

Abstract
Apoptosis is postulated to be involved as an anti-viral immune mechanism by killing infected cells before viral replication has occurred. The Fas–Fas ligand interaction is a powerful regulator of T cell apoptosis and could potentially act as a potent anti-viral immune mechanism against T cell tropic virus such as human immunodeficiency virus (HIV). We investigated the status of Fas ligand in peripheral blood mononuclear cells (PBMCs) obtained from persons infected with HIV. We found that monocytes in freshly isolated PBMCs from healthy individuals possess cell surface Fas ligand. In contrast, monocytes in freshly isolated PBMCs from HIV-infected patients had no detectable Fas ligand on the cell surface. Consistent with these findings of surface expression, Fas ligand activity was deficient in the cells from HIV-infected persons. The effect of replacing Fas ligand activity on HIV production by patients’ cells was assessed in an in vitro assay. The addition of a functional anti-Fas antibody to PBMCs from HIV-infected individuals inhibited viral production by greater than 90% without affecting lymphocytic function. These findings suggest the possibility of a new therapeutic modality for the treatment of HIV-infected individuals based on the reconstitution of Fas ligand activity.
Viruses are obligate intracellular parasites. They require the machinery of cells for their own replication. Consequently, apoptosis of a cell after infection but before viral replication may limit the production of infectious virus and thereby act as an anti-viral immune mechanism. The Fas–Fas ligand interaction regulates a major pathway of apoptosis, and we (1) and others (2) have proposed that this pathway may play an important role in mediating an anti-viral effect.
Viruses have developed many different mechanisms to subvert cellular suicide. The selection and maintenance of these mechanisms in a wide variety of viruses represents support for the concept that apoptosis is an important anti-viral defense mechanism. Viruses that inhibit apoptosis of infected cells include (i) Epstein–Barr virus that encodes homologs of interleukin 10 (3) and bcl-2 (4), both of which inhibit the apoptosis of infected cells; (ii) adenovirus that encodes E1B-19K, E3–14.7K, and E3–10.4K/14.5K, all of which inhibit apoptosis mediated by tumor necrosis factor (5), and the E1B-55K protein, which inactivates p53, an endogenous molecule that can induce apoptosis (6); (iii) cowpox virus that encodes serpin (from the crmA gene) that inactivates the interleukin 1β-converting enzyme, a molecule in the apoptotic pathway (7); (iv) myxoma virus that encodes for T2, a soluble homolog of the tumor necrosis factor receptor, which acts to inhibit apoptosis of infected cells (8); (v) papillomavirus that encodes the E6 protein, which inactivates p53 (9); and (vi) parainfluenza virus type 3 that induces interleukin 10 production thereby mediating an anti-apoptotic effect (10).
Recently, we have shown (1) that herpes simplex virus type 2 infection of a cell inhibits its capacity to mediate Fas ligand activity. Fas ligand mRNA was not affected by the infection, and protein was efficiently produced. Nevertheless, the Fas ligand produced remained intracellular and was not expressed on the cell surface. This defect was specific for Fas ligand: four other proteins were expressed in herpes simplex virus type 2-infected cells to a level commensurate with the level in uninfected cells. Moreover, we demonstrated that inhibition of Fas ligand activity had profound implications for viral replication.
To our knowledge, the possibility that viral inhibition of apoptosis is operative in HIV pathogenesis has not been previously considered. Other investigators have shown that inhibition of T cell apoptosis in vitro enhances the production of HIV and thereby facilitates persistent infection (11, 12). Moreover, peripheral blood mononuclear cells (PBMCs) from HIV-infected patients demonstrate enhanced levels of Fas expression that is correlated with enhanced susceptibility to the induction of apoptosis with anti-Fas antibodies (13–15). We sought to determine the status of Fas ligand in these patients.
MATERIALS AND METHODS
Fas Ligand Activity.
