

Abstract
The urbs1 gene encodes a transcriptional regulator of siderophore biosynthesis in Ustilago maydis. Biological and DNA-binding activities of the two putative zinc-finger motifs of Urbs1 were studied by analyzing mutants containing altered finger domains. The mutated urbs1 alleles from three previously described N′-methyl-N′-nitro-N-nitrosoguanidine (NTG) mutants were mapped and cloned by a gap-repair procedure. Sequence analyses revealed single amino acid substitutions in two of the NTG mutants. Both mutations (G-507 to D in urbs1–1 and P-491 to L in urbs1–3), which are located in the Urbs1 C-terminal finger domain, reduced DNA-binding activity by 10-fold and were sufficient to confer a urbs1–minus phenotype. The third NTG urbs1 mutant (urbs1–2) also contained a mutation in one of the conserved amino acids (P-518 to S) in the C-terminal finger domain, but this mutation alone was not sufficient to confer a urbs1–minus phenotype. A second frame shift mutation was identified in urbs1–2 and is necessary for the urbs1–minus phenotype. In an analysis of the function of the N-terminal finger of Urbs1, the conserved amino acid Arg-350 was mutated to leucine. A Urbs1 protein with this mutation complemented a urbs1 null mutant strain. By contrast, a similar mutation in the C-terminal domain abolished the ability of Urbs1 to regulate siderophore biosynthesis and greatly reduced its ability to bind target DNA.
Keywords: siderophore, transcription factor, corn smut, epitope, gel shift assay
Iron is one of the essential elements required by virtually all life forms (1). Since most iron exists in extremely insoluble complexes of ferric hydroxide in nature, the amount of iron that is biologically available to organisms is very limited (2, 3). In addition, too much iron in the cell can be detrimental, since in the presence of oxygen, iron can catalyze the production of cell damaging hydroxyl radicals (4). To balance the need for iron and its potentially harmful effects, living organisms have developed various elaborate iron uptake and storage systems (2, 3).
In response to iron starvation, Ustilago maydis produces two types of siderophores (microbial iron transport agents), ferrichrome and ferrichrome A, to harvest iron from the environment (5, 6). The first committed step in the siderophore biosynthesis pathway in U. maydis is the hydroxylation of l-ornithine to δ-N-hydroxyornithine catalyzed by the enzyme l-ornithine N5-oxygenase. sid1, the gene encoding this enzyme, has been previously characterized (7). urbs1, a gene encoding a regulator of siderophore biosynthesis belonging to the GATA family of transcription factors, has also been characterized (8). Recent genetic and molecular studies have shown that Urbs1 acts as a transcriptional repressor of sid1 by directly binding to GATA motifs in the sid1 promoter region (9).
Molecular characterization of urbs1 has revealed that it contains two putative zinc-finger motifs (8). With the exception of gaf2 from Schizosaccharomyces pombe (10) and SreP (11) from Penicillium chrysogenum, Urbs1 is the only reported GATA family transcription factor from fungi that has two-finger motifs (8, 12–17). Unlike the other double-finger-containing proteins from avians, mammals, and worms, in which the two fingers follow one another directly, the two-finger domains in Urbs1 are separated by 94 amino acids (8, 18–24). The finger domains of gaf2 and Srep are separated by 110 and 92 amino acids, respectively, and considerable identity exists over a 30 amino acid stretch in the respective intervening regions of Urbs1, gaf2, and SreP (8, 10, 11).
Mutagenesis and deletion analysis of mouse GATA-1 (25) and chicken GATA-1 (26) proteins has shown that the two fingers have distinct functions; the carboxyl finger is required for DNA binding, whereas the amino finger cooperates with the C-terminal finger to provide full stability and specificity of binding. These findings are supported by studies using mixtures of synthetic oligonucleotides as probes, in which the two finger domains of GATA-1 were shown to have different DNA-binding specificities (27, 28). Recently, Trainor et al. (29) demonstrated that the N-terminal finger of GATA-1 is required for stable association with a palindromic GATA motif of the chicken and mouse GATA-1 promoter. These palindromic GATA sites are required for optimal expression of GATA-1. Consistent with this finding, Visvader et al. (30) found that the C-terminal zinc finger of human GATA-1 and GATA-2 is sufficient to induce megakaryocytic differentiation of an early myeloid cell line, whereas the N-terminal finger affects the level of differentiation. While not required for binding to the T cell receptor promoter, the N-terminal finger of human GATA-3, along with sequences immediately flanking this finger, are required for nucleus localization (31). Thus, the N-terminal finger of GATA factors can have subtle to profound affects on the level of target gene activation. Interestingly, the Aspergillus nidulans AreA-300 didactyly mutant, which contains a 417-bp duplication of the single-finger motif, has an altered specificity of gene activation (32). Studying the functions of the uniquely organized fingers of Urbs1 may shed light on the relationship of structure/function and evolution of the finger domains of GATA family transcription factors.
In this report we describe the analysis of biological and DNA-binding activities of chemically induced and site-directed finger mutants of Urbs1. Our results demonstrate that the C-terminal finger of Urbs1 is essential for DNA binding and iron-mediated regulation of siderophore production in U. maydis. By contrast, a mutation at one of the conserved amino acids in the N-terminal finger domain of Urbs1 had no discernible effect on its biological function in siderophore gene regulation when expressed on a multicopy plasmid despite reducing its DNA-binding activity.
MATERIALS AND METHODS
Reagents.
