

Abstract
Chronic lymphocytic leukemia (CLL) is the most common form of adult leukemia in Western countries, and there is significant variability in survival within CLL clinical stages. Earlier studies showed that CLL cells produce and are usually growth inhibited by transforming growth factor β type 1 (TGF-β1), suggesting a mechanism for the clinically indolent course of most CLL. Here we studied the mechanism by which CLL cells from about one-third of the patients are insensitive to TGF-β1. Of the 13 patients studied, CLL cells isolated from the peripheral blood of 8 patients were sensitive to growth inhibition by TGF-β1, as determined by incorporation of tritiated thymidine, whereas those from 5 patients were completely resistant to TGF-β1. As judged by binding of radiolabeled TGF-β1 followed by cross-linking and immunoprecipitation with anti-receptor antisera, CLL cells sensitive to TGF-β1 exhibited normal cell surface expression of both types 1 and 2 TGF-β receptors. In contrast, all CLL cells resistant to TGF-β1 exhibited no detectable surface type I receptors able to bind TGF-β1, but normal expression of type II receptors. Both TGF-β1-sensitive and TGF-β1-resistant CLL cells contained normal amounts of both type 1 and type 2 receptor mRNAs. Specific loss of type 1 receptor expression represents a new mechanism by which cells acquire resistance to TGF-β1-mediated growth inhibition in the development and progression of human lymphoproliferative malignancies.
In Western countries, chronic B-cell lymphocytic leukemia (CLL) is the most common adult leukemia. The diagnosis and clinical staging of CLL is usually straightforward, but there is wide variability in the rate of progression and the frequency of disease-related complications. Some CLL patients live for 20 years or longer, while others succumb to the disease within a few months. At present, the clinical stage of CLL probably remains the strongest predictor of patient survival, but there remains significant variability in survival within a given clinical stage. This variability has led to the search for biological mechanisms that may underlie the progression of CLL (1–3).
Endogenous production of transforming growth factor β type 1 (TGF-β1) has been proposed as one of the factors that contributes to the low proliferative rate of CLL cells. In short term cultures of freshly isolated CLL cells, the majority of patient samples exhibit increased proliferation in the presence of neutralizing antibodies to TGF-β1 in a specific and dose-dependent manner. Similarly, in the majority of patients, exogenous TGF-β1 inhibits proliferation of CLL cells induced by phorbol esters, anti-CD40, anti-IgM, interleukin (IL)-2, IL-10, or anti-TGF-β1 (4, 5).
The three major cell surface receptors for TGF-β are termed types I, II, and III (TβRI, II, and III, respectively) (6, 7). TβRI and TβRII are the signaling receptors, while TβRIII appears to promote ligand binding to TβRI and TβRII (6–13). The type I and II receptors are serine/threonine kinases with approximately 40% identity in their kinase domains (6–8, 11, 12); like their ligands, they are members of a large superfamily (6). Both endogenous and exogenous TGF-β1 mediate growth inhibition by interacting with a functional complex of TβRI and TβRII. TGF-β1 binds to TβRII and recruits TβRI into a functional complex, which leads to transphosphorylation of TβRI by TβRII and generation of an anti-proliferative signal (13–16).
Loss of growth inhibition by TGF-β is thought to contribute to the development and progression of a variety of tumors. In retinoblastoma, colon and gastric cancer, hepatoma, and some T cell malignancies, loss of TGF-β responsiveness correlates with loss of cell surface TβRII expression due to mutations or deletions in the TβRII gene (17–23). A recent report implicated genetic changes in TβRI with insensitivity to TGF-β1 in the human prostate cancer cell line, LNCaP, but there are no reports of a specific TβRI defect associated with resistance to TGF-β1 in human lymphoproliferative malignancies or primary tumor samples (24).
In approximately one-third of patients with CLL, cultures of freshly isolated CLL cells are resistant to TGF-β1-mediated growth inhibition (5). The mechanism of TGF-β1 resistance in these patients is unknown and TGF-β receptor expression in CLL has not been analyzed (4, 5). To this end, we compared surface expression of TβRI and TβRII in both TGF-β1-sensitive and -resistant CLL cells obtained from patient peripheral blood samples. Resistance to TGF-β1 was associated with loss-of-functional surface TβRI; surface TβRII was normal. Thus, loss-of-functional TβRI on the cell surface may represent a new mechanism of TGF-β1 resistance involved in the pathogenesis and progression of human CLL.
