

Abstract
Cyclic ADP ribose (cADPR) has been shown to trigger Ca2+ release from intracellular stores through ryanodine receptor/channel. In our previous study we observed that all-trans-retinoic acid stimulates cADPR synthesis by ADP ribose cyclase (ADPR cyclase) in cultured epithelial cells. We have now investigated whether cADPR may play a signaling role in action of β-estradiol (E2), an archetypal steroid superfamily hormone, upon its major target organ, uterus, in vivo. Administration of E2 to gonadectomized rats (0.2 mg/kg per day for 7 days) resulted in an ≈Δ + 300% increase of ADPR cyclase activity in extracts from uterus, but in liver, brain, or skeletal muscle ADPR cyclase was unchanged. Most of the E2-stimulated uterine ADPR cyclase was associated with membranes. The higher ADPR cyclase activity in response to E2 was due to the increase of VMAX without change in Km. Simultaneous administration of estrogen antagonist tamoxifen (8 mg/kg per day) with E2 (0.2 mg/kg per day) prevented an increase in ADPR cyclase. In uterine extracts from E2-treated rats, the rate of cADPR inactivation by cADPR hydrolase and the activity of NADase was increased, but to a much lesser degree than activity of ADPR cyclase. Our results indicate that E2, via action to its nuclear receptors in vivo, increases ADPR cyclase activity in uterus. We propose that some of the estrogen effects, and by extension the effects of other steroid superfamily hormones, upon specialized cellular functions and upon hormone-induced gene expression in target cells, are mediated by cADPR-Ca2+ release pathway.
Transient release of Ca2+ from intracellular vesicular stores into cytoplasm is a widespread signal transduction mechanism in action of many hormones upon target cells (1). Various hormones and autacoids stimulate generation of second messenger, inositol-1,4,5-trisphosphate (InsP3), that binds to a specific InsP3 receptor/channel within membranes of endoplasmic reticulum and triggers release of Ca2+ (2). Another mechanism for intracellular Ca2+ [Ca2+]i release, distinct from that triggered by InsP3, is regulated/modulated by recently discovered adenine dinucleotide cyclic ADP ribose (cADPR) (3, 4). Current experimental evidence shows that cADPR binds on its specific receptor, which is associated within the membrane of the endoplasmic reticulum with ryanodine receptor/channel (RyR) and triggers Ca2+-induced Ca2+ release through RyR (4, 5). Biosynthesis of cADPR from β-NAD is catalyzed by ADP ribose cyclase (ADPR cyclase) and cADPR is hydrolyzed by cADPR hydrolase to ADP ribose (noncyclic), a nucleotide devoid of Ca2+-releasing activity (3–5). Enzymes of cADPR metabolism appear to be ubiquitous in mammalian cells and tissues, a feature that may suggest that cADPR-elicited [Ca2+]i release mechanism may play a role as a second messenger in intracellular signal transduction (3, 4). However, unlike as with InsP3 (2), the putative role of cADPR as a second messenger in the action of hormones upon mammalian cells and tissues, namely in vivo, remains unexplored (4, 5).
In the course of our recent in vitro studies of cADPR metabolism in cultured epithelial cells, we discovered that all-trans-retinoic acid (atRA) elicits a manyfold increase of ADPR cyclase activity in LLCPKi cells, whereas activity of cADPR hydrolase was not detectably changed (6). The delayed onset of atRA action as well as other features of the stimulatory effect of atRA upon ADPR cyclase all indicate that atRA acts via genomic mechanism (6). Retinoids, calcitriol, steroid, and thyroid hormones (“steroid superfamily hormones”) all act on target cells by binding to structurally related nuclear receptors and regulate transcription of specific genes (7). Our finding that atRA enhances synthesis of cADPR (4, 6) suggested to us that the cADPR system may play a signaling role in the action of steroid superfamily hormones.
To examine experimentally this novel concept, we set out to investigate the action of a typical hormone of steroid superfamily upon cADPR generation in target cells that are endowed with a well-established specific functional response elicited by [Ca2+]i release. According to recent reports, at least in some types of smooth muscle cells (SMC), cADPR can elicit the [Ca2+]i release (8, 9) from sarcoplasmic reticulum and cell contraction (9). As in other muscle cells, the contraction of uterine myometrial smooth muscle cells (SMC) is regulated by increase in cytosolic Ca2+ in part due to intracellular Ca2+ release (10), and RyR was detected in myometrial SMC (11). An archetypal hormone of steroid superfamily, β-estradiol (E2), enhances [Ca2+]i release-mediated contractility of myometrial SMC in response to several hormones and autacoids (12).
As a first step in testing our hypothesis, which posits that some of the tissue responses to E2, such as enhancement of myometrial SMC contractility, are mediated by stimulation of cADPR synthesis and cADPR-facilitated [Ca2+]i release, we studied in vivo effects of E2 upon enzymes of cADPR metabolism in rat uterus. Results presented herein demonstrate experimental evidence in support of our hypothesis.
