

Abstract
High-density oligonucleotide microarrays enable simultaneous monitoring of expression levels of tens of thousands of transcripts. For accurate detection and quantitation of transcripts in the presence of cellular mRNA, it is essential to design microarrays whose oligonucleotide probes produce hybridization intensities that accurately reflect the concentration of original mRNA. We present a model-based approach that predicts optimal probes by using sequence and empirical information. We constructed a thermodynamic model for hybridization behavior and determined the influence of empirical factors on the effective fitting parameters. We designed Affymetrix GeneChip probe arrays that contained all 25-mer probes for hundreds of human and yeast transcripts and collected data over a 4,000-fold concentration range. Multiple linear regression models were built to predict hybridization intensities of each probe at given target concentrations, and each intensity profile is summarized by a probe response metric. We selected probe sets to represent each transcript that were optimized with respect to responsiveness, independence (degree to which probe sequences are nonoverlapping), and uniqueness (lack of similarity to sequences in the expressed genomic background). We show that this approach is capable of selecting probes with high sensitivity and specificity for high-density oligonucleotide arrays.
Keywords: design, modeling, microarray design
High-density oligonucleotide arrays (1, 2) have revolutionized the study of gene expression. The technology enables researchers to detect and quantify tens of thousands of transcripts in a single experiment and has become a standard for the discovery of gene functions, drug evaluation, pathway dissection, and classification of clinical samples (3). With the availability of mRNA sequences for a large subset of the draft of the human genome (4, 5) microarrays provide the potential to simultaneously monitor the whole expressed human genome. The expression profile of the whole human genome will allow a detailed and comprehensive view of cellular processes, responses, and their functional consequences.
Quantitative detection of transcripts requires that microarray probes exhibit a sensitive and predictable response to concentrations of specific targets of the probes. This response must occur in the presence of a complex mixture of nonspecific targets. The previous probe selection method for GeneChip expression microarrays was to select candidate probes based on a set of heuristic rules (1). The rules act as filters to remove extreme sequence features that were known to degrade probe performance. However, probes passing the filters were treated as being of equal quality. To select optimal probe sets, it is essential to establish a continuous metric that distinguishes superior probes from merely good probes. Several theoretical studies of microarray probe selection (6, 7) were based on solution thermodynamics. No experimental data were produced to demonstrate that the theoretical predictions for hybridization in solution approximate hybridization behavior of immobilized probe systems, where hybridization behavior is more complex (8). Tobler et al. (9) showed the use of machine learning approaches to model the fluorescent intensity of a probe, and they defined good probes as those with intensities above a threshold. However, as discussed in this article, high intensity alone does not ensure that probes are responsive to specific targets.
The study presented here describes a model-based approach for prediction of optimal probe sets. We designed Affymetrix GeneChip probe arrays that contained all 25-mer probes for hundreds of human and yeast transcripts and collected data over a 4,000-fold concentration range. Multiple linear regression (MLR) models were built to predict hybridization intensities of each probe at given target concentrations, and each intensity profile is summarized by a continuous probe response metric that measures the intensity (I) response on the Ln-Ln (natural logarithm) scale to target concentration. This model-based probe selection system selects probe sets that are optimized with regard to this response metric and to uniqueness and independence. Our method combines a formal thermodynamic model with empirically derived parameters to fundamentally change the method of designing expression arrays.
Methods
Cloning and Target Preparation. Yeast ORFs were purchased from Invitrogen and then topoisomerase-cloned into the vector pCR4-TOPO (Invitrogen). Human cDNA fragments were cloned from cDNA made from a human lymphoblast cell line. Colony PCR products were used directly as in vitro transcription templates to produce biotinylated antisense RNA, and labeled RNAs were purified and fragmented following the method described in the Affymetrix gene expression manual. The complex background for yeast test experiments consisted of labeled mRNA from four human tissues: fetal brain, liver, lung and testis. The complex background of human test experiments consisted of labeled mRNA from human heart tissue where the target transcripts were knocked out in vitro (see Supporting Text, which is published as supporting information on the PNAS web site, www.pnas.org).
