as well as in the cytoplasm of a cell. Out of six clones demonstrating G418 resistance, four showed up-regulation of nexin-I mRNA, indicating that the fusion transcript was present in adequate concentrations in both nucleus and cytoplasm. When RNA from the two remaining clones was probed for neomycin resistance gene sequences, fusion transcripts were detected at reduced levels compared with RNA from the clones showing nexin-I up-regulation (unpublished observations). These results suggest that stronger selection pressure (i.e., higher G418 concentration during selection) might increase the proportion of clones with effective nuclear concentrations of triplex sequences.
C6(t1) cells transfected with pTH-AG-IGFIR exhibited dramatically suppressed tumor growth as compared with pTH-CT-IGFIR-transfected cells or cells transfected with vector sequences only, until the animals were sacrificed 2 weeks postinjection. This result strongly suggests continuous and/or stable delivery of the purine effector sequence to its nuclear target for a period of 2 weeks in the absence of selective pressure. Therefore, vector-mediated delivery of triplex-forming ribonucleotide effector sequences appears to be superior to the use of exogenously added oligonucleotides, which may lose their biological activity after several days in cultured cells (37). Furthermore, it was reported that triplexes that contain RNA rather than DNA as the Hoogsteen paired third strand are more stable, possibly through an additional hydrogen bond between the 2′ hydroxyl proton of DNA and a phosphate oxygen on the backbone of the purine RNA strand (38).
Although the structural and physicochemical properties of triplex formation have been extensively studied in vitro, the experimental conditions required differ considerably from those in the nucleus of an intact cell. In particular, pH constrains the formation of pyrimidine-directed triple-helical complexes, which require cytosine protonation for stabilization. Therefore, this type of triple helix is not stable at physiological conditions of pH between 7.4 and 7.8. In contrast, purine triplex structures are essentially insensitive to pH over a range of at least 5.5 to 8.3 because the proposed triplets in these structures do not involve ionized bases (39, 40). Our results are consistent with these observations since IGF-IR suppression occurred only with the polypurine triplex expression vector in C6(t1) rat glioblastoma cells. Indeed, to our knowledge, triplex formation in intact cells using unmodified ODNs has been demonstrated exclusively with guanosine-rich effector sequences (26, 37, 41–43). Pyrimidine-mediated triplex formation could be demonstrated in cells only with modified ODNs (e.g., ODNs containing a 5-methylcytosine substitution, which has been shown to reduce the stringency of pH requirements; ref. 44). Modified ODNs may possess binding affinities that are different from those of their unmodified counterparts in vitro (45). However, our goal was to investigate triplex formation leading to suppression of IGF-IR in a biological system.
We compared the phenotype of C6(t1) cells, in which IGF-IR was suppressed by transfection with the purine triplex expression vector pTH-AG-IGFIR, with the phenotype of cells following suppression of IGF-IR by the independent method of antisense inhibition. Both approaches resulted in down-regulation of IGF-I, the ligand of the IGF-IR. In contrast, C6(t1) cells transfected with either the pyrimidine triplex expression vector pTH-CT-IGFIR, or with control plasmids pTH-CMV (transcribing no potential triplex sequence) or pMT-EP (transcribing no antisense sequence) contained IGF-I transcript levels comparable to untransfected cells. In addition, because we observed that inhibition of IGF-I expression by either purine RNA triplex or antisense resulted in elevation of protease inhibitor nexin-I transcripts in C6(t1) cells (26), we examined pTH-AG-IGFIR-transfected and vector-transfected cells for nexin-I expression. Only C6(t1) cells transfected with the purine triplex expression vector pTH-AG-IGFIR exhibited up-regulation of nexin-I mRNA. Moreover, only C6(t1) transfectants, which showed elevated nexin-I levels, produced suppressed tumor growth in nude mice. These data, together with the fact that poly(A)+ RNA from pTH-AG-IGFIR-transfected cells displayed reduced levels of IGF-IR transcripts, provide support for the specificity of triple-helix-mediated suppression of the IGF-IR gene.