MATERIALS AND METHODS
Experiments were conducted on surgically ovariectomized mature female Sprague–Dawley rats (200–250 g body weight); rats were also thyroparathyrectomized to control for effects of thyroid hormones upon E2 receptors (13). Rats had access to ad lib standard rat chow and drinking water supplemented with 0.35% Ca2+ gluconate for 2–3 weeks prior to the experiment. Animals were then administered subcutaneously 0.2 mg/kg per day of E2 dissolved in sesame oil for 7 consecutive days and control rats received only the solvent. In each experiment, the control and E2-treated rats entered experimental protocol and were sacrificed on the same day. Rats were anesthetized with ether, blood drawn, and uterus, brain cortex, liver, and skeletal muscle (psoas) were quickly dissected and chilled in ice-cold 0.9% NaCl. Plasma level of immunoreactive E2 at the day of sacrifice in E2-treated rats (275 ± 53 pg/ml, mean ± SEM, n = 6) was >10-fold higher (P < 0.01, t test) then in controls (14 ± 5 pg/ml, n = 6). The tissues were minced in a solution containing 0.25 M sucrose and 20 mM Tris⋅HCl (pH 7.2), homogenized (1:4, wt/vol) in a Dounce homogenizer using 8–10 strokes and the resulting homogenates were centrifuged at 2000 × g for 10 min at 4°C. The supernatant, denoted further as “extract,” was collected and used, unless specified otherwise in Results, for determination of enzymatic activities. With respect to the experiments shown in Table 3, the homogenates were first centrifuged at 2,000 × g for 10 min, the pellet was discarded, and the supernatant was further centrifuged at 30,000 × g for 60 min. Resulting supernatant and resuspended pellet, denoted as “membrane fraction,” were assayed for ADPR cyclase activity. In some experiments, as indicated in Results, the endometrium was scraped off from split-open uterine horns with a cotton-tipped swab and a razor blade.
Table 3.
ADPR cyclase activity measured with NGD substrate in membrane fraction and in supernatant of homogenates (30,000 × g per 1 h) from uteri of control and E2-treated rats
| n | Control | E2 treated | |
|---|---|---|---|
| Membrane fraction | 3 | 0.52 ± 0.02* | 1.76 ± 0.01*† |
| Supernatant | 3 | 0.15 ± 0.03 | 0.25 ± 0.02 |
Values denote mean ± SEM of ADPR cyclase activity (nmol cGDPR per min per mg protein) from n = 3 experiments.
Significantly different from cytosol (P < 0.01, t test).
Significantly different from controls (P < 0.01, t test).
ADPR Cyclase Activity.
ADPR cyclase activity was assayed with use of β-NAD as substrate, as in our previous studies (6, 14). Tissue extracts (1 mg protein per ml) were incubated in a medium containing 0.5 mM β-NAD, 0.25 M sucrose, and 20 mM Tris⋅HCl (pH 7.2) at 37°C for 60 min (or other time periods specified). Aliquot samples of incubate from the 0 time and at the end of incubation were assayed for cADPR content using sea urchin egg homogenate Ca2+ release bioassay (6, 14, 15) to determine the increment of cADPR (Figs. 1A and 2); specific activity was expressed as nanomoles cADPR per mg of protein per 60 min. Alternatively, the ADPR cyclase activity was determined by the conversion of NAD analog nicotinamide guanine dinucleotide (NGD+) to the fluorescent product cyclic GDP ribose (cGDPR) (16, 17). Tissue extracts were incubated in a thermostated 250 μl cuvette, the contents of which were continuously mixed with a magnetic stirring bar at 37°C with 0.4 mM NGD (or as specified in Results), and generation of cGDPR was continuously monitored using a Hitachi spectrofluorimeter (F-2000) at 300 nm excitation and 410 nm emission (Fig. 1B). The ADPR cyclase activity was then calculated using a standard curve of authentic cGDPR, and the specific activity was expressed as nanomoles cGDPR per mg of protein per minute.
Figure 1.
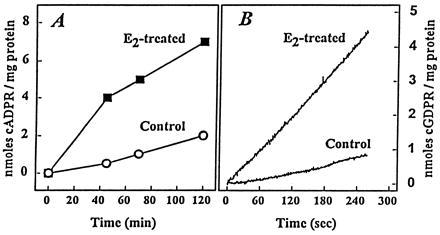
Time course of ADPR cyclase activity in extracts of uteri from control rats (○) and E2-treated rats (▪) determined with the use of β-NAD as substrate (A Left) or with NGD as substrate (B Left).
cADPR Hydrolase Activity.
cADPR hydrolase activity was assayed by incubating extracts for 60 min in a medium containing (in final concentrations): 50 μM cADPR, 0.25 M sucrose, and 20 mM Tris⋅HCl (pH 7.4), and the cADPR contents at time 0 and at 60 min were determined in aliquots of incubate by Ca2+ release bioassay. The specific activity of cADPR hydrolase is expressed as nanomoles cADPR hydrolyzed to ADPR per mg of protein per 60 min.