Probe Selection Test (PST) Set. Yeast and human cRNA transcripts (the targets) were spiked into labeled complex human backgrounds at known concentrations, and hybridization intensities were obtained for yeast test chips (YTC) and human test chips (HTC). Target groups were arranged in a classic Latin square design (10) so that each hybridization mixture contained at least one target at each chosen target concentration [T]. Ninety-nine yeast cRNA targets for YTC experiments were spiked at 14 concentrations ranging from 0.25 to 1,024 pM in 2-fold dilution steps and included 0 pM (no target present). Ninety cRNA targets for HTC experiments were spiked at 16 concentrations that included 0.0, 0.25, 0.50, 0.75, 1.00, 1.50, 2, 3, 4, 6, 8, 12, 16, 32, 128, and 512 pM. HTC targets included 6 bacterial and 84 human cRNAs. Hybridization and wash conditions were the same as indicated in the Affymetrix gene expression manual. Hybridization intensities were generated for the experiments according to the standard procedures for GeneChip expression probe arrays.
MLRs. MLRs (11) were used to fit the weight coefficients of Eqs. 4, 5, or 12. Values for dependent variables, Ln(I) or Y (Eq. 2), were computed from hybridization intensities produced by targets spiked at a common concentration. HC and HN are counts of the longest runs of consecutive and nonconsecutive base pairs in hairpin structures, respectively. Examples of consecutive and nonconsecutive hairpins are shown below.
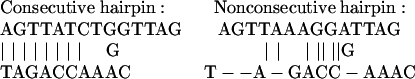 |
Qb, Qm, and Qe count the number of runs of four G bases in regions: b = 1–7, m = 8–15, and e = 16–22. Other independent variables are described in Results. All correlation coefficients for predicted vs. observed results were generated by using the standard cross-validation method (11), where test cases were held out of the cases used to train the model.
Linear and Sigmoid Model Equations. Eq. 11 [(Ln(I) = Ln(β + αC*[T][P]) + (–1/(RT*))ΔGd] predicts that Ln(I) increases linearly with ΔGd. However, when using predicted ΔGd from the first approximation, Eq. 12, or even a rough estimation of ΔGd based on the GC content of the probe, we observe that the relationship between Ln(I) and ΔGd is more closely approximated by a sigmoid function. Using a sigmoid equation to relate Ln(I) to ΔGd gives
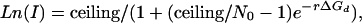 |
[1] |
where N0, r, and ceiling are constants of the sigmoid equation, and ceiling (the Ln intensity value at chemical saturation) = 8.5. Rearranging gives
 |
[2] |
where
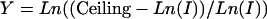 |
[3] |
and C3 = –r and C4 = Ln((Ceiling – N0)/N0) are constants. We add four quadratic terms to Eq. 8 (for ΔGd):
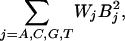 |
where B is the number of base type j, and then substitute into Eq. 2 to obtain the sigmoid model equation for MLR:
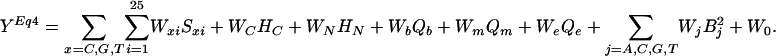 |
[4] |
And the predicted Ln(I)Eq4 equal Ceiling/(1 + eYEq4).
We add the quadratic terms:
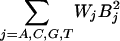 |
to Eq. 8 and substitute into Eq. 11 to obtain the linear model equation for MLR:
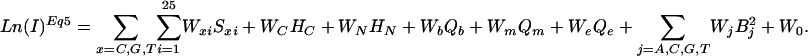 |
[5] |
Results
The Physical Model. In microarray systems, a probe (P) with one end tethered to a surface interacts with a target (T) in solution, forming a duplex (T·P) when there is a favorable free energy change (ΔGd). In this study, the probe sequence is DNA and target sequence is RNA. Throughout the article, thymine (T) and uridine (U) are used for probe and target sequence, respectively. The stability of the duplex depends on the free energy change, which is comprised of a collection of favorable and unfavorable interactions, among which are stacking energies, hydrogen bonding, secondary structure in both probe and target, and a variety of additional effects (12). We also postulate a dependence of the free energy on the position within the probe of each base and produce a model for the sequence dependence of ΔGd, which takes into account the positional contributions of each base to duplex stability and a subset of the possible unfavorable interactions.