Restriction endonucleases and DNA modifying enzymes were from New England Biolabs. Reagents from other sources are identified in the text. Low iron, high iron, and complete media were prepared as described (33, 34). Transformation agar consisted of complete medium containing 1 M sorbitol and 300 μg of hygromycin B per ml. Siderophore detection medium consisted of transformation agar containing 1 mM FeSO4. Potato dextrose agar medium was from Difco. Siderophore concentration was determined by using the ferric perchlorate assay (35). Synthetic oligonucleotide primers were made by the Applied Biosystems 392 DNA/RNA synthesizer.
Strains.
Escherichia coli DH5α (Bethesda Research Laboratories) was used for all DNA manipulations. U. maydis strains used in this paper are listed in Table 1. N-methyl-N′-nitro-N-nitrosoguanidine (NTG) urbs1 mutant strains (UMC002, UMC005, and UMC007) have been described (8). UMC015, a gene disruption mutant of urbs1, was constructed as follows: The internal 2-kb StuI fragment of urbs1 in pSC3 (Fig. 1A) (8) was replaced by blunt-end ligation with the 2-kb XbaI/HindIII fragment of pUble20 (36), which contains the Tn5-phleo U. maydis expression cassette (conferring resistance to phleomycin) to give pUWAN41. The XbaI/BamHI fragment in the urbs1 locus (Fig. 1A) in the genome of strain 518 was then recombinationally replaced with the XbaI/BamHI fragment of pUWAN41 containing the Tn5-phleo cassette. Gene replacements were scored by screening for the orange colony phenotype of urbs1 mutants on siderophore detection medium containing 1 mM FeSO4 (8) and confirmed by Southern hybridization analysis.
Table 1.
U. maydis strains used in this study
| Strain | Relevant characteristics |
|---|---|
| 518* | Wild-type a2b2 |
| UMC015† | a2b2 urbs1::phleor |
| UMC002‡ | a2b2 urbs1-1 NTG mutant of 518 |
| UMC005‡ | a2b2 urbs1-2 NTG mutant of 518 |
| UMC007‡ | a2b2 urbs1-3 NTG mutant of 518 |
| UMC002-1† | a2b2 urbs1-1 gene replacement mutant |
| UMC005-1† | a2b2 urbs1-2 gene replacement mutant |
| UMC007-1† | a2b2 urbs1-3 gene replacement mutant |
Obtained from R. Holliday (Commonwealth Scientific and Industrial Research Organization, Laboratory for Molecular Biology, Sydney).
Constructed by gene replacement as described in the text.
NTG urbs1 mutants of 518 (8).
Figure 1.
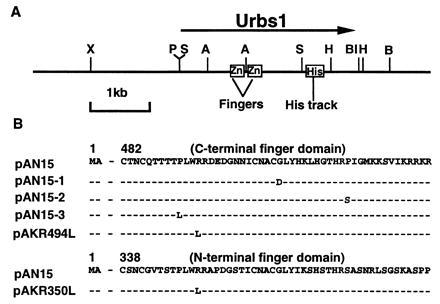
(A) urbs1 locus of U. maydis. Horizontal arrow represents the urbs1 open reading frame (ORF). Restriction sites: A, AsuII; B, BamHI; BI, BglII; H, HindIII; P, PstI; S, StuI; X, XbaI. (B) Epitope-tagged urbs1 alleles. pAN15 contains the wild-type allele. pAN15–1, pAN15–2, and pAN15–3 contain the NTG-induced urbs1 mutations contained in the 2-kb StuI fragment from UMC002, UMC005, and UMC007, respectively. pAKR494L and pAKR350L contain single amino acid mutations introduced via site-directed mutagenesis.
DNA Procedures.
A microfuge tube-based (8, 37) procedure was employed for U. maydis transformation. Plasmid DNA was prepared from E. coli by the boiling minipreparation method (38) or by the ammonium acetate differential precipitation method (39). U. maydis genomic DNA was isolated by the glass-bead protocol (40) or the protoplast lysis method (37). DNA transformation of E. coli, Southern hybridization, and other DNA procedures was carried out as described by Sambrook et al. (41) or as modified by Mei et al. (7). DNA sequencing was by the dideoxy method using Sequenase (United States Biochemical).
Mapping and Cloning of the Mutated urbs1 Loci from Three NTG-Induced Mutants by a Gap-Repair Procedure.
The mutations of two NTG-induced urbs1 mutants, UMC002 and UMC007, were determined by a gap-repair procedure (42) with two gapped plasmids pSC3ΔAsu and pSC3ΔStu. These plasmids were constructed by deleting the internal 0.7-kb AsuII or 2-kb StuI fragment of pSC3 (8), a self-replicating plasmid that contains the two putative zinc-finger motifs of Urbs1 (Fig. 1A). To generate gap-repaired plasmids, pSC3ΔAsu or pSC3ΔStu DNA was linearized at the AsuII or StuI site and then introduced into UMC002 and UMC007 by transformation selecting for resistance to hygromycin B. A mutation(s) was mapped by testing the ability of gapped DNA to complement the urbs1–minus phenotype as determined using the siderophore detection medium (8). If all of the transformants were orange (no white colonies) on this medium, the mutation(s) most likely mapped within the segment of DNA corresponding to the gapped region. Genomic DNA from selected orange transformants was isolated and digested with either BamHI or XbaI to release the repaired plasmid that was circularized by intramolecular ligation and used to transform E. coli. No transformants were obtained when untreated genomic DNA was used directly to transform E. coli. The vector appeared to have integrated as shown by Southern hybridization analysis of uncut and XbaI-digested genomic DNA (data not shown). Plasmid DNA was isolated from ampr E. coli transformants, and plasmids carrying the putative mutated urbs1 loci were then introduced into respective U. maydis urbs1 mutants for complementation testing using the siderophore detection agar. Mutations were identified within the repaired DNA