MATERIALS AND METHODS
Patients.
Thirteen unselected patients with a diagnosis of CLL were studied; determination of the clinical stage (Table 1) was according to the Rai classification (1, 2). All patients had a sustained lymphocytosis in excess of 20 × 109/liter; values ranged from 37–119 × 109/liter. Morphologic analysis of peripheral blood films showed small, mature appearing lymphocytes having condensed chromatin, indistinct nucleoli, and scant cytoplasm; there were fewer than 10% prolymphocytes. The morphologic diagnosis was substantiated by immunophenotypic analysis using flow cytometry. Peripheral blood mononuclear cells were purified by Ficoll density gradient separation and analyzed for the expression of the markers CD45, HLA-DR, CD19, CD20, CD22, CD2, CD3, CD4, CD5, CD7, CD8, CD23, CD10, CD11c, CD25, κ and λ light chains, IgM, IgD, and IgG. In all patients, the lymphocytes had an immunophenotype characteristic of B-CLL that included expression of HLA-DR, the pan B cell antigens CD19 and CD20, CD23, the T cell marker CD5, monotypic immunoglobulin light chains, and dim expression of surface immunoglobulin (25). In all patients, CLL cells accounted for more than 95% of the total cell population .
Table 1.
Summary of responsiveness to TGF-β1 and cell surface TβRI and TβRII expression in CLL cells
| Patient | CS | Growth in presence of TGF-β1 and
|
Growth in presence of anti-TGF-β1 and
|
Cell surface expression of
|
|||||
|---|---|---|---|---|---|---|---|---|---|
| PMA | IL-2 | IL-10 | PMA | IL-2 | IL-10 | TβRI | TβRII | ||
| 1 | 3 | 0.58 | 0.68 | 0.73 | 1.36 | 1.61 | 1.67 | + | + |
| 2 | 3 | 0.95 | 1.05 | 0.97 | 1.02 | 1.00 | 1.11 | − | + |
| 3 | 2 | 1.04 | 1.01 | 0.98 | 0.87 | 0.94 | 0.98 | − | + |
| 4 | 1 | 0.99 | 1.03 | 1.04 | 1.12 | 0.98 | 1.06 | − | + |
| 5 | 2 | 0.21 | 0.14 | 0.29 | 2.77 | 2.25 | 1.14 | + | + |
| 6 | 4 | 0.46 | 0.72 | 0.49 | 1.59 | 2.32 | 1.47 | + | + |
| 7 | 3 | 1.00 | 1.03 | 0.98 | 1.06 | 0.91 | 0.95 | − | + |
| 8 | 3 | 0.91 | 0.98 | 1.04 | 0.96 | 1.02 | 0.98 | − | + |
| 9 | 2 | 0.22 | 0.74 | 0.35 | 1.51 | 1.63 | 1.85 | + | + |
| 10 | 0 | 0.67 | 0.71 | 0.51 | 1.38 | 1.58 | 1.35 | + | + |
| 11 | 3 | 0.49 | 0.72 | 0.68 | 1.11 | 1.27 | 1.59 | ND | ND |
| 12 | 2 | 0.30 | 0.55 | 0.69 | 1.37 | 1.19 | 3.04 | ND | ND |
| 13 | 2 | 0.56 | 0.43 | 0.57 | 1.77 | 1.29 | 1.89 | ND | ND |
CLL cells were stimulated to proliferate in the presence of anti-IgM and combinations of PMA, IL-2, or IL-10. Responsiveness to TGF-β1 was determined by comparing proliferation in the presence or absence of added TGF-β1 and the presence or absence of anti-TGF-β1. Growth in the presence of TGF-β1 is expressed as the ratio of proliferation in the presence of TGF-β1 to proliferation in the absence of TGF-β1 for each of the stimulation conditions. Growth in the presence of anti-TGF-β1 is the expressed as the ratio of proliferation in the presence of anti-TGF-β1 to proliferation in the presence of a control antibody for each of the stimulation conditions. CS, clinical stage of disease; PMA, phorbol 12-myristate 13-acetate; ND, not determined.