We model ΔGd as the sum of contributions from each base at each position. Using the approach introduced by Hacia et al. (13), we model the relative free energy change relative to the free energy resulting from an A base at each position, ΔΔGd,
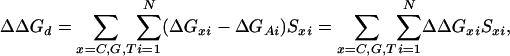 |
[6] |
where ΔGxi is the contribution of base x at position i to ΔGd and N is probe length and Sxi is the occupation variable,
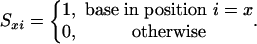 |
[7] |
We can then recover ΔGd as ΔΔGd + C0, where
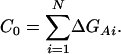 |
We consider three specific unfavorable interactions: consecutive hairpins (ΔGC), nonconsecutive hairpins (ΔGN), which are diagramed in Methods, and G quartets, which are hydrogen-bonded G tetraplexes (12). The contribution to the free energy is considered separately for the presence of a G quartet in the beginning (ΔGb), middle (ΔGm), and end (ΔGe) of the probe sequence. We combine terms for these unfavorable interactions with ΔΔGd and express ΔGd as
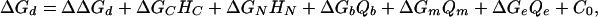 |
[8] |
where HN and HC are variables for the potential of the probe sequence to form nonconsecutive and consecutive hairpins, respectively (see Methods). The G quartet variables, Qb, Qm, and Qe, are counts of runs of four G bases in the beginning, middle, and end of the probe sequence (see Methods).
The physical model for the concentration of the target-probe duplex is then
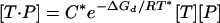 |
[9] |
in which [T] and [P] are the total concentrations of target and probe, R is the molar gas constant, T* is the temperature, and C* is a constant. Detailed derivations of Eq. 9 are described in Supporting Text. We further model fluorescent intensities as linear in [T·P],
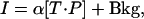 |
[10] |
where Bkg is the background hybridization of nonspecific targets to a given probe. The background (i.e., intensity measured in the absence of the specific target) increases with probe affinity. We empirically find that the variation in the background is reasonably explained by a Boltzmann-like model, Bkg = βe–ΔGd/RT* (see Fig. 5, which is published as supporting information on the PNAS web site). Because probe performance is fundamentally limited by background hybridization, we do not subtract background in the following equations. As discussed below, we select probes with stronger response to specific target than to the background. Using Eqs. 9 and 10 for a given target concentration [T], we have
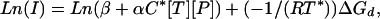 |
[11] |
where Ln(β + αC*[T][P]) and –1/(RT*) are constants for fixed target concentrations.
Linear Prediction of Intensity Values. Based on the physical model, we observe that the log of microarray intensity data are linear in all sequence-dependent terms. We can therefore build MLR models relating the log of intensity at a fixed concentration to our observed sequence. In particular, substituting Eq. 8 into Eq. 11, and simplifying terms, we obtain the following model (for 25-mer probes),
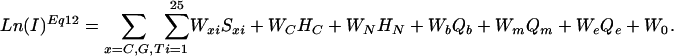 |
[12] |
The weights Wxi are the effective –ΔΔGxi values for the contribution to duplex stability of each base, x, in each position, i; and the weights, Wc, WN, Wb, Wm, and We are the negatives of the effective ΔGc, ΔGN, ΔGb, ΔGm, and ΔGe values, respectively. Eq. 12 serves as a first approximation model equation for MLR analysis (see Methods), and Ln(I)Eq12 refers to intensities predicted by a MLR solution to Eq. 12.