NADase (NAD Glycohydrolase).
NADase activity was determined by incubating tissue extracts with the fluorogenic substrate 0.1 mM 1,N6-etheno-NAD (ɛ-NAD), and the product was determined fluorometrically at 300 nm excitation and 410 nm emission (19, 20).
Sea Urchin Egg Homogenate Ca2+ Release Bioassay.
Homogenates from sea urchin eggs (Lytechinus pictus) were prepared as described (6, 14, 15, 16). Fluorescence of Ca2+ probe Fluo-3 was monitored at 490 nm excitation and 535 nm emission in a 250-μl cuvette at 17°C with a circulation water bath and continuously mixed with a magnetic stirring bar, using Hitachi spectrofluorimeter (Fig. 2). The bioassay was calibrated using authentic cADPR (6, 14, 16).
Figure 2.
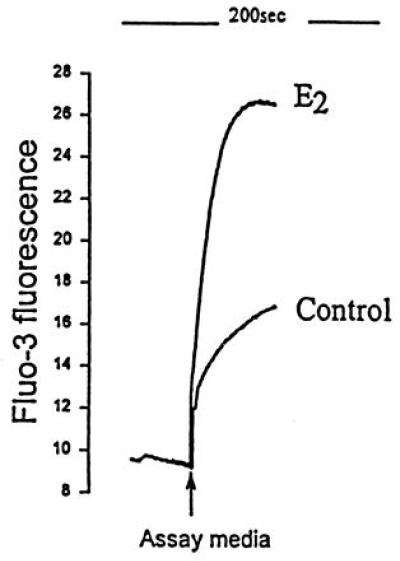
Accumulation of cADPR after incubation of β-NAD with extracts from uteri of E2-treated rats and control rats. The content of cADPR in incubate was determined by sea urchin homogenate Ca2+ release bioassay (4, 6, 14, 16). E2, sample of incubate from E2-treated rats; Control, sample of incubate from control rats. Ordinate denotes Fluo-3 fluorescence of Ca2+. The Ca2+ release bioassay was calibrated with cADPR as described (6, 15, 16).
HPLC Purification of Nucleotides.
Conversions of β-NAD to cADPR, NGD, or cGDPR, catalyzed by ADPR cyclase, were verified by HPLC anion exchange chromatography analysis using AG MP-1 (Bio-Rad) column (1 × 10 cm) eluted with nonlinear gradient of trifluoroacetic acid, as described (6, 14, 16). The peaks of interest, detected by UV absorption at 254 nm, were determined by coelution with authentic standards (6, 14, 18). To verify identity of the cADPR-induced Ca2+ release activity, the incubate from ADPR cyclase assay (with use of β-NAD) was fractionated, and collected HPLC fractions were concentrated by Speed-Vac prior to sea urchin egg homogenate Ca2+ release bioassay (14–16). The protein content in extracts was determined by the method of Lowry et al. (21). The results were evaluated statistically by Student’s t test.
Materials.
Authentic cADPR was biosynthesized enzymatically from β-NAD by ADPR cyclase from ovotestis of Aplysia californica (20), as described before (14, 15). Nicotinic acid adenine dinucleotide phosphate (NAADP) was synthesized via the base-exchange reaction catalyzed by NAD(P)-glycohydrolase (15). The nucleotides were purified by HPLC anion exchange (6, 15); cADPR and NAADP were at least 98% pure as determined by HPLC.
L. pictus and A. californica were obtained from Marinus (Long Beach, CA). Fluo-3 was purchased from Molecular Probes and InsP3 was from Calbiochem. cGDPR, NGD, ɛ-NAD, 8-Br-cADPR, E2, tamoxifen, thionicotinamide-NADP (thio-NADP), and all other reagents (all of the highest purity grade available), were supplied by Sigma. E2 in serum was determined by the radioimmunoassay kit “Coat-a-Count” purchased from Diagnostic Products (Los Angeles).