Cell Preparation.
Ten to 20 ml of heparinized whole blood was obtained from each of the patients following informed consent. The blood was diluted 1:2 with Hanks’ balanced salt solution (HBSS; GIBCO), overlaid on Ficoll–Hypaque (Pharmacia), and centrifuged at 500 g for 30 minutes at room temperature. Cells at the interface were harvested and washed twice in HBSS, suspended in aliquots of HBSS containing 10% DMSO and 40% FCS, and cryopreserved in liquid nitrogen until needed. The viability of cryopreserved cells was assessed by trypan blue dye exclusion immediately prior to use in all assays and typically exceeded 95%.
Proliferation Assays.
Responsiveness to TGF-β1 was measured using a tritiated thymidine incorporation assay (4, 5, 26). CLL cells (500,000) were suspended in 200 μl of RPMI medium 1640 containing 10% fetal calf serum, and placed in wells of a 96-well microtiter plate in the presence or absence of recombinant human TGF-β1 (R & D Systems; 10 ng/ml), or in the presence of a neutralizing antibody to human TGF-β1 (R & D Systems; 10 μg/ml), or in the presence of a control antibody. Cells were stimulated to proliferate by adding goat-anti-human IgM F(ab′)2 antibodies (Cappel, NC; 10 μg/ml) in combination with phorbol 12-myristate 13-acetate (Sigma; 1 ng/ml), recombinant human IL-2 (R & D Systems; 100 units/ml), or recombinant human IL-10 (R & D Systems; 10 ng/ml). All assay conditions were performed in triplicate. Cells were incubated with [3H]thymidine (1 μCi per well; 1 Ci = 37 GBq) during the final 18 hr of a 5-day culture period and collected using an automated harvester. Radioactivity incorporated by the cells was measured by liquid scintillation counting. Growth inhibition by TGF-β1 was calculated as the ratio of the mean value of radioactivity incorporated in the presence of TGF-β1 to that in its absence. Growth stimulation by anti-TGF-β1 was calculated as the ratio of the mean value of radioactivity incorporated in the presence of a neutralizing antibody to TGF-β1 to that in the presence of a control antibody.
Measurement of Cell Surface TGF-β Receptors.
Radioiodination of TGF-β1 was performed according to ref. 27. Cryopreserved CLL cells were thawed and washed twice in PBS. Ten million cells were suspended in KRH binding buffer [Krebs–Ringer salts buffered with 25 mM Hepes, pH 7.5/5 mM MgSO4/0.5% bovine serum albumin (BSA)] and incubated for 2–3 hr at 4°C with 250 pM 125I-TGF-β1. Unbound ligand was removed by repeated centrifugation with binding buffer in silicon-treated tubes. Bound 125I-TGF-β1 was then cross-linked to cells in 5 ml of KRH buffer containing 50 μl of a 10 mg/ml stock solution of disuccinimidyl suberate for 15 min at 4°C and the cells were lysed in 1 ml of lysis buffer (10 mM Tris, pH 7.5/1 mM EDTA, pH 7.5/1% Triton X-100). Cell lysates were incubated with 10 μl of specific antisera (8, 10) against the C terminus of TβRII or the juxtamembrane region of TβRI for 4 hr at 4°C. Protein A Sepharose (50 μl) was then added for 90 min and the precipitate was collected by centrifugation and washed twice with lysis buffer. TGF-β receptors were analyzed by SDS/PAGE followed by exposure to x-ray films or to a phosphoimager screen for detection in a Fuji Phosphoimager.
Northern Blot Analysis.