We applied MLR, using Eq. 12, to intensity data generated from two custom GeneChip microarrays, YTC and HTC. These custom arrays contain all 25-mer probes covering 600- to 1,000-bp regions of 99 YTC and 90 HTC transcripts, respectively. Two types of probe sequences covered each position in each transcript sequence: a perfect match (PM) probe with sequences exactly matching the cloned sequence, and a mismatch (MM) probe with a single substitution at the central position. Using multiple experiments, we obtained intensity data for each probe at known concentrations covering a 4,000 fold-range. Details of this PST set are in Methods.
Result of Linear Predictions of Intensities. Fig. 1 shows profiles of the effective –ΔΔGxi values for C, G, and T bases at each base position in PM probes (Fig. 1 A) and MM probes (Fig. 1B). The C base profile is the highest, which is consistent with the higher stability of GC base pairs. The lower height of the G base profile, relative to the C base, might be caused by the interference of labels on the C bases of target since when we changed the labeling method from fragment body labeling to fragment end labeling, this difference was observed only at end of fragments (data not shown). The –ΔΔG values decrease at the 3′ and 5′ ends of the probe, suggesting that the bases at ends of the probe have decreased contributions to duplex stability. The result is consistent with the cooperative behavior of duplex formation (14) and observation by Tobler et al. (9). The MM position at the center of the probe did not contribute to duplex stability, as expected (Fig. 1B). In addition, the –ΔΔG contributions of bases in noncentral positions of MM probes are decreased. These observations suggest that the center position contributes significantly to duplex stability. Thus, the fitted weights produced by MLR solution to Eq. 12 reflect our understanding of hybridization behavior.
Fig. 1.

The effective –ΔΔG values for the 25 probe base positions. The fitted weights, Wxi (Eq. 12), are the effective –ΔΔGxi values for the bases: C (red curve), G (green curve), and T (yellow curve) in each sequence position, i (i = 1 to 25 from the 3′ end of the probe), relative to the reference base, A, in the same position. (A) –ΔΔG values for PM probes. (B) –ΔΔG values for MM probes.
When MLR solutions to Eq. 12 are used to predict Ln(I)Eq12 values, there is good correlation with observed Ln(I) values. Profiles of observed Ln(I) and predicted Ln(I)Eq12 values for probes that cover a target sequence are shown for a representative yeast and human target spiked at 8 pM (Fig. 2 A and B). The correlation coefficients are 0.85 and 0.90 for yeast and human targets, respectively. The high correlation coefficients hold a >4,000-fold concentration range as shown in Fig. 2C. In addition, we found the hairpins terms contribute positively to ΔGd because of interference with target binding. In contrast, G quartets contribute negatively to ΔGd because of increase nonspecific target binding. Because only small percentage of the probe contains the hairpin and G quartet structures, the influence of the structures on overall correlation coefficients are small: 3% for hairpins and 2% for G quartets. Most of the variance in Ln(I) is explained by the first 75 terms of Eq. 12.
Fig. 2.

The predicted and observed Ln(I) values, given 8 pM target. Note that the errors in prediction are typically much smaller than the overall variation in intensity. Profiles are for predicted (blue line) Ln(I)Eq12 and observed (red dots) Ln(I) values for consecutive 25-mer probes covering the base positions of the target sequence. Predicted Ln(I)Eq12 values were computed by using MLR solutions to Eq. 12. (A) Ln(IPM) values for yeast target, GenBank accession no. YCL055W. Training set consisted of ≈98,000 probe sequences covering 98 YTC targets (excluding GenBank accession no. YCL055W). (B) Ln(IPM) values for human target, GenBank accession no. X51688_2844. Training set consisted of probes covering of ≈50,000 probe sequences covering 89 HTC targets (excluding GenBank accession no. X51688_2844). (C) Average correlation coefficients for predicted Ln(I)Eq12 vs. observed Ln(IPM) at each target concentration, [T], for ≈99,000 probes covering all 99 yeast targets. Predicted Ln(I)Eq12 intensities are produced by cross-validation, where probes for each target are held out of the training set used to produce the prediction model.