RESULTS
The specific activity of ADPR cyclase, determined as the rate of β-NAD conversion to cADPR, was severalfold higher in uterine extracts from E2-treated rats than in controls, and the rate of cADPR generation was proportional to incubation time (see Fig. 1A and Table 2). The authenticity of cADPR generated by incubation of β-NAD with extracts from uteri of E2-treated rats and determined by Ca2+ release bioassay (Fig. 2) was affirmed by several key criteria (4, 6, 14, 15). First, Ca2+-releasing activity that accumulated after incubation of uterine extracts from E2-treated rats with β-NAD, when tested in sea urchin homogenate bioassay (Fig. 2), was inhibited by specific antagonists (4, 14, 15, 22) of cADPR-triggered Ca2+ release (Table 1). On the other hand, neither heparin, a selective inhibitor of InsP3, nor thio-NADP, a selective inhibitor of NAADP-triggered Ca2+ release (4, 15), blocked Ca2+ release (Table 1). Identity of cADPR in incubate was also documented, as in our previous studies (4, 14, 15, 16, 22), by criteria of homologous desensitization with cADPR, and the lack of heterologous desensitization of sea urchin egg Ca2+ release bioassay with InsP3 and NAADP, as well as by HPLC analysis (data not shown). Similarly, as in our study of cADPR metabolism in kidney (14), synthesis of cADPR from β-NAD was inhibited by addition of 1 mM DTT (−97%) or by 1 mM nicotinamide (−98%); furthermore, when 0.5 mM β-NAD was replaced by 0.5 mM α-NAD, a stereoisomer that does not serve as substrate for ADPR cyclase (3, 4), no cADPR generation was detected, similar to our previous studies (4, 6, 14).
Table 2.
Activities of ADPR cyclase, cADPR hydrolase, and NADase in extracts from uteri of control and E2-treated rats
| Control | E2 treated | Δ,* % | |
|---|---|---|---|
| ADPR cyclase (nmol cADPR/mg protein per 60 min) | 1.5 ± 0.1 | 6.5 ± 0.5† | +330 |
| cADPR hydrolase (nmol ADPR/mg protein per 60 min) | 3.0 ± 0.05 | 7.5 ± 0.1† | +150 |
| NADase (Δ fluorescence per min) | 1.2 ± 0.1 | 2.18 ± 0.2† | +81 |
Values denote mean ± SEM from four experiments.
Denotes relative percent difference between E2 treated and controls.
E2-treated value significantly (P < 0.01, t test) higher than controls.
Increment in ADPR cyclase activity from E2-treated compared with control is significantly (P < 0.01, t test) higher than increments of cADPR hydrolase and NADase activities.
Table 1.
Effect of specific antagonists upon the Ca2+ release activity generated by incubaiton of uterine extract from E2-treated rats with β-NAD
| Inhibitor | Ca2+ release activity, % |
|---|---|
| None (controls) | 100 |
| 8-Br-cADPR (40 μM) | 5 ± 1 |
| Spermine (3 mM) | 4 ± 1 |
| Procaine (1 mM) | 3 ± 2 |
| Mg2+ (10 mM) | 5 ± 1 |
| Heparin (1 mg/ml) | 106 ± 2 |
| thio-NADP (10 μM) | 98 ± 3 |
Data are expressed (mean ± SEM, n = 3) as percent of control.
ADPR cyclase activity, when determined by conversion of NGD to cGDPR (17, 18), showed similar differences between uterine extracts from E2-treated rats and extracts from controls (Figs. 1B and 3) as found in assay using β-NAD substrate (Fig. 1A and Table 2). Kinetic analysis shows that higher ADPR cyclase activity in uterine extracts from E2-treated rats was due solely to increase of VMAX without detectable change in KmNGD (Fig. 4). Centrifugal separation of uterine extracts (30,000 × g per 60 min) shows that specific activity of ADPR cyclase is severalfold higher in pelleted membrane fraction than in supernatant of extracts from both E2-treated and control rats (Table 3). Moreover, marked increase (+228%) of ADPR cyclase activity was found only in membrane fraction of E2-treated rats, whereas little or no increase was detected in supernatant fraction (Table 3). Activity of ADPR cyclase in extract from whole uteri (1.5 ± 0.1 nmol cGDPR/min per mg protein; mean ± SEM, n = 4) did not differ significantly from ADPR cyclase activity in extracts from uteri with endometrium removed by scraping (1.39 ± 0.08 nmol cGDPR/min per mg protein; mean ± SEM, n = 4), thus indicating that treatment with E2 increased specific activity of ADPR cyclase in myometrium.
Figure 3.
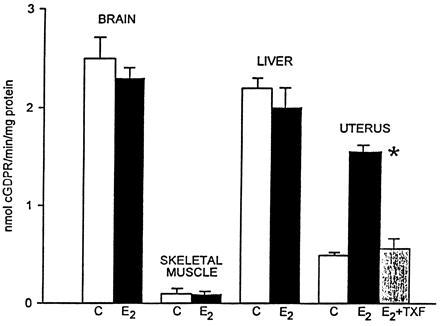
ADPR cyclase activity in extracts from brain cortex, skeletal muscle, liver, and uterus of E2-treated rats (E2, solid bars) as compared with controls (C, open bar) and the effect of administration of E2 (0.2 mg/kg per day) together with tamoxifen (8 mg/kg per day) (E2 + TXF, shaded bar) for 7 days upon ADPR-cyclase activity in uterus. The activity was determined with NGD as substrate. ∗, Denotes value significantly different (P < 0.01, t test) from uteri of both control (C) and E2 + TXF-treated rats). Each bar denotes mean ± SEM, n = 3–4 experiments.