The effector sequence selected to target IGF-IR gene sequences (Fig. 2) differs in 2 out of 20 nucleotides from the effector strand employed to suppress expression of the IGF-I gene, as reported recently (26). A mismatch occurs at position 6 and a deletion at position 17 relative to the IGF-IR gene sequence. Because a minimum length of 8–14 nucleotides is required for an ODN to form a triple-helical structure (17), the IGF-I effector sequence might form a partly mismatched triple-helical structure with sequences of the IGF-IR gene. However, the resulting mismatch (CG*G to CG*A, whereby “*” denotes Hoogsteen bonding) at position 6 of the IGF-I effector ribonucleotide sequence is predicted to reduce the half-dissociation temperature as indicated in the detailed study of Mergny et al. (46), and would be expected to disrupt stable triplex formation. Conversely, the presence of TA*G instead of TA*A at position number 6, which would occur upon binding of the IGF-IR effector strand to sequences of the IGF-I gene, would also be predicted to destabilize triplex formation. Deletion of a guanosine residue at position 17 in the IGF-IR effector sequence would be expected to further destabilize triplex formation with sequences of the IGF-I gene. The internal positions of both nucleotide deviations are predicted to be particularly disruptive to triple-helical structures, according to the study of Mergny et al. (46).
IGF-I effector sequence/IGF-IR target sequence crossreactivity was tested in a biological context by measuring transcript levels of IGF-IR in C6(t1) cells transfected with constructs encoding IGF-I triplex effector sequences. IGF-IR mRNA was not reduced in C6(t1) cells transfected with the IGF-I triplex effector sequence. These data therefore provide strong support for the specificity of the effector sequences used. We could not investigate crossreaction of the IGF-IR effector sequence with the IGF-I gene, because endogeneous IGF-I transcripts are not detectable in C6(t1) cells in which IGF-IR transcripts are repressed. As discussed above, however, the nature and position of sequence deviations between the IGF-IR effector sequence and the IGF-I target sequence suggest that such crossreaction would be unlikely to occur. In addition, the fact that suppression of IGF-IR expression in C6(t1) cells by the two independent methods of antisense and triplex both result in IGF-I deficiency argues that IGF-I suppression in IGF-IR down-regulated cells is a biological phenomenon rather than a result of crossreaction.
Because the polypurine effector strand was targeted to sequences of the IGF-IR gene that are located in the 3′ untranslated region, our results suggest that the mechanism of transcriptional inhibition is based on interruption of transcription elongation. It was shown that blocking of RNA polymerase II in vitro by triple-helical complexes was transient unless the triplex was stably crosslinked to the target DNA (22, 23). The authors proposed that stalling of the polymerase complex occurred in a region of a triple helix that seems to mediate triplex dissociation, allowing transcription elongation to ensue. In our experiments, we observed a low level of IGF-IR transcripts in C6(t1) cells transfected with the vector pTH-AG-IGFIR, suggesting that triple-helix-mediated suppression was not complete. An alternative explanation for the low transcript level of IGF-IR observed in pTH-AG-IGFIR-transfected cells may be that C6(t1) cells that are completely deficient in IGF-IR would fail to grow in culture since a functional IGF-IR promotes growth of C6 cells (reviewed in ref. 47).
The down-regulation of IGF-I as a consequence of IGF-IR suppression raises interesting questions. It is known that as a result of IGF-IR suppression the expression of transcription factors such as c-fos and c-jun are decreased (48–51). Possibly one of their targets may be the IGF-I gene itself. Although not the scope of this study, possible molecular mechanisms regulating the IGF-IR and its ligand IGF-I clearly await further investigation. However, our results suggest that one common mechanism could account for decreased tumorigenicity of C6(t1) glioblastoma cells (3, 32) regardless of whether IGF-I or IGF-IR expression is inhibited.
Acknowledgments
We appreciate the technical expertise provided by Lijuan Yi. This work was supported by the National Institutes of Health Grants CA 59926 to Joseph Ilan and HD-28451 to Judith Ilan and by the Deutsche Forschungsgemeinschaft Grant RI 792/1–1 to F.R.