Heparinized peripheral blood was obtained from HIV-infected patients and healthy volunteers. Fas ligand activity was measured with a DNA fragmentation assay (1). Jurkat cells (106 cells per ml of RPMI 1640 medium containing 10% fetal bovine serum) were labeled with tritiated thymidine (2.5 μCi/ml; 1 Ci = 37 GBq) for 4 h at 37°C. Either an anti-Fas IgG1 monoclonal antibody that inhibits apoptosis mediated through the Fas–Fas ligand pathway (Immunotech, Westbrook, ME) or an isotype-matched control monoclonal antibody was added at 2 μg/ml to the Jurkat cells during the last hour of this incubation. The target cells were washed and added to PBMCs along with antibody at 500 ng/ml. After a 14-h incubation at 37°C, wells were harvested, and the tritiated thymidine content of the DNA was determined by liquid scintillation. Percent specific apoptotic death was calculated by subtracting the experimental cpm from the spontaneous cpm, dividing this number by the spontaneous cpm, and multiplying by 100.
Human Fas Ligand Transfectants.
K562 cells were infected with a recombinant retrovirus, derived from pLXSN supplied by Dusty Miller (University of Washington, Seattle). An expression construct for human Fas ligand was produced by recombining the coding sequence (obtained in Bluescript from Shigekazu Nagata, Osaka Bioscience Institute) with pLXSN by using the EcoRI–BamHI site. The packaging cell lines PE501 and PA317 were used consecutively to obtain recombinant infectious virus encoding for human Fas ligand. Infected K562 cells were obtained by culturing the cells in neomycin.
Fas Ligand Reverse Transcription-Coupled PCR (RT–PCR).
The RT–PCR method for identification of human Fas ligand message was a modification of a published technique for identification of murine Fas ligand message (16). Total RNA was isolated from freshly isolated PBMCs and reverse-transcribed to cDNA by using an oligo(dT) primer and Moloney murine leukemia virus reverse transcriptase (GIBCO/BRL) at 37°C for 60 min. RNA at 1.6, 0.8, and 0.4 μg was amplified by PCR specific for Fas ligand using the following primers: forward direction, 5′-CAGCTCTTCCACCTACAGAAGG; reverse, 5′-AGATTCCTCAAAATTGACCAGAGAGAG. β-Actin primers were also used. Nucleotides at 10 mM were added to the RNA and the primers, and 2.5 units of Taq polymerase were added to the reaction after the reaction reached 94°C. The conditions for the PCR are as follow: 40 cycles of 94°C for 1.5 min, 60°C for 1 min, and 72°C for 2 min. The products were resolved on 1.2% agarose gels and visualized with ethidium bromide under UV irradiation.
Analysis of Cells Treated with a Fas Agonist.
PBMCs from HIV-infected patients were plated at 1 × 106 cells per ml and cultured in the presence of either anti-Fas IgM or IgM isotype control antibody (100 ng/ml) for 2–3 days. After the incubation period, cells were recovered, washed, and replated at 104–106 viable cells per ml. Phytohemagglutinin (PHA) blasts derived 3 days prior from a healthy volunteer were added to the HIV-infected PBMCs at 0.5 × 106 cells per ml. At the indicated time points, 1 ml of supernatant was removed, frozen at −70°C and replaced with fresh medium supplemented with recombinant interleukin 2 (Chiron) at 20 units/ml. The supernatants were analyzed for HIV p24 content with the HIV-1 p24 antigen assay kit (Immunotech, Westbrook, ME). After the patient samples were treated with anti-Fas IgM or control IgM for 2–3 days, the cells were also tested for proliferative responsiveness to PHA. The cells were washed and replated at 105 cells per well in 96-well flat-bottom plates. PHA was added at 2 μg/ml and cells were cultured at 37°C for 3 days. Wells were then pulse-labeled with tritiated thymidine (0.5 μCi per well), and incorporation of tritiated thymidine into cellular DNA was assessed 18 h later.
Flow Cytometric Analysis for Fas Ligand.
Freshly isolated PBMCs were stained with biotinylated NOK-1 (PharMingen), a murine IgG1 anti-human Fas ligand monoclonal antibody (17) or a biotinylated control murine IgG1. After washing, the cells were incubated with streptavidin-RED670 (GIBCO/BRL) and then analyzed by flow cytometry on a FACScan (Becton Dickinson). Monocytes were identified by their characteristic profile on a side-scatter/forward-scatter histogram. Their identity as monocytes was confirmed by staining with anti-CD14 (Dako). Specific inhibition of staining was accomplished by preincubation of NOK-1 with a soluble form of Fas ligand. The soluble form of Fas ligand was produced in Pichia pastoris (Invitrogen).