Total cellular RNA was isolated by the RNAzol B method (Cinna/Biotecx Laboratories, Friendswood, TX), and poly(A)+ RNA was purified using a Poly(A)+ Tract Isolation System kit (Promega) according to the manufacturers’ instructions. Thereafter, 4 μg of poly(A)+ RNA was resolved on a 1% agarose/2.2 M formaldehyde gel before being blotted onto a nylon membrane (Hybond N, Amersham). The filter was hybridized overnight in 5× SSC buffer containing 50% formamide at 45°C. Blots were washed at 50°C in 0.2× SSC containing 0.1% SDS and exposed to x-ray film. A 642-bp PCR-amplified cDNA product between nt 870 and 1512 of TβRI (8) and a 902-bp PCR-amplified cDNA product between nt 802 and 1704 of TβRII (11) were labeled by the random priming method and used as probes.
RESULTS
Tritiated thymidine incorporation assays allowed us to determine whether the malignant B cells from individual CLL patients were sensitive or resistant to growth inhibition by TGF-β1. As summarized in Table 1, 8 of the 13 CLL patients examined displayed sensitivity to TGF-β1. This was evidenced both by a decrease in proliferation when grown in the presence of TGF-β1 and by an increase in proliferation in the presence of a neutralizing antibody to TGF-β1. This effect was consistently seen with all combinations of agents used to stimulate the proliferation of CLL cells. In five of the patients (2, 3, 4, 7, and 8), the malignant B cells showed complete resistance to TGF-β1-mediated growth inhibition, again regardless of the proliferative stimulus applied to the cells.
To determine the basis of the resistance to TGF-β1, we examined CLL cells from the five TGF-β1-resistant patients and five of the TGF-β1-sensitive patients for surface expression of TGF-β receptors. We employed binding of radiolabeled TGF-β and cross-linking, followed by immunoprecipitation with antisera specific for the cytoplasmic domains of TβRI and TβRII. As exemplified by Patient 1 in Fig. 1, cells sensitive to growth inhibition by TGF-β showed, as expected, normal surface expression of both TβRI and TβRII. In contrast, as exemplified by Patients 2 and 3 in Fig. 1, all CLL patients resistant to growth inhibition by TGF-β1 showed a complete lack of expression of cell surface TβRI able to bind and become cross-linked to radioiodinated TGF-β1. This was evidenced by the amount of 125I-TGF-β1 bound to surface TβRI and immunoprecipitated with anti-TβRI serum. In all patients, TβRII surface expression was normal, as demonstrated by the amount of 125I-TGF-β1 bound to surface TβRII and immunoprecipitated by anti-TβRII. In the case of Patient 3, no 125I-TGF-β1 bound to TβRII could be immunoprecipitated with the anti-TβRI antiserum, suggesting that either there is no TβRI on the cell surface or that surface TβRI cannot be incorporated into a complex with TβRII. In contrast, with cells from Patient 2, 125I-TGF-β1 bound to surface TβRII can be immunoprecipitated with the TβRI-specific antiserum. Thus, with Patient 2, TβRI is expressed on the cell surface and can form a complex with TβRII, but cannot bind or be cross-linked to the 125I-TGF-β1 ligand. Presumably Patient 2 has a mutation(s) in the extracellular domain of TβRI that affects binding to TGF-β.
Figure 1.
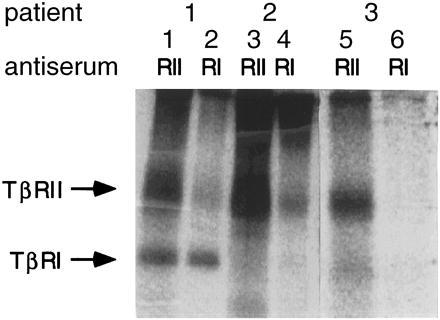
Binding and cross-linking of 125I-TGF-β1 to freshly isolated B-CLL cells followed by immunoprecipitation with anti-TβRI and anti-TβRII antisera. Shown are the results from three representative patients. Lanes 1 and 2 correspond to Patient 1 of Table 1, lanes 3 and 4 correspond to Patient 2, and lanes 5 and 6 correspond to Patient 3. CLL cells from Patient 1 exhibit surface expression of TβRII and TβRI, whereas cells from Patients 2 and 3 exhibit normal surface expression of TβRII but no TβRI is able to bind TGF-β.