Probe Response Metric. The ability to predict Ln(I) values from probe sequence provides a foundation for probe selection. One essential criterion of probe selection for a quantitative expression analysis is that hybridization intensities of the selected probes have a predictable response to target concentration, [T]. In this section we present the rationale and results for a probe response metric.
Our first pass through our physical model led us to conclude that the log of intensity was a linear function of the free energy difference, holding the concentration constant (Eq. 11). Because we are interested in the response across concentrations, this equation can be rearranged to isolate the dependence on concentration. Neglecting background, we rewrite Eq. 11 as
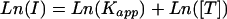 |
[13] |
with the apparent affinity constant Kapp = αC*e–ΔGd/RT*[P]. We observe the data better fits the form
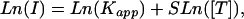 |
[14] |
where 0 < S < 1. This improved fit is primarily caused by the onset of chemical saturation (all available probe binding sites are occupied) of the probe feature (an area of the microarray that is covered by a set of oligonucleotides with a common sequence). Ln(Kapp) is the intercept and S is the slope (the Ln-Ln slope) of the line that relates Ln(I) to Ln([T]) (Fig. 3A, black line). As the Ln-Ln slope approaches one, the relationship between I and [T] approaches the ideal linear form, I = Kapp[T]. Selection of probes with Ln-Ln slopes closest to one maximizes the linearity of the relationship between intensity and target concentration. Therefore, we set the Ln-Ln slope, S, to be the probe response metric.
Fig. 3.

(A) Profiles of observed Ln(I) vs. Ln([T]) values for six probes. The Ln(Kapp) (intercept) values for the six probes range from 2.0 to 7.3 (shown in the label for each curve). The solid black line is the best fit (least squares) line that relates Ln(I) to Ln([T]) for the given probe. (B) Predicted (red dots) and observed (blue triangles) Ln-Ln slopes. Ln-Ln slopes are shown for probes covering a HTC target, GenBank accession no. AL049450_1209. MLR training sets consisted of the PST data sets for the remaining 89 HTC target clones. Ln-Ln slopes* are predicted by the prediction model. The errors in prediction are much smaller than the overall variation seen in the observed values.
The empirical data show that there are two classes of unresponsive probes, those whose hybridization affinities are either too high or too low. Probes with low Ln(Kapp) values also have low Ln-Ln slopes. Such probes (Fig. 3A, brown and green) are useless because of low hybridization affinities. As Ln(Kapp) increases, Ln-Ln slopes increase (Fig. 3A, pink and red) and probe intensity changes strongly with concentration. However, probes whose Ln(Kapp) values exceed a threshold exhibit decreasing Ln-Ln slopes with increasing Ln(Kapp) values (Fig. 3A, blue). These high-affinity probes become unresponsive to specific target because they cross-hybridize to nonspecific targets in the complex genomic background and saturate their binding sites.
We predict Ln-Ln slopes* and Ln(Kapp)* values by fitting a line through a set of points (Ln(I)*t, [T]t), where t = 1, P, consecutive target concentrations from the PST data set (see Methods). Ln(I)*t refers to the probe intensity predicted by the MLR solution to Eq. * (* = Eqs. 4, 5, or 12), at a fixed target concentration, [T]t. Thus, a set of P concentration-specific MLR models are used to predict a set of P Ln(I)*t values. We will refer to Ln-Ln slope and Ln(Kapp) values without the superscript (* = Eqs. 4, 5, or 12) as observed values. Profiles of observed Ln-Ln slope and predicted Ln-Ln slope* values for probes that cover a target sequence are shown for a representative human transcript (Fig. 3B).