RESULTS
Deficient Fas Ligand Activity in HIV-Infected Patients.
As a measure of Fas ligand functional activity, we used a cytotoxicity assay that measures DNA fragmentation, a hallmark of apoptotic cell death (1). PBMCs were tested for their ability to kill Fas-sensitive Jurkat target cells. Inhibition of cytotoxicity of the target cells pretreated with an inhibitory anti-Fas IgG1 monoclonal antibody was used to ascertain Fas ligand specific killing. The difference between the killing of target cells pretreated with the control antibody and the killing of the target cells pretreated with the specific anti-Fas inhibitory antibodies is used as a measure of Fas ligand-dependent cytotoxicity.
The validity of our assay system was confirmed by the demonstration that the inhibitory anti-Fas IgG1 antibody used in the functional assay blocked killing by an apoptosis-inducing anti-Fas IgM antibody, as well as killing mediated by Fas ligand expressed on KFL cells. KFL cells were produced by infecting K562 cells with a recombinant retrovirus encoding the production of human Fas ligand. Additionally, we determined that the anti-Fas IgG1 inhibitory antibody had no effect on killing mediated by PBMCs against the Fas-insensitive K562 target cells. Cytotoxicity of Jurkat targets by PBMCs was partially blocked by inhibitory anti-Fas IgG1 (Fig. 1). The residual killing in these cultures not inhibited by the antibody was due to granular exocytosis by natural killer cells. Depletion of natural killer cells totally eliminated residual cytotoxicity (data not shown).
Figure 1.
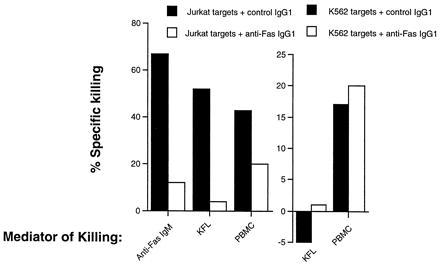
Bioassay for Fas ligand activity. The inhibitory anti-Fas IgG1 monoclonal antibody used to identify the component of the killing dependent on Fas ligand was tested for its capacity to inhibit apoptosis of Jurkat cells mediated by an anti-Fas IgM monoclonal antibody that induces apoptosis, KFL cells, or PBMCs. Moreover, the effects of this antibody on killing of K562 cells, which are not susceptible to lysis by ligation of Fas, were assessed with KFL and PBMC effectors. For PBMC effectors against Jurkat targets the effector/target ratio was 20:1. For KFL effectors against Jurkat and K562 targets, the effector/target ratio was 5:1. For PBMC effectors against K562 targets, the effector/target ratio was 50:1.
PBMCs from patients infected with HIV and from healthy volunteers were isolated and tested for Fas ligand activity without in vitro activation. Representative results (Fig. 2a) demonstrate that PBMCs from six patients infected with HIV were markedly depressed in their capacity to mediate Fas ligand-dependent cytotoxicity compared with PBMCs from three healthy volunteers. In aggregate, PBMCs from HIV+ patients were approximately 80% decreased in Fas ligand activity (Fig. 2b). More than half of the HIV-infected patients demonstrated a complete absence of this cytotoxic activity. The deficiency in Fas ligand activity was not a consequence of the clinical deterioration of the patients or the loss of CD4+ T lymphocytes since 44% of the patients with greater than 400 CD4+ T cells per mm3 demonstrated complete absence of Fas ligand-mediated cytotoxicity.
Figure 2.