To determine whether a loss of surface TβRI expression in CLL patients was due to an absence of TβRI mRNA, we used Northern blot analysis to evaluate mRNA expression. As shown in Fig. 2, we detected normal levels of both TβRI and TβRII mRNA in all freshly isolated CLL cells, including those that did not exhibit TβRI surface expression. This finding suggests that a defect in the level of transcription of the TβRI or TβRII genes was not responsible for the absence of functional surface TβRI in the five patients resistant to TGF-β1-mediated growth inhibition.
Figure 2.
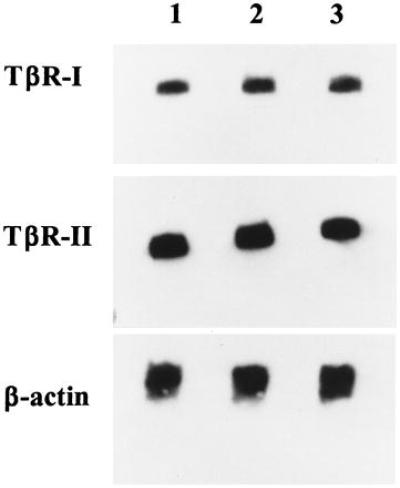
Northern blot analysis of TβRII and TβRI expression in freshly isolated B-CLL cells. Shown are the results from three representative CLL patients. Lanes 1, 2, and 3 correspond to Patients 1, 2, and 3, respectively. The same blot was hybridized to a 32P-labeled probe specific for TβRI, stripped and reprobed using a 32P-labeled probe specific for TβRII, and stripped and reprobed with a 32P-labeled probe to β-actin to verify equal loading of mRNA.
DISCUSSION
Our results demonstrate a correlation between sensitivity to growth inhibition by TGF-β1 and expression of functional TβRI in primary tumor samples obtained from CLL patients. All TGF-β1-sensitive CLL cells express both TβRI and TβRII, while CLL cells that are insensitive to TGF-β1 lack functional cell surface TβRI able to bind and be cross-linked to the TGF-β1 ligand.
A heterodimeric complex of TβRI and TβRII is required for TGF-β1 signaling, and loss of expression of either of these receptors may render cultured cells or tumor cells insensitive to TGF-β1-mediated growth inhibition (13, 15, 18, 22–24). Acquired resistance to TGF-β has been observed in many human malignancies, and in all patients studied in detail these tumor cells have lost expression of cell surface TβRII. Because binding of TGF-β to cell surface TβRI requires the presence of TβRII (6–8, 13, 15, 16), these tumors might exhibit normal cell surface TβRI, which cannot be detected by standard 125I-TGF-β binding and cross-linking assays. Importantly, loss of TβRII can account for the observed resistance to TGF-β. The mechanisms responsible for TβRII loss are varied and include deletions and rearrangements of the gene leading to loss of mRNA expression, and abnormal cytoplasmic sequestration of the protein (17, 19, 20, 28). Missense mutations within highly conserved regions of the serine/threonine kinase domain of TβRII have been implicated in TGF-β resistance in two human head and neck squamous carcinoma cell lines. In one of these cell lines, SqCC/Y1, the missense mutation resulted in defective TβRII autophosphorylation and transphosphorylation of TβRI. In the other cell line, A253, both TβRII autophosphorylation and transphosphorylation of TβRI were dramatically increased, suggesting constitutive activation of the mutant TβRII kinase. Resistance to TGF-β1 in A253 cells might be explained by desensitization of TβRI receptors due to high levels of TβRI phosphorylation independent of TGF-β1 treatment (29).
Recently we reported the identification of a dominant inhibitory mutation in the cytoplasmic domain of TβRII isolated from a TGF-β-resistant human cutaneous T cell lymphoma cell line (23). The mutant receptor was expressed, together with its wild-type counterpart, only in an advanced malignant tumor; a clonally related TGF-β-sensitive cell line isolated from the same patient at an earlier, clinically indolent disease stage showed expression only of the functional wild-type TβRII. Unlike dominant inhibitory mutants of growth factor receptors, which inhibit activation of the cell surface ligand-induced oligomer (30–33), the dominant inhibitory TβRII inhibited cell surface expression of the wild-type TβRII. This was the first description of a dominant inhibitory TβRII isolated from a TGF-β-resistant tumor cell line.