Figure 4.
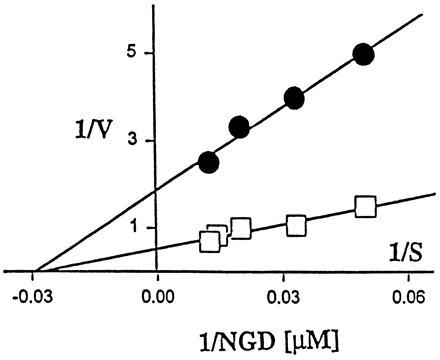
Kinetic parameters of ADPR cyclase from E2-treated rats (•) and from the control rats (□) evaluated by the Lineweaver–Burk double-reciprocal plot. ADPR cyclase was measured with NGD as a substrate in concentration range 10–400 mM NGD. KmNGD from E2-treated rats (30 μM) was not different from KmNGD of controls (33 μM). VMAX value from E2-treated uterine extract (1.76 nmol cGDPR/min per mg protein) was >3 times higher than VMAX in extracts from controls (0.52 nmol cGDPR/min per mg protein).
ADPR cyclase activity was increased severalfold in uterine extracts from E2-treated animals, but no difference in ADPR cyclase activity between E2-treated and control animals was found in extracts from brain cortex, liver, or skeletal muscle (Fig. 3). Unlike in rats treated with E2 alone, in uterine extracts from rats that were simultaneously administered E2 and the estrogen receptor antagonist tamoxifen, the ADPR cyclase activity was not increased and was indistinguishable from controls (Fig. 3). Finally, we determined in uterine extracts from E2-treated rats and controls three related enzymatic activities: ADPR cyclase, cADPR hydrolase, and NADase (4, 20, 23–25). Although all three enzyme activities were significantly elevated in extracts from uteri of E2-treated rats (Table 2), the extent of increase of ADPR cyclase activity (Δ + 330%) was much higher (P < 0.01, t test) than the increment of cADPR hydrolase (Δ + 150%) or of NADase activity (Δ + 81%), respectively (Table 2).
DISCUSSION
The principal finding in our study is a discovery that E2, an archetypal hormone of steroid superfamily, resulted in an increase of ADPR cyclase activity in uterus (Figs. 1, 2, 3), one of the key target organs for estrogens (7). When assayed either with natural substrate β-NAD (Fig. 1A) or with the use of fluorogenic substrate NGD (Fig. 1B), the activities of ADPR cyclase were severalfold higher in extracts from uteri of E2-treated rats (Figs. 1 and 3, Table 2). The authenticity of cADPR product generated from β-NAD by incubation with extract from E2-treated rats was verified by key established criteria; the effect of specific inhibitors upon cADPR-triggered Ca2+ release, and the presence of homologous and the absence of heterologous desensitization in sea urchin egg bioassay, as well as by HPLC analysis (3, 4, 14, 15, 22). Concordance of increases of ADPR cyclase activity in uterine extracts in response to E2, as measured by two independent methods (6, 14, 15, 17), argues for identity of an enzyme that catalyzes cyclization of both NAD and NGD (4, 6, 18).
Several characteristics of uterine ADPR cyclase activity, which is enhanced in response to E2, are noteworthy. ADPR cyclase that is increased by E2 treatment is predominantly membrane-bound enzyme (Table 3) and is inhibited by DTT, similar to ADPR cyclases from mammalian tissues (4, 14, 20, 23–25). This is in contrast with ADPR cyclase from ovotestis of invertebrate Aplysia (20), which is soluble protein and also is insensitive to inhibition of DTT (4, 20).
ADPR cyclase most frequently found in vertebrate tissues and cells is a bi- or multifunctional enzyme, in many, if not all, instances identical to lymphocytic antigen CD38, the “CD38 ADPR cyclase” (3, 4). In CD38 enzyme either cloned from HL-60 promyelocytic cell line (20) or isolated from erythrocyte membranes (24), the ratio of the three enzyme activities, ADPR cyclase/cADPR hydrolase/NADase, is about 1:10:100 (20, 24). In contrast in epithelial LLCPKi cells, incubation with atRA increased only ADPR cyclase activity (6). Treatment of HL-60 cells with atRA enhanced ADPR cyclase and cADPR hydrolase activities to the same degree, so that the ratio of these enzyme activities (1:10) was preserved (17, 20). Our findings show that in extracts from uteri of control rats, the ratio of ADPR cyclase/cADPR hydrolase/NADase was 1.25:2.5:1, and in E2-treated rats, the ratio was 3:3.5:1 (Table 2), a spectrum of activities that is different from both CD38 ADPR cyclase (20) and ADPR cyclase found in atRA-treated LLCPKi cells (6).