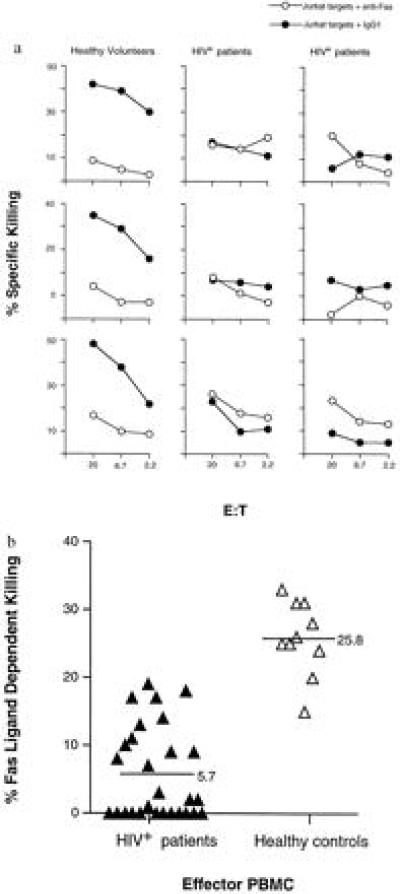
Defective Fas ligand activity in PBMCs of HIV-infected individuals. (a) The percent specific killing mediated by PBMCs from three healthy controls and six HIV+ patients are shown. Solid symbols indicate the killing in the presence of the isotype control antibody, and open symbols indicate the killing in the presence of the anti-Fas inhibitory antibody. (b) Fas ligand dependent killing at an effector/target ratio of 20:1 was compared for 10 healthy controls and 28 HIV-infected individuals. Average Fas-dependent killing of the two groups of samples is indicated by horizontal lines. A two-tailed t test was performed that indicated statistical significance, P < 0.001. Percent Fas ligand-dependent killing was determined by subtracting the percent of background killing seen with anti-Fas inhibitory IgG1 antibody from the percent of killing seen in the presence of the isotype control IgG1.
Deficient Fas Ligand Surface Expression on Monocytes of HIV-Infected Patients.
The cytotoxic activity mediated by PBMCs from healthy controls is consistent with the presence of Fas ligand mRNA from freshly isolated PBMCs (Fig. 3a). Total RNA from unstimulated Jurkat cells was used as a negative control for the RT–PCR, and total RNA from the KFL cell line was used as a positive control. To determine the status of Fas ligand mRNA in HIV-infected patients, we analyzed RNA from freshly isolated PBMCs by semiquantitative RT–PCR. We found that there was no decrease in Fas ligand mRNA in freshly isolated PBMCs of HIV-infected patients compared with PBMCs from healthy volunteers (Fig. 3b). Moreover, equivalent levels of Fas ligand mRNA were present from patient and control samples after overnight culture (data not shown). Thus, the mechanism of deficient Fas ligand activity in these patients could not be explained by an absence of message or the loss of effector cells during the assay.
Figure 3.
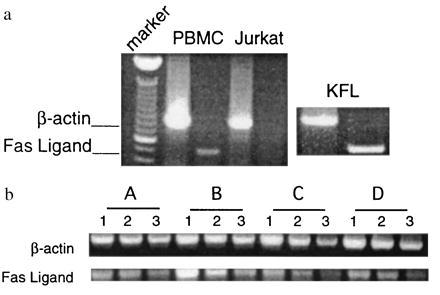
Fas ligand mRNA in PBMCs from healthy volunteers and HIV-infected persons. (a) Fas ligand and β-actin mRNA were analyzed by RT-PCR from total RNA obtained from freshly isolated PBMCs of a healthy volunteer, from Jurkat cells that do not express Fas ligand activity, and from KFL cells that express abundant Fas ligand. (b) Fas ligand and β-actin mRNA were analyzed by RT–PCR from total RNA obtained from freshly isolated PBMCs of a healthy volunteer (lane A) and three HIV-infected individuals (lanes B–D). The PBMCs from these patients exhibited no Fas ligand activity (data not shown). Lanes 1–3 for each sample represent the analyses of 1.6, 0.8, and 0.4 μg of total RNA, respectively.
Human Fas ligand activity has been shown to be mediated by activated T lymphocytes (17–19), activated monocytes (20), and activated natural killer cells (21), as well as freshly isolated natural killer cells (22). To determine which cell population among unstimulated PBMCs expressed Fas ligand activity in our analyses, we stained freshly isolated PBMCs with anti-Fas ligand monoclonal antibody (17) and analyzed the cells by flow cytometry. Fas ligand was not expressed on any cells within the characteristic lymphocyte gates that include lymphocytes and natural killer cells; however, we were consistently able to detect moderate levels of Fas ligand on the cells falling within the characteristic monocyte gates (Fig. 4a), and this staining was inhibited with soluble Fas ligand (Fig. 4b). These cells were verified as monocytes by staining with anti-CD14 (data not shown). Thus, monocytes have the capacity to express the Fas ligand activity that we have observed in unstimulated PBMCs.