In contrast, there is little evidence implicating loss of or defects in TβRI expression as an important mechanism for acquired TGF-β1 resistance in cancer. In this regard, there is only a single report that correlates loss of TβRI expression with insensitivity to TGF-β1; in the human prostate cancer cell line LNCaP there is a defect in TβRI protein expression due to a structural alteration in the TβRI gene that led to a loss of mRNA expression (24). Several types of mutations in the TβRI gene were generated when mink lung epithelial cells were treated with the mutagen ethylmethane sulfonate and selected for growth in the presence of TGF-β. All could be complemented by expression of wild-type TβRI. One class (R) lacked cell surface TβRI (15, 34). Three TGF-β-resistant cell lines (S mutants) expressed surface TβRI able to bind TGF-β. One TβRI mutant, (G217E) within the ATP binding site, resulted in a kinase-dead polypeptide, whereas two (G261E and G322D) resulted in nontransphosphorylatable TβRI polypeptides. Coexpression of G217E with G322D was found to complement and restore TGF-β-mediated gene induction and growth arrest (35). These results are consistent with the notion that TβRI exists as a homodimer, and that the protein kinase domain of one TβRI polypeptide interacts with the membrane-proximal GS domain of another, enabling them to be phosphorylated and activated by TβRII polypeptides.
Our study of CLL, which used primary tumor samples and not cell lines, correlates insensitivity to TGF-β1 with defects in functional TβRI surface expression, but not loss of either TβRI or TβRII mRNA. In at least one patient (Patient 2), some TβRI is expressed on the cell surface and can form a complex with TβRII, but cannot bind or be cross-linked to TGF-β1. Our results indicate that loss of surface TβRI receptor proteins able to bind TGF-β and form a signaling complex with TβRII may be another common mechanism of acquired TGF-β1 resistance in human cancer. It is likely that defects in functional cell surface TβRI are responsible for the resistance to TGF-β1 observed in some cases of CLL (5, 8, 13, 14, 24). In support of this, sensitivity to TGF-β-mediated growth inhibition was restored in the LNCaP cell line by transfection with TβRI cDNA (24).
Because our work involves nonimmortal primary tumor samples, we are unable to transfect these cells with wild-type TβRI cDNA and select those cells that express the wild-type protein. However, cultured tumor cell lines may have acquired secondary mutations that are not relevant to growth control of the primary tumor cells. Unlike LNCaP cells (24), CLL cells lacking functional surface TβRI express normal amounts of TβRI mRNA. The absence of functional TβRI protein on the surface of CLL cells, despite normal mRNA expression, might be explained by point mutations in TβRI that lead to premature stop codons and truncated proteins, or point mutations that generate mutant TβRI proteins that are unstable or incapable of appearing on the plasma membrane or that cannot bind ligand or interact with TβRII. Since we have not yet cloned and sequenced the TβRI mRNAs from the TGF-β-resistant CLL cells, we do not know whether these cells contain wild-type as well as mutant TβRI mRNA. If both wild-type and mutant mRNAs are present, it would imply that a mutant TβRI protein can dominantly inhibit the function of the wild-type protein in CLL, analogous to the dominant inhibitory TβRII protein we described in the malignant progression of a cutaneous T cell lymphoma (23). The significance of a dominant inhibitory mutant TGF-β receptor in the development of human cancer is that only one of the two alleles encoding the receptor must have been mutated. With the exception of the cutaneous T cell lymphoma we studied (23), when TβRII expression is lost in a tumor both TβRII alleles have undergone loss or mutation.
Our study, which included 13 patients representing all stages of CLL, did not show any obvious correlation between loss of TβRI surface-protein expression and stage of disease. It will be of interest to study other patients with CLL to determine if loss of TβRI surface expression is a predictor of outcome in this most common adult leukemia, or if it contributes to the histologic transformation to an aggressive large cell lymphoma observed in some patients (1, 2).