Conceivably, uterine ADPR cyclase may be a single multifunctional enzyme, sui generis, and E2 can alter, directly or indirectly, the ratio of enzyme activities with dominant increase of ADPR cyclase. Alternatively, uterine extract may contain two or more NAD-metabolizing enzymes that may be located in different subcellular fractions, and E2 may enhance uniquely, or preferentially, ADPR cyclase activity in one of the enzymes. These mechanisms ought to be clarified in future studies. However, it is important to recall that even the increase in multifunctional ADPR cyclase enzyme activity leads to a manyfold increase of cellular levels of cADPR (26).
Observation that stimulation of ADPR cyclase by E2 is prevented by estrogen receptor blocker tamoxifen (27) suggest that the stimulatory effect of E2 requires binding to nuclear estrogen receptors (7, 27). This notion is also consistent with the findings that ADPR cyclase activity was stimulated by E2 in uterus but not in other studied tissues. Uterus is one of the primary target tissues for estrogens and has at least a 3 times higher density of nuclear E2 receptors than other studied tissues (28). The E2-elicited increase in ADPR cyclase activity is due to the rise of VMAX without change of KmNGD, a feature consistent with the proposition that E2 increased gene expression and de novo synthesis of ADPR cyclase enzyme (Fig. 4). However, it cannot be excluded that E2 may have increased ADPR cyclase activity indirectly by stimulating de novo synthesis of an as yet unknown activator of ADPR cyclase. Clearly, the exact molecular mechanism by which E2 increases uterine ADPR cyclase activity remains to be deciphered in future studies.
It should be briefly considered whether and how cADPR may play a signaling role as a second messenger in action of E2. ADPR cyclase activity increased in response to E2 and most likely causes elevation of cADPR level (26) in cells populating uterus. The only biologic activity of cADPR known to date is facilitation of Ca2+-induced Ca2+ release (3–5). In view of this, at least two signaling roles of cADPR can be envisioned (Fig. 5).
Figure 5.
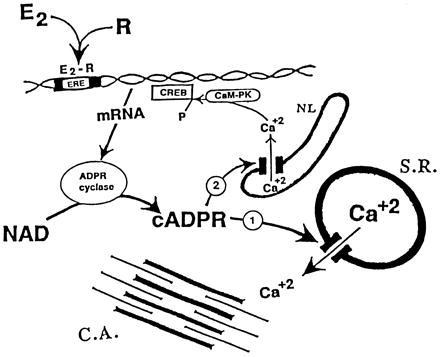
The scheme of the proposed second messenger/modulator role of cADPR in action of E2 upon uterine smooth muscle cell. E2-receptor complex (E2-R) binds to estrogen-responsive element and enhances de novo synthesis of ADPR cyclase. Increased activity of ADPR cyclase catalyzes generation of cADPR from β-NAD and the increased level of cADPR, which then (i) facilitates [Ca2+]i release from sarcoplasmic reticulum (S.R.) and enhances Ca2+-dependent responses of a contractile apparatus (C.A.); (ii) cADPR triggers Ca2+ release from nucleolemmal (NL) space into nucleoplasm, Ca2+ activates calmodulin-dependent protein kinase IV (CaM-PK), which phosphorylates the cAMP responsive element binding protein (CREB) (and other transcription factors). Phosphorylated CREB enhances expression of genes that are regulated by estrogen. cADPR may play similar signaling roles in the action of other hormones of steroid superfamily.
First, cADPR-facilitated Ca2+ release may modulate/regulate specialized functional responses in target cells (Fig. 5). In myometrial SMC, for example, the major functional Ca2+-dependent response is cell contraction that is, at least in part, elicited by Ca2+-induced Ca2+ release from sarcoplasmic reticulum (10, 29). RyR were detected in myometrial SMC (11), and E2-stimulated ADPR cyclase is present in the myometrial layer of uterine wall (see Results); active ADPR cyclase was found in vascular SMC (14). Therefore, increased cADPR levels, due to enhanced E2-dependent ADPR cyclase activity, can facilitate Ca2+-dependent contraction of myometrial SMC in response to various polypeptide hormones and autacoids (10), and the increased Ca2+ release by elevated cADPR may account for the sensitizing effect of E2 upon myometrium (12).