Figure 4.
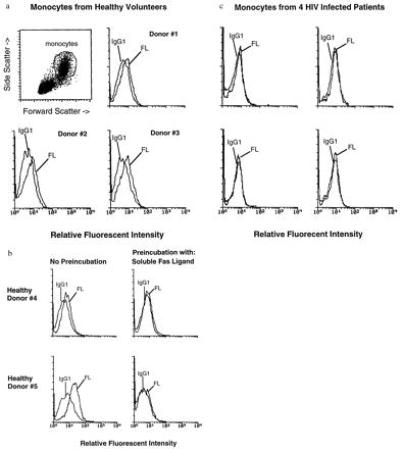
Defective Fas ligand surface expression on monocytes from HIV-infected persons. (a and b) PBMCs were isolated from five healthy volunteers and analyzed by flow cytometry after staining with antibodies to Fas ligand (FL) or control IgG1 (IgG1). (a) Forward scatter and side scatter were analyzed for all donors. Shown is the analysis for donor 1. The monocytes were identified in this histogram and this population was gated for analysis. The analyses of three healthy donors is shown. (b) Monocytes from two healthy donors were stained and analyzed for Fas ligand expression (Left). Inhibition of the staining reaction was accomplished by preincubating the anti-Fas ligand monoclonal antibody with soluble Fas ligand for 15 min prior to adding the mixture to the cells. (c) PBMCs from four HIV-infected individuals were isolated and stained with anti-Fas ligand antibodies (FL) or control IgG1 (IgG1). Analyses of the cells falling within the monocytic gates are shown. The abscissa for the histograms is logarithm of fluorescence intensity, and the ordinate is relative cell number for all analyses shown.
Monocytes from HIV-infected persons were analyzed to determine whether deficient natural Fas ligand activity mediated by their PBMCs was related to decreased levels of Fas ligand surface expression. We found that monocytes in freshly isolated PBMCs from four HIV-infected patients were totally lacking surface Fas ligand (Fig. 4c). Moreover, monocytes isolated by adherence from healthy donors but not from HIV-infected individuals demonstrated Fas ligand activity, consistent with our surface staining results (data not shown). Consequently, the defect in Fas ligand activity seen in PBMCs from HIV-infected persons can be explained, at least in part, by a defect in surface Fas ligand expression on the patients’ monocytes.
Effects of Reconstituting Fas Ligand Activity on HIV Replication.
We have demonstrated that the Fas–Fas ligand interaction has a powerful anti-viral effect for herpes simplex virus type 2 (1). Consequently, we considered the possibility that HIV production might also be affected by a Fas–Fas ligand anti-viral mechanism. PBMCs from four HIV-infected patients were treated with anti-Fas IgM or control IgM. The anti-Fas IgM is an agonist for Fas-induced apoptosis so that treatment with this antibody represents a reconstitution of Fas ligand activity in the patients’ PBMCs. PBMCs were incubated with anti-Fas IgM for 2–3 days, washed, and subsequently mixed with PHA blasts from healthy volunteers to test for HIV production by a p24 assay (Fig. 5a). In the samples treated with anti-Fas IgM, HIV production was markedly inhibited.
Figure 5.
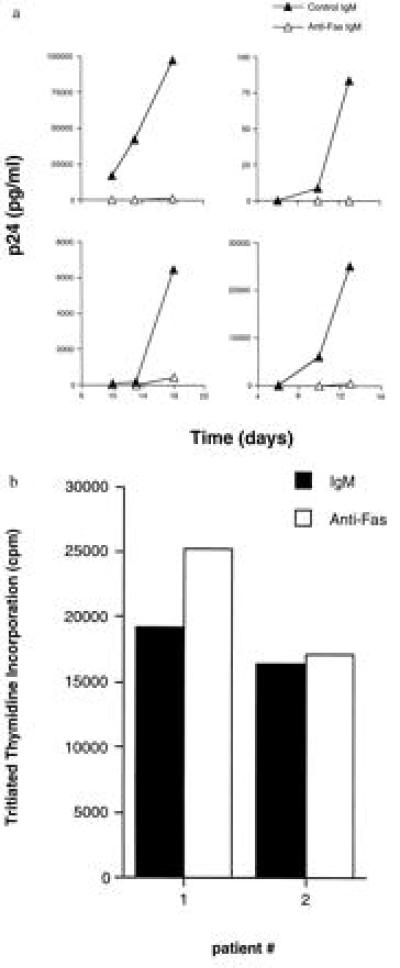
Reduction of virus production but not proliferation in PBMCs from HIV-infected individuals treated with anti-Fas IgM. (a) PBMCs from four HIV-infected patients were treated with either anti-Fas IgM or IgM isotype control antibody for 2–3 days. PHA blasts derived 3 days prior from a healthy volunteer were added to the HIV-infected PBMCs and at various times supernatant was removed and analyzed for HIV p24 content. (b) PBMCs from two of the HIV-infected patients were tested for PHA-stimulated proliferation after treatment with anti-Fas IgM or control IgM for 2–3 days. Cells were plated in 96-well flat-bottom plates, and PHA was added. Three days later incorporation of tritiated thymidine into cellular DNA was assessed.