Empirical Adjustments for Nonlinear Behavior. Eq. 12 does not account for chemical saturation, and therefore this model cannot predict the decreasing response of increasingly high affinity probes above a threshold. Instead it predicts that Ln-Ln slopesEq12 continue to increase, which is the behavior that would be observed in the absence of chemical saturation. We observe that a sigmoid model, which incorporates the existence of a ceiling for predicted Ln(I)* values caused by chemical saturation, gives a better fit than Eq. 12. The addition of quadratic terms,
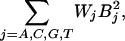 |
where B is the number of base type j, reduces the overprediction of slopes of high-affinity, GC-rich probes. One possible explanation is that these terms compensate for missing higher-order sequence interaction terms, such as nearest neighbor interactions and/or runs of poly G and poly C. Such runs have been empirically observed to influence hybridization behavior on microarrays (data not shown). Inclusion of the quadratic terms and use of a sigmoid function to relate Ln(I) to ΔGd gives the sigmoid model (Eq. 4, see Methods). This model predicts that Ln-Ln slopesEq4 decrease when Ln(Kapp)Eq4 values exceed a threshold.
As summarized in Table 1, the ability of models to generalize was tested by making predictions for HTC PST data set based on YTC models and vice versa. Sigmoid models created from one data set, correctly predicts for other data set, the trend of decreasing slope for probes with high Ln(Kapp)Eq4. However, some bias (either under or over prediction) is introduced in the slope predictions for probes with Ln(Kapp)Eq4 values below the threshold. We find that this data set bias is greatly reduced when using a version of the first approximation, the linear model (Eq. 5 and see Methods) for these lower-affinity probes. We therefore combined both model equations into the prediction model by using the sigmoid model equation for probes whose predicted Ln(Kapp)Eq5 values exceed a threshold, and the linear model equation for probes whose predicted Ln(Kapp)Eq5 values fall below a threshold. The average correlation coefficient for slope predictions of HTC probes based on an YTC model improved from 0.65 to the value of 0.73 reported in Table 1. The relationships between observed Ln-Ln slopes and predicted Ln(Kapp)* values are shown graphically in Fig. 6, which is published as supporting information on the PNAS web site.
Table 1. Correlation coefficients for predicted vs. observed values for YTC (99 yeast test array genes) and HTC (90 human test array genes) within the PST data set, using the prediction model.
| Correlation coefficients§
|
|||
|---|---|---|---|
| Model* | Target† | Ln-Ln slopes | Ln(IPM)‡ |
| HTC | HTC | 0.76 ± 0.10 | 0.83 ± 0.06 |
| YTC | YTC | 0.73 ± 0.09 | 0.85 ± 0.04 |
| YTC | HTC | 0.74 ± 0.12 | 0.85 ± 0.04 |
| HTC | YTC | 0.73 ± 0.08 | 0.82 ± 0.07 |
Array type of data set used to train the MLR models.
Array type of data set over which correlation coefficients were computed.
Ln(IPM) in the presence of 8 pM target concentration.
Correlation coefficient values for rows 1 and 2 were based on full cross-validation, where probes for a given target gene were withheld from the model used to predict the target. The process was repeated for every target in the data set.
Table 1 summarizes average correlation coefficients (averaged over all transcripts) between predicted and observed values, using the prediction model and a threshold of 5.7, and data for 90 HTC and 99 YTC transcripts. Average correlation coefficients are broken out according to the array type of the data used to train the model and the array type of the data predicted by the model. Full cross-validation (11) was used when the data set used for training the prediction model was the same as that used as the target of the model (rows one and two in Table 1). Average correlation coefficients for the four rows in Table 1 are ≈0.84 for Ln(IPM) values and 0.74 for slope values. Slope values are predicted with lower correlation because the fit is through a set of predicted Ln(IPM)* points and covers the lower range (0.25–16 pM) of target concentration. The prediction model appears to generalize well because models trained on YTC data can be used to predict HTC data and vice versa (Table 1, rows three and four).
Probe Selection System. Our model-based probe selection system takes the transcript sequence as input and uses a dynamic programming approach to select a probe set that optimizes a probe set score. The probe set score combines the continuous value of probe response Si, which in this case is the Ln-Ln slope (described above), and two penalty metrics into a single value for a set of N probes.