Acknowledgments
We thank Adriana Naumov for assistance in obtaining patient material used in this study. J.F.D. and R.L. are Fellows of the Medical Research Council of Canada. M.E.K. was supported by National Institutes of Health Grant CA 54062 and the Beth Israel Hospital Pathology Foundation. P.I.K. was supported by an AIDS Research Fund postdoctoral fellowship from the German Cancer Research Institute (Heidelberg). This work was also supported in part by National Institutes of Health Grant R01 CA-636260 to H.F.L.
ABBREVIATIONS
- CLL
chronic lymphocytic leukemia
- TGF-β1
transforming growth factor β type 1
- IL
interleukin
References
- 1.Montserrat E, Rozman C. Baillieres Clin Haematol. 1993;6:849–866. doi: 10.1016/s0950-3536(05)80179-9. [DOI] [PubMed] [Google Scholar]
- 2.Rozman C, Montserrat E. N Engl J Med. 1995;333:1052–1057. doi: 10.1056/NEJM199510193331606. [DOI] [PubMed] [Google Scholar]
- 3.Dohner H, Fischer K, Bentz M, Hansen K, Benner A, Cabot G, Diehl D, Schlenk R, Coy J, Stilgenbauer S, Volkmann M, Galle P R, Poustka A, Hunstein W, Lichter P. Blood. 1995;85:1580–1589. [PubMed] [Google Scholar]
- 4.Lotz M, Ranheim E, Kipps T J. J Exp Med. 1994;179:999–1004. doi: 10.1084/jem.179.3.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lagneaux L, Delforge A, Bron D, Strykmans P. Blood. 1994;81:526. (abstr.). [Google Scholar]
- 6.Massague J, Attisano L, Wrana J L. Trends Cell Biol. 1994;4:172–178. doi: 10.1016/0962-8924(94)90202-x. [DOI] [PubMed] [Google Scholar]
- 7.Massague J. Cell. 1992;69:1067–1070. doi: 10.1016/0092-8674(92)90627-o. [DOI] [PubMed] [Google Scholar]
- 8.Franzen P, ten Dijke P, Ichijo H, Yamashita H, Schulz P, Heldin C, Miyazono K. Cell. 1994;75:681–692. doi: 10.1016/0092-8674(93)90489-d. [DOI] [PubMed] [Google Scholar]
- 9.Lopez-Casillas F, Wrana J L, Massague J. Cell. 1993;73:1435–1444. doi: 10.1016/0092-8674(93)90368-z. [DOI] [PubMed] [Google Scholar]
- 10.Moustakas A, Lin H Y, Henis Y I, Plamondon J, O’Connor-McCourt M D, Lodish H F. J Biol Chem. 1993;268:22215–22218. [PubMed] [Google Scholar]
- 11.Lin H-Y, Wang X, Ng-Eaton E, Weinberg R A, Lodish H F. Cell. 1992;68:775–785. doi: 10.1016/0092-8674(92)90152-3. [DOI] [PubMed] [Google Scholar]
- 12.ten Dijke P, Yamashita H, Ichijo H, Franzen P, Laiho M, Miyazono K, Heldin C-H. Science. 1994;264:101–104. doi: 10.1126/science.8140412. [DOI] [PubMed] [Google Scholar]
- 13.Wrana J L, Attisano L, Weiser R, Ventura F, Massague J. Nature (London) 1994;370:341–347. doi: 10.1038/370341a0. [DOI] [PubMed] [Google Scholar]
- 14.Chen F, Weinberg R A. Proc Natl Acad Sci USA. 1995;92:1565–1569. doi: 10.1073/pnas.92.5.1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Laiho M, Weis F M B, Boyd F T, Ignotz R A, Massague J. J Biol Chem. 1991;266:9108–9112. [PubMed] [Google Scholar]
- 16.Wrana J L, Attisano L, Carcamo J, Zentella A, Doody J, Laiho M, Wang X, Massague J. Cell. 1992;71:1003–1114. doi: 10.1016/0092-8674(92)90395-s. [DOI] [PubMed] [Google Scholar]
- 17.Kimchi A, Wang X F, Weinberg R A, Cheifetz S, Massague J. Science. 1988;240:196–199. doi: 10.1126/science.2895499. [DOI] [PubMed] [Google Scholar]