Consistent with the results of others (13), treatment of the patients’ PBMCs with anti-Fas IgM produced variable amounts of cell death. To ascertain whether the surviving cells were functional, we obtained viable cells after the anti-Fas IgM or control IgM treatments. These cells were replated in the presence of PHA and assessed for proliferation (Fig. 5b). Treatment with anti-Fas IgM did not affect the capacity of the PBMCs to proliferate. Thus, replacement of Fas ligand activity significantly inhibited HIV production but not lymphocytic proliferation.
DISCUSSION
Physiological cell death has been proposed to be a potent mechanism for limiting viral replication (1, 2). The evolution and maintenance in many different viruses of various strategies to circumvent apoptosis lend support to this concept (3–10). We suggest that the deficiency in Fas ligand activity may play an important role in the pathogenesis of HIV disease. It has been shown that inhibition of T cell apoptosis enhances HIV replication and facilitates persistent infection (11, 12). Moreover, HIV-infected tumor cell lines show enhanced susceptibility to Fas-mediated cell death (23). We propose that HIV infection of patients depresses Fas ligand expression on monocytes in vivo and thereby enhances the survival of cells that actively produce virus.
Our results show that the monocytes of healthy volunteers display Fas ligand, a powerful inducer of apoptosis, but the monocytes from HIV-infected individuals are deficient in the expression of this molecule. Others have found that CD4 cross-linking in the presence of HIV tat induces PBMCs to express Fas ligand mRNA and presumably Fas ligand protein (24). Moreover, another group of investigators have indicated that HIV infection of human monocytes from PBMCs induced Fas ligand expression in these cells that were then capable of killing activated uninfected T lymphocytes (20). It should be noted that both of these investigations used PBMCs from healthy volunteers but they did not study PBMCs from HIV-infected individuals.
Our findings of deficient Fas ligand surface expression on monocytes from HIV-infected patients are not necessarily contradictory to the previous findings that HIV induces Fas ligand expression in PBMCs and monocytes from healthy volunteers (20, 24). It is possible that induction of Fas ligand on an acute basis, as demonstrated in vitro, actually results in a chronic deficiency in vivo. According to this model, cells from HIV-infected patients who are chronically exposed to a stimulus that has the capacity to induce Fas ligand expression become refractory to the stimulus. Consequently, the inability of the cell to maintain Fas ligand upregulation in the face of chronic stimulation results in a deficiency of Fas ligand expression.
To our knowledge, the finding of constitutive Fas ligand expression on monocytes from freshly isolated PBMCs has not been previously reported; however, this finding is consistent with the prior demonstration of Fas ligand message in freshly isolated human monocytes (19). The constitutive expression of Fas ligand on monocytes may play an important role in viral immunity. Specifically, it may provide an innate mechanism to quickly and efficiently eliminate infected cells prior to the onset of viral production.
Others have recently demonstrated that freshly isolated human natural killer cells possess Fas ligand activity and message (22). Since we have examined Fas ligand in PBMCs, which contain both monocytes and natural killer cells, it is possible that the deficiency in Fas ligand activity by PBMCs from HIV-infected individuals represents effects on both monocytes and natural killer cells.