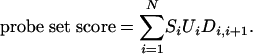 |
[15] |
The uniqueness penalty, U, identifies whether a probe is likely to cross-hybridize to other known expressed sequences in the genomic background. In this study, U is either zero (not unique) or one (unique), based on a sequence similarity rule (see Supporting Text). Additionally, we expect probes whose sequences overlap to be vulnerable to similar systematic errors. We hence introduce a multiplicative penalty term, D, based on the distance between the positions, P, in the target sequence that align with the centers of two consecutive probes, i and i + 1 (see Supporting Text). With a penalty term of this form (only dependent on consecutive probes) and exploiting the overall additive score, optimal probe sets can be generated by dynamic programming that finds maximal scoring sets by efficient recursion instead of by enumeration of all possible sets.
An MM probe (if desired) is then generated for each chosen PM probe. The system has been used for large-scale probe selections of whole organism expressed genomes, including the Hg-U133 human genome GeneChip microarray set. Fig. 4 gives a representative example of probes selected by the heuristic and model-based systems. The models and selection criteria achieve both the goal of selecting probes with higher Ln-Ln slopes, on average, than those selected by the previous heuristic system and better spacing between probes for more independent sampling of the target. The detailed comparison of performance of probe sets is described in Supporting Text (see Figs. 7–9, which are published as supporting information on the PNAS web site).
Fig. 4.

Example of probe selection. Observed slopes of all 25-mer probes are shown for one HTC target. Large blue circles enclose the Ln-Ln slopes of 11 probes selected by our model system, and small yellow circles enclose the Ln-Ln slopes of 16 probes selected by the previous heuristic system.
Discussion
We have demonstrated the ability of the probe selection system to model microarray hybridization intensities and built a prediction model that captures the sequence dependence of the complex hybridization behavior of immobilized probes in the presence of whole genomic background. The prediction model generates a continuous and quantitative metric for probe response. The combination of this response metric and the uniqueness and independence criteria enables selection of optimal probe sets in a systematic and large-scale manner.
Results presented here show that the prediction model produced good correlation coefficients for predicted vs. observed values, including Ln(I) and Ln-Ln slopes. However, the prediction model requires two different model equations, the sigmoid model equation (Eq. 4) for high-affinity probes and the linear equation (Eq. 5) for lower-affinity probes. The sigmoid model captures the nonlinear relationship between Ln(I) and ΔGd and assumes that Ln(I) values will approach a ceiling. This ceiling is more likely to be approached by high-affinity probe sequences, which become chemically saturated by nonspecific target. The sigmoid model was found to be less accurate than the linear model with lower-affinity probes. This discrepancy may be caused by the assumption of a single ceiling value required by the sigmoid model. However, the linear model results in overprediction of the Ln-Ln slopes of high-affinity probes. Thus the best prediction results were achieved by combining the two models. A promising direction for future work comes in applying a Langmuir adsorption isotherm (15, 16). The Langmuir isotherm naturally accounts for (i) the linear behavior in [T] and Kapp at low concentrations and hybridization affinities and (ii) nonlinear chemical saturation behavior as a function of [T] and Kapp at high concentrations and affinities.
Second, refinements also are needed on the model equations to account for remaining variations between predicted and observed Ln(I) values. Hybridization free energy depends not only on Watson–Crick base-pair interactions but also on stacking effects between neighboring bases. Therefore, one source of improvement is to add terms for nearest-neighbor interactions (17), which have been shown to adequately describe oligonucleotide duplex formation in solution. In addition, higher-order sequence interactions, such as runs of G and C bases, may be included in a systematic way. Finally, it is possible to reduce the number of fitting parameters by replacing the set of ΔΔG values for each base in each sequence position with a smooth function of probe base position. Fig. 1 A shows that positional contribution of each base, x, to ΔGd can be approximated by parabolic function of i, the distance from the center of the probe: Wxi = W0x + W1x*i + W2x*i2. Thus the 75 fitting parameters of Eq. 6 can be reduced to nine by replacing Eq. 6 with:
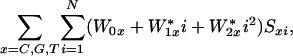 |
where Sxi is the indicator variable (Eq. 7).