ABBREVIATIONS
- IGF-I
insulin-like growth factor type I
- IGF-IR
IGF receptor
- ODN
oligodeoxyribonucleotides
References
- 1.Rubin R, Baserga R. Lab Invest. 1995;73:311–331. [PubMed] [Google Scholar]
- 2.LeRoith D, Werner H, Beitner-Johnson D, Roberts C T. Endocr Rev. 1995;16:143–163. doi: 10.1210/edrv-16-2-143. [DOI] [PubMed] [Google Scholar]
- 3.Resnicoff M, Sell C, Rubini M, Coppola D, Ambrose D, Baserga R, Rubin R. Cancer Res. 1994;54:2218–2222. [PubMed] [Google Scholar]
- 4.Resnicoff M, Weping L, Basak S, Herlyn D, Baserga R, Rubin R. Cancer Immunol Immunother. 1996;42:64–68. doi: 10.1007/s002620050252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pass H I, Mew D J, Carbone M, Matthews W A, Donington J S, Baserga R, Walker C, Resnicoff M, Steinberg S M. Cancer Res. 1996;56:4044–4048. [PubMed] [Google Scholar]
- 6.Burfeind P, Chernicky C, Rininsland F, Ilan J, Ilan J. Proc Natl Acad Sci USA. 1996;93:7263–7268. doi: 10.1073/pnas.93.14.7263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pietzrkowski Z, Mulholland G, Gomella L, Jameson B A, Wernicke D, Baserga R. Cancer Res. 1993;53:1102–1106. [PubMed] [Google Scholar]
- 8.Arteaga C L, Kitten L J, Coronado E B, Jacobs S, Kull F C, Allred D C, Osborne C K. J Clin Invest. 1989;84:1418–1423. doi: 10.1172/JCI114315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gansler T, Furlanetto R, Gramling T S, Robinson K A, Blocker N, Buse M G, Sense D A, Garvin A J. Amer J Pathol. 1989;135:961–966. [PMC free article] [PubMed] [Google Scholar]
- 10.Kalebic T, Tsokos M, Helman L J. Cancer Res. 1994;54:5531–5534. [PubMed] [Google Scholar]
- 11.Le Doan T, Perrouault L, Praseuth D, Habhoub N, Decout J L, Thuong N T, Lhomme J, Helene C. Nucleic Acids Res. 1987;15:7749–7760. doi: 10.1093/nar/15.19.7749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moser H, Dervan P B. Science. 1987;238:645–650. doi: 10.1126/science.3118463. [DOI] [PubMed] [Google Scholar]
- 13.Wells R D, Collier D A, Hanvey J C, Shimizu M, Wohlrab F. FASEB J. 1988;2:2939–2949. [PubMed] [Google Scholar]
- 14.Helene C. Anticancer Drug Des. 1991;6:569–584. [PubMed] [Google Scholar]
- 15.Maher L J, Dervan P B, Wold B J. Biochemistry. 1990;29:8820–8826. doi: 10.1021/bi00489a045. [DOI] [PubMed] [Google Scholar]
- 16.Mergny J L, Sun J S, Rougee M, Montenay-Garestier T, Barcelo F, Chomilier J, Helene C. Biochemistry. 1991;30:9791–9798. doi: 10.1021/bi00104a031. [DOI] [PubMed] [Google Scholar]
- 17.Maher L J., III Bioessays. 1992;14:807–815. doi: 10.1002/bies.950141204. [DOI] [PubMed] [Google Scholar]
- 18.Gueiysse A L, Praseuth D, Francois J C, Helene C. Biochem Biophys Res Commun. 1995;217:186–194. doi: 10.1006/bbrc.1995.2762. [DOI] [PubMed] [Google Scholar]
- 19.Hacia J G, Wold B J, Dervan P B. Biochemistry. 1994;33:5367–5369. doi: 10.1021/bi00184a002. [DOI] [PubMed] [Google Scholar]
- 20.Samadashwily G M, Day A, Mirkin S. EMBO J. 1993;12:4975–4983. doi: 10.1002/j.1460-2075.1993.tb06191.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Birg F, Praseuth D, Zerial A, Thuong N T, Asseline U, LeDoan T, Helene C. Nucleic Acids Res. 1990;18:2901–2908. doi: 10.1093/nar/18.10.2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Young S L, Krawczyk S H, Matteucci M D, Toole J J. Proc Natl Acad Sci USA. 1991;88:10023–10026. doi: 10.1073/pnas.88.22.10023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rando R F, DePaolis L, Durland R H, Jayaraman K, Kessler D J, Hogan M E. Nucleic Acids Res. 1994;22:678–685. doi: 10.1093/nar/22.4.678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Escude C, Giovannangel C, Sun J S, Lloyd D H, Chen J K, Gryaznov S M, Garestier T, Helene C. Proc Natl Acad Sci USA. 1996;93:4365–4369. doi: 10.1073/pnas.93.9.4365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Loke S L, Stein C A, Zhang X H, Morik K, Nakanishi M, Subsinghe C, Cohen J S, Neckers L M. Proc Natl Acad Sci USA. 1989;86:3474–3478. doi: 10.1073/pnas.86.10.3474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shevelev A, Burfeind P, Schulze E, Rininsland F, Johnson T R, Trojan J, Chernicky C L, Helene C, Ilan J, Ilan J. Cancer Gene Ther. 1997;4:105–112. [PubMed] [Google Scholar]