Second, cADPR-facilitated release of [Ca2+]i may modulate E2-elicited gene expression in target cells. According to recent reports, cADPR can trigger Ca2+ release from lumen of nuclear envelope, which is an extension of the endoplasmic reticulum lumen, into nucleoplasma (30). As a consequence, increased Ca2+ concentration within nucleus can activate Ca2+ calmodulin-dependent protein kinase IV (CaM-PK IV), an intranuclear enzyme that phosphorylates and activates CREB, which functions as a Ca2+-responsive transcription factor (31–33); Ca2+-dependent phosphorylations can also activate other nuclear transcription factors, such as serum responsive element (34). By this mechanism, cADPR-elicited rise of nuclear Ca2+ can, via phosphorylation of CREB, enhance the transcription rate of genes that are activated by E2. Such regulatory action of E2, cADPR-dependent Ca2+ release that causes activation of transcription factors and gene expression, may be operant in any cell type that is sensitive to genomic actions of estrogens, regardless of its differentiation and functional specialization. Hypothetical signaling roles of cADPR in E2 actions is outlined schematically on Fig. 5.
To sum up, this study documents that E2 enhances in vivo biosynthesis of cADPR in uterus, an organ that is the primary target tissue for estrogens. We propose that cADPR-triggered Ca2+ release may be, at least in part, the underlying mechanism by which E2 enhances Ca2+-dependent contractile responses of myometrial SMC, and that cADPR-triggered Ca2+ release into nucleoplasm (30) activates Ca2+-dependent phosphorylation of nuclear transcription factors (31–34), and thereby enhances genomic actions of estrogens (Fig. 5). Such a putative signaling role of cADPR may not be unique for E2, since other hormones of the steroid superfamily, namely atRA (4, 6) and triiodothyronine (35), also stimulate ADPR-cyclase activity in their respective target cells and organs.
Acknowledgments
Ms. Carol A. Davidson provided excellent secretarial assistance. This study was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grants DK-30759 and DK-16105 and the Mayo Foundation. E.N.C. is the recipient of a postdoctoral research fellowship from the National Kidney Foundation and F.G.S.d.T. is a postdoctoral research fellow supported by the Mayo Foundation.
ABBREVIATIONS
- ADPR cyclase
ADP ribose cyclase
- RyR
ryanodine receptor/channel
- atRA
all-trans-retinoic acid
- cADPR
cyclic ADP ribose
- E2
β-estradiol
- SMC
smooth muscle cells
- CREB
cAMP responsive element binding protein
- InsP3
inositol-1,4,5-trisphosphate
- NGD
nicotinamide guanine dinucleotide
- cGDPR
cyclic GDP ribose
- NAADP
nicotinic acid adenine dinucleotide phosphate
References
- 1.Rasmussen H. N Engl J Med. 1986;314:1094–1101. doi: 10.1056/NEJM198604243141707. [DOI] [PubMed] [Google Scholar]
- 2.Berridge M J, Irvine R F. Nature (London) 1989;341:197–205. doi: 10.1038/341197a0. [DOI] [PubMed] [Google Scholar]
- 3.Lee H C. Recent Prog Horm Res. 1996;51:355–389. [PubMed] [Google Scholar]
- 4.Dousa T P, Chini E N, Beers K W. Am J Physiol. 1996;271:C1007–C1024. doi: 10.1152/ajpcell.1996.271.4.C1007. [DOI] [PubMed] [Google Scholar]
- 5.Galione A. Trends Pharmacol Sci. 1992;13:304–306. doi: 10.1016/0165-6147(92)90096-o. [DOI] [PubMed] [Google Scholar]
- 6.Beers K W, Chini E N, Dousa T P. J Clin Invest. 1995;95:2385–2390. doi: 10.1172/JCI117932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jensen E V. In: Nuclear Hormone Receptors. Parker M G, editor. San Diego: Academic; 1991. pp. 1–13. [Google Scholar]
- 8.Kannan M S, Fenton A M, Prakash Y S, Sieck G C. Am J Physiol. 1995;270:H801–H806. doi: 10.1152/ajpheart.1996.270.2.H801. [DOI] [PubMed] [Google Scholar]