Supplementary Material
Abbreviations: MLR, multiple linear regression; PST, probe selection test; YTC, yeast test chips; HTC, human test chips; PM, perfect match; MM, mismatch.
References
- 1.Lockhart, D. J., Dong, H., Byrne, M. C., Follettie, M. T., Gallo, M. V., Chee, M. S., Mittmann, M., Wang, C., Kobayashi, M., Horton, H. & Brown, E. L. (1996) Nat. Biotechnol. 14, 1675–1680. [DOI] [PubMed] [Google Scholar]
- 2.Lipshutz, R. J., Fodor, S. P. A., Gingeras, T. R. & Lockhart, D. J. (1999) Nat. Genet. 21, 20–24. [DOI] [PubMed] [Google Scholar]
- 3.Lander, E. S. (1999) Nat. Genet. 21, 3–4. [DOI] [PubMed] [Google Scholar]
- 4.Lander, E. S., Linton, L. M., Birren, B., Nusbaum, C., Zody, M. C., Baldwin, J., Devon, K., Dewar, K., Doyle, M. & FitzHugh, W. (2001) Nature 409, 860–921. [DOI] [PubMed] [Google Scholar]
- 5.Venter, J. C., Adams, M. D., Myers, E. W., Li, P. W., Mural, R. J., Sutton, G. G., Smith, H. O., Yandell, M., Evans, C. A. & Holt, R. A. (2001) Science 291, 1304–1351. [DOI] [PubMed] [Google Scholar]
- 6.Li, F. & Stormo, G. D. (2001) Bioinformatics 17, 1067–1076. [DOI] [PubMed] [Google Scholar]
- 7.Rouillard, J., Herbert, C. J. & Zucher, M. (2001) Bioinformatics 18, 486–487. [DOI] [PubMed] [Google Scholar]
- 8.Forman, J. E., Walton, I. D., Stern, D., Rava, R. P. & Trulson, M. O. (1998) Molecular Modeling of Nucleic Acids (Am. Chem. Soc., Washington, DC).
- 9.Tobler, J. B., Molla, M. N., Nuwaysir, E. F., Green, R. D. & Shavlik, J. W. (2002) in Bioinformatics Proceedings of the Tenth International Conference on Intelligent Systems for Molecular Biology, ed. Sander, C. (Oxford Univ. Press, Oxford, U.K.), S164–S171.
- 10.Box, G. E. P., Hunter, W. G. & Hunter, J. S. (1978) Statistics for Experimenters: An Introduction to Design, Data Analysis, and Model Building (Wiley, New York).
- 11.Hastie, T., Tibshirani, R. & Freidman, J. (2001) The Elements of Statistical Learning, Data Mining, Inference, and Prediction (Springer, New York).
- 12.Turner, D. H. (2000) in Nucleic Acids Structure, Properties, and Functions, eds. Bloomfield, V. A., Crothers, D. M. & Tinoco, I. (University Science Books, Sausalito, CA), pp. 259–334.
- 13.Hacia, J. G., Sun, B., Hunt, N., Edgemon, K., Mosbrook, D., Robbins, C., Fodor, S. P. A., Tagle, D. A. & Collins, F. S. (1998) Genome Res. 8, 1245–1258. [DOI] [PubMed] [Google Scholar]
- 14.Bloomfield, V. A., Crothers, D. M. & Tinoco, I. (2000) in Nucleic Acids Structure, Properties, and Functions, eds. Bloomfield, V. A., Crothers, D. M. & Tinoco, I. (University Science Books, Sausalito, CA), pp. 259–334.
- 15.Hekstra, D., Taussig, A. R., Magnasco, M. & Naef, F. (2003) Nucleic Acids Res. 31, 1962–1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Masel, R. I. (1996) Principles of Adsorption and Reaction on Solid Surfaces (Wiley, New York).
- 17.SantaLucia, J. (1998) Proc. Natl. Acad. Sci. USA 95, 1460–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.