- 27.Benda P, Lightbody J, Sato G, Levine L, Sweet W. Science. 1968;161:370–371. doi: 10.1126/science.161.3839.370. [DOI] [PubMed] [Google Scholar]
- 28.Chomczynski P, Sacchi N. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 29.Feinberg A P, Vogelstein B. Anal Biochem. 1983;132:6–13. doi: 10.1016/0003-2697(83)90418-9. [DOI] [PubMed] [Google Scholar]
- 30.Murphy L J, Bell G I, Duckworth M L, Friesen H G. Endocrinology. 1987;121:684–691. doi: 10.1210/endo-121-2-684. [DOI] [PubMed] [Google Scholar]
- 31.Cleveland D W, Lopata M A, Macdonald R J, Cowan N J, Rutter W J, Kirschner M W. Cell. 1980;20:95–105. doi: 10.1016/0092-8674(80)90238-x. [DOI] [PubMed] [Google Scholar]
- 32.Trojan J, Blossey B K, Johnson T R, Rudin S D, Tykocinski M L, Ilan J, Ilan J. Proc Natl Acad Sci USA. 1992;89:4874–4878. doi: 10.1073/pnas.89.11.4874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Han H, Dervan P B. Proc Natl Acad Sci USA. 1993;90:3806–3810. doi: 10.1073/pnas.90.9.3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roberts R W, Crothers D M. Science. 1992;258:1463–1466. doi: 10.1126/science.1279808. [DOI] [PubMed] [Google Scholar]
- 35.Escude C, Francois J, Sun J C, Ott G, Spinzel M, Garestier T, Helene C. Nucleic Acids Res. 1993;21:5547–5553. doi: 10.1093/nar/21.24.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rees S, Coote J, Stables J, Goodson S, Harris S, Lee M G. BioTechniques. 1996;20:102–110. doi: 10.2144/96201st05. [DOI] [PubMed] [Google Scholar]
- 37.Porumb H, Gousset H, Letellier R, Salle V, Briane D, Vassy J, Amor-Gueret M, Israel L, Taillandier E. Cancer Res. 1996;56:515–522. [PubMed] [Google Scholar]
- 38.Holland J A, Hoffman D W. Nucleic Acids Res. 1996;24:2841–2848. doi: 10.1093/nar/24.14.2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Evans T, Efstratiadis A. J Biol Chem. 1986;261:14771–14780. [PubMed] [Google Scholar]
- 40.Htun H, Dahlberg J E. Science. 1988;241:1791–1796. doi: 10.1126/science.3175620. [DOI] [PubMed] [Google Scholar]
- 41.Durland R H, Kessler D J, Gunnell S, Duvic M, Pettitt B M, Hogan M E. Biochemistry. 1991;30:9246–9255. doi: 10.1021/bi00102a017. [DOI] [PubMed] [Google Scholar]
- 42.Postel E H, Flint S J, Kessler D J, Hogan M E. Proc Natl Acad Sci USA. 1991;88:8227–8231. doi: 10.1073/pnas.88.18.8227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McShan W M, Rossen R D, Laughter A H, Trial J, Kessler D J, Zendegui J G, Hogan M E, Orson F M. J Biol Chem. 1992;267:5712–5721. [PubMed] [Google Scholar]
- 44.Lee J S, Woodsworth M L, Latimer L J, Morgan A R. Nucleic Acids Res. 1984;12:6603–6614. doi: 10.1093/nar/12.16.6603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Noonberg S B, Francois J C, Praseuth D, Guieysse-Peugeot A L, Lacoste J, Garestier T, Helene C. Nucleic Acids Res. 1995;23:4042–4049. doi: 10.1093/nar/23.20.4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mergny J L, Sun J S, Rougee M, Montenay-Garestier T, Barcelo F, Chomilier J, Helene C. Biochemistry. 1991;30:9791–9798. doi: 10.1021/bi00104a031. [DOI] [PubMed] [Google Scholar]
- 47.Baserga R, Sell C, Porcu P, Rubini M. Cell Prolif. 1994;27:63–70. doi: 10.1111/j.1365-2184.1994.tb01406.x. [DOI] [PubMed] [Google Scholar]
- 48.Wieland M, Bahr F, Hohne M, Schurmann A, Ziehm D, Joost H G. J Cell Physiol. 1991;419:428–435. doi: 10.1002/jcp.1041490311. [DOI] [PubMed] [Google Scholar]
- 49.Heidenreich K A, Zeppelin T, Robinson L J. J Biol Chem. 1993;268:14662–14670. [PubMed] [Google Scholar]
- 50.Damante G, Cox F, Rapoport B. Biochem Biophys Res Commun. 1988;151:1194–1199. doi: 10.1016/s0006-291x(88)80492-3. [DOI] [PubMed] [Google Scholar]
- 51.Chiow S T, Chang W C. Biochem Biophys Res Commun. 1992;183:524–531. doi: 10.1016/0006-291x(92)90513-k. [DOI] [PubMed] [Google Scholar]