- 9.Kuemmerle J F, Makhlouf G M. J Biol Chem. 1995;270:25488–25494. doi: 10.1074/jbc.270.43.25488. [DOI] [PubMed] [Google Scholar]
- 10.Arnaudeau S, Lepretre N, Mironneau J. Pflügers Arch. 1994;428:51–59. doi: 10.1007/BF00374751. [DOI] [PubMed] [Google Scholar]
- 11.Lynn S, Morgan J M, Lamb H K, Meissner G, Gillespie J I. FEBS Lett. 1995;372:6–12. doi: 10.1016/0014-5793(95)00924-x. [DOI] [PubMed] [Google Scholar]
- 12.Romanini C. In: The Uterus. Chard T, Grudzinskas J G, editors. Cambridge, U.K.: Cambridge Univ. Press; 1994. pp. 337–355. [Google Scholar]
- 13.Eriksson H A, Freyschuss B. J Steroid Biochem. 1988;29:401–405. doi: 10.1016/0022-4731(88)90249-x. [DOI] [PubMed] [Google Scholar]
- 14.Chini E N, Klener P, Jr, Beer K W, Chini C C S, Grande J P, Dousa T P. Kidney Int. 1997;51:1500–1506. doi: 10.1038/ki.1997.206. [DOI] [PubMed] [Google Scholar]
- 15.Chini E N, Beers K W, Dousa T P. J Biol Chem. 1995;270:3216–3223. doi: 10.1074/jbc.270.7.3216. [DOI] [PubMed] [Google Scholar]
- 16.Beers K W, Chini E N, Lee H C, Dousa T P. Am J Physiol. 1995;268:C741–C746. doi: 10.1152/ajpcell.1995.268.3.C741. [DOI] [PubMed] [Google Scholar]
- 17.Graeff R M, Mehta K, Lee H C. Biochem Biophys Res Commun. 1994;205:722–727. doi: 10.1006/bbrc.1994.2725. [DOI] [PubMed] [Google Scholar]
- 18.Graeff R M, Walseth T F, Fryxell K, Brandton W D, Lee H C. J Biol Chem. 1994;269:30260–30267. [PubMed] [Google Scholar]
- 19.Kontani K, Nishina H, Ohoka Y, Takahashi K, Katada T. J Biol Chem. 1993;268:16895–16898. [PubMed] [Google Scholar]
- 20.Inageda K, Takahashi K, Tokita K, Nishina H, Kanaho Y, Kukimoto I, Kontani K, Hoshino S, Katada T. J Biochem. 1995;117:125–131. doi: 10.1093/oxfordjournals.jbchem.a124698. [DOI] [PubMed] [Google Scholar]
- 21.Lowry O H, Rosebrough N J, Farr A L, Randall R J. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 22.Chini E N, Dousa T P. Am J Physiol. 1996;270:C530–C537. doi: 10.1152/ajpcell.1996.270.2.C530. [DOI] [PubMed] [Google Scholar]
- 23.Kim H, Jacobson E L, Jacobson M K. Science. 1993;261:1330–1333. doi: 10.1126/science.8395705. [DOI] [PubMed] [Google Scholar]
- 24.Zocchi E, Franco L, Guida L, Benatti U, Bergellesi A, Malavasi F, Lee H C, De Flora A. Biochem Biophys Res Commun. 1993;196:1459–1465. doi: 10.1006/bbrc.1993.2416. [DOI] [PubMed] [Google Scholar]
- 25.Takasawa S, Tohgo A, Noguchi N, Koguma T, Nata K, Sugimoto T, Yonekura H, Okamoto H. J Biol Chem. 1993;268:26052–26054. [PubMed] [Google Scholar]
- 26.Takahashi K, Kukimoto I, Tokita K, Inageda K, Inoue S, Kontani K, Hoshino S, Nishina H, Kanaho Y, Katada T. FEBS Lett. 1995;371:204–208. doi: 10.1016/0014-5793(95)00914-u. [DOI] [PubMed] [Google Scholar]
- 27.Jordan V C. Endocr Rev. 1984;11:578–612. doi: 10.1210/edrv-11-4-578. [DOI] [PubMed] [Google Scholar]
- 28.Eisenfeld A J, Axelrod J. Endocrinology. 1966;79:38–42. doi: 10.1210/endo-79-1-38. [DOI] [PubMed] [Google Scholar]
- 29.Morgan J M, Gillespie J I. FEBS Lett. 1995;369:295–300. doi: 10.1016/0014-5793(95)00771-z. [DOI] [PubMed] [Google Scholar]
- 30.Gerasimenko O V, Gerasimenko J V, Tepikin A V, Petersen O H. Cell. 1995;80:439–444. doi: 10.1016/0092-8674(95)90494-8. [DOI] [PubMed] [Google Scholar]
- 31.Tokumitsu H, Enslen H, Soderling T R. J Biol Chem. 1995;270:19320–19324. doi: 10.1074/jbc.270.33.19320. [DOI] [PubMed] [Google Scholar]
- 32.Cruzalegui F H, Means A R. J Biol Chem. 1993;268:26171–26178. [PubMed] [Google Scholar]
- 33.Matthews R P, Guthrie C R, Wailes L M, Zhao X, Means A R, McKnight G S. Mol Cell Biol. 1994;14:6107–6116. doi: 10.1128/mcb.14.9.6107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hardingham G E, Chawla S, Johnson C M, Bading H. Nature (London) 1997;385:260–265. doi: 10.1038/385260a0. [DOI] [PubMed] [Google Scholar]
- 35.Chini E N, de Toledo F G S, Dousa T P. J Invest Med. 1997;45:230. (abstr.). [Google Scholar]