- 18.Markowitz S, Wang J, Myeroff L, Parsons R, Sun L, Lutterbaugh J, Fan R S, Zborowska E, Kinzler K W, Vogelstein B, Brattain M, Willson J K V. Science. 1995;268:1336–1338. doi: 10.1126/science.7761852. [DOI] [PubMed] [Google Scholar]
- 19.Myeroff L L, Parsons R, Kim S-J, Hedrick L, Cho K R, Orth K, Mathis M, Kinzler K W, Lutterbaugh J, Park K, Bang Y-J, Lee H Y, Park J-G, Lynch H T, Roberts A B, Vogelstein B, Markowitz S D. Cancer Res. 1995;55:5545–5547. [PubMed] [Google Scholar]
- 20.Park K, Kim S-J, Bang Y-J, Park H-G, Kim N K, Roberts A B, Sporn M B. Proc Natl Acad Sci USA. 1994;91:8772–8776. doi: 10.1073/pnas.91.19.8772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Inagaki M, Moustakas A, Lin H-Y, Lodish H F, Carr B I. Proc Natl Acad Sci USA. 1993;90:5359–5363. doi: 10.1073/pnas.90.11.5359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kadin M E, Cavaille-Coll M W, Gertz R, Massague J, Cheifetz S, George D. Proc Natl Acad Sci USA. 1994;91:6002–6006. doi: 10.1073/pnas.91.13.6002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Knaus P I, Lindemann D, DeCoteau J F, Perlmann R, Yankelev H, Hille M, Kadin M E, Lodish H F. Mol Cell Biol. 1996;16:3480–3488. doi: 10.1128/mcb.16.7.3480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim I-Y, Ahn H-J, Zelner D J, Shaw J W, Sensibar J A, Kim J-H, Kato M. Cancer Res. 1996;56:44–48. [PubMed] [Google Scholar]
- 25.Matutes E, Owusu-Ankomah K, Morilla R, Garcia Marco J, Houlihan A, Que T H, Catovsky D. Leukemia. 1994;8:1640–1645. [PubMed] [Google Scholar]
- 26.Newcom S R, Tagra K K, Kadin M E. Am J Pathol. 1992;140:709–718. [PMC free article] [PubMed] [Google Scholar]
- 27.Cheifetz S, Andres J L, Massague J. J Biol Chem. 1988;263:16984–16991. [PubMed] [Google Scholar]
- 28.Capocasale R J, Lamb R J, Vonderheid E C, Fox F E, Rook A H, Nowell P C, Moore J S. Proc Natl Acad Sci USA. 1995;92:5501–5505. doi: 10.1073/pnas.92.12.5501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Garrigue-Antar L, Munoz-Antonia T, Antonia S J, Gesmonde J, Velluci V F, Reis M. Cancer Res. 1995;55:3982–3987. [PubMed] [Google Scholar]
- 30.Kashles O, Yarden Y, Fischer R, Ullrich A, Schlessinger J. Mol Cell Biol. 1991;11:1454–1463. doi: 10.1128/mcb.11.3.1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tan J C, Nocka K, Ray P, Traktman P, Besmer P. Science. 1990;247:209–212. doi: 10.1126/science.1688471. [DOI] [PubMed] [Google Scholar]
- 32.Ueno H, Colbert H, Escobedo J A, Williams L T. Science. 1991;252:844–848. doi: 10.1126/science.1851331. [DOI] [PubMed] [Google Scholar]
- 33.Watowich S S, Hilton D J, Lodish H F. Mol Cell Biol. 1994;14:3535–3549. doi: 10.1128/mcb.14.6.3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Franzen P, ten Dijke P, Ichijo H, Yamashita H, Schulz P, Heldin C H, Miyazono K. Cell. 1993;75:681–692. doi: 10.1016/0092-8674(93)90489-d. [DOI] [PubMed] [Google Scholar]
- 35.Weis-Garcia F, Massague J. EMBO J. 1996;15:276–289. [PMC free article] [PubMed] [Google Scholar]