We speculate that reconstitution of Fas agonism in vitro selectively induces the apoptosis of cells actively producing HIV, thereby accounting for the precipitous fall in viral production that we observed after a single treatment. Fas ligand deficiency may contribute to the pathogenesis of HIV disease by failing to eliminate a safe haven for the virus. Recent studies have shown that the level of virus in infected individuals is an important variable in the progression of the disease (25–28). Consequently, effective therapies have been directed at decreasing the production of virus, such as protease and reverse transcriptase inhibitors. Nevertheless, the capacity of HIV to mutate may limit the usefulness of drugs directed at specific viral components.
We propose an adjunctive therapy that is not predicated on inhibiting the activity of a viral protein. Since the deficiency in Fas ligand seems to favor viral production, the replacement of Fas ligand activity in HIV-infected individuals may decrease the amount of virus in vivo and thereby provide benefit to patients.
Others have shown that Fas antigen expression on CD4+ and CD8+ T lymphocytes increases with HIV disease progression and that these cells are susceptible to Fas-mediated cell death (13–15, 29). Consequently, therapy based on reconstituting Fas ligand activity in HIV-infected patients would optimally be instituted early in the disease process before the accumulation of significant numbers of susceptible uninfected T lymphocytes.
Hepatocytes constitutively express Fas and are also highly susceptible to apoptotic cell death. Anti-Fas monoclonal antibodies given intraperitoneally to mice have been shown to cause fatal hepatic necrosis (30); however, functional soluble Fas ligand has been given to mice intraperitoneally without toxicity (31). Potential Fas ligand replacement therapy will have to be formulated so that damage to the liver is minimized.
Acknowledgments
We thank Drs. K. Smachlo, N. Greenspan, S. Vande Pol, M.-S. Sy, H. Boom, and M. Lamm for their comments and the members of the Diagnostic Immunology Lab, the Special Immunology Clinic, and Drs. John Carey and Nashant Ramzi for their participation in obtaining patient samples.
ABBREVIATIONS
- PBMC
peripheral blood mononuclear cell
- PHA
phytohemagglutinin
References
- 1.Sieg S, Yildirim Z, Smith D, Kayagaki N, Yagita H, Huang Y, Kaplan D. J Virol. 1996;70:8747–8751. doi: 10.1128/jvi.70.12.8747-8751.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vaux D, Haecker G, Strasser A. Cell. 1994;76:777–779. doi: 10.1016/0092-8674(94)90350-6. [DOI] [PubMed] [Google Scholar]
- 3.Moore K, Vieira P, Fiorentino D, Trounstine M, Khan T, Mosmann T. Science. 1990;248:1230–1234. doi: 10.1126/science.2161559. [DOI] [PubMed] [Google Scholar]
- 4.Henderson S, Huen D, Rowe M, Dawson C, Johnson G, Rickinson A. Proc Natl Acad Sci USA. 1993;90:8479–8483. doi: 10.1073/pnas.90.18.8479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Krajcsi P, Dimitrov T, Hermiston T, Tollefson A, Ranheim T, Vande Pol S, Stephenson A, Wold W. J Virol. 1996;70:4904–4913. doi: 10.1128/jvi.70.8.4904-4913.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yew P, Berk A. Nature (London) 1992;357:82–85. doi: 10.1038/357082a0. [DOI] [PubMed] [Google Scholar]
- 7.Ray C, Black R, Kronheim S, Greenstreet T, Sleath P, Salvesen G, Pickup D. Cell. 1992;69:597–604. doi: 10.1016/0092-8674(92)90223-y. [DOI] [PubMed] [Google Scholar]
- 8.Macen J, Graham K, Lee S, Schreiber M, Boshkov L, McFadden G. Virology. 1996;218:232–237. doi: 10.1006/viro.1996.0183. [DOI] [PubMed] [Google Scholar]
- 9.Scheffner M, Takahashi T, Huibregtse J, Minna J, Howley P. J Virol. 1992;66:5100–5105. doi: 10.1128/jvi.66.8.5100-5105.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sieg S, King C, Huang Y, Kaplan D. J Virol. 1996;70:4845–4848. doi: 10.1128/jvi.70.7.4845-4848.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Antoni B, Sabbatini P, Rabson A, White E. J Virol. 1995;69:2384–2392. doi: 10.1128/jvi.69.4.2384-2392.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sandstrom P, Pardi D, Goldsmith C, Chengying D, Diamond A, Folks T. J Virol. 1996;70:4617–22. doi: 10.1128/jvi.70.7.4617-4622.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Katsikis P, Wunderlich E, Smith C, Herzenberg L, Herzenberg L. J Exp Med. 1995;181:2029–2036. doi: 10.1084/jem.181.6.2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gehri R, Hahn S, Rothen M, Steuerwald M, Nuesch R, Erb P. AIDS. 1996;10:9–16. [PubMed] [Google Scholar]
- 15.Estaquier J, Tanaka M, Suda T, Nagata S, Golstein P, Ameisen J. Blood. 1996;87:4959–4966. [PubMed] [Google Scholar]
- 16.Ettinger R, Panka D, Wang J, Stanger B, Ju S, Marshak-Rothstein A. J Immunol. 1995;154:4302–4308. [PubMed] [Google Scholar]
- 17.Kayagaki N, Kawasaki A, Ebata T, Ohmoto H, Ikeda S, Inoue S, Yoshino K, Okumura K, Yagita H. J Exp Med. 1995;182:1777–1783. doi: 10.1084/jem.182.6.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alderson M, Tough T, Davis-Smith T, Braddy S, Falk B, Schooley K, Goodwin R, Smith C, Ramsdell F, Lynch D. J Exp Med. 1995;181:71–77. doi: 10.1084/jem.181.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu M, Ao Z, Hegen M, Morimoto C, Schlossman S. J Immunol. 1996;157:707–713. [PubMed] [Google Scholar]
- 20.Badley A, McElhinney J, Leibson P, Lynch D, Alderson M, Paya C. J Virol. 1996;70:199–206. doi: 10.1128/jvi.70.1.199-206.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eischen C, Schilling J, Lynch D, Krammer P, Leibson P. J Immunol. 1996;156:2693–2699. [PubMed] [Google Scholar]
- 22.Oshimi Y, Shoji O, Honda Y, Nagata S, Miyazaki S. J Immunol. 1996;157:2909–2915. [PubMed] [Google Scholar]
- 23.Kobayashi N, Hamamoto Y, Yamamoto N, Ishii A, Yonehara M, Yonehara S. Proc Natl Acad Sci USA. 1990;87:9620–9624. doi: 10.1073/pnas.87.24.9620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Westendorp M, Frank R, Ochsenbauer C, Stricker K, Dhein J, Walczak H, Debatin K, Krammer P. Nature (London) 1995;375:497–500. doi: 10.1038/375497a0. [DOI] [PubMed] [Google Scholar]
- 25.Wei X, Ghosh S, Taylor M, Johnson V, Emini E, Deutsch P, Lifson J, Bonhoeffer S, Nowak M, Hahn B, Saag M, Shaw G. Nature (London) 1995;373:117–122. doi: 10.1038/373117a0. [DOI] [PubMed] [Google Scholar]
- 26.Ho D, Neumann A, Perelson A, Chen W, Leonard J, Markowitz M. Nature (London) 1995;373:123–126. doi: 10.1038/373123a0. [DOI] [PubMed] [Google Scholar]
- 27.Saksela K, Stevens C, Rubenstein P, Baltimore D. Proc Natl Acad Sci USA. 1994;91:1104–1108. doi: 10.1073/pnas.91.3.1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Furtado M, Kingsley, Wolinsky S. J Virol. 1995;69:2092–2100. doi: 10.1128/jvi.69.4.2092-2100.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Silvestris F, Cafforio P, Frassanito M, Tucci M, Romito A, Nagata S, Dammacco F. AIDS. 1996;10:131–141. doi: 10.1097/00002030-199602000-00002. [DOI] [PubMed] [Google Scholar]
- 30.Ogasawara J, Watanabe-Fukunaga R, Adachi M, Matsuzawa A, Kasugai T, Kitamura Y, Itoh N, Suda T, Nagata S. Nature (London) 1993;364:806–809. doi: 10.1038/364806a0. [DOI] [PubMed] [Google Scholar]
- 31.Rensing-Ehl A, Frei K, Flury R, Matiba B, Mariani S, Weller M, Aebishcher P, Krammer P, Fontana A. Eur J Immunol. 1995;25:3353–2258. doi: 10.1002/eji.1830250821. [DOI] [PubMed] [Google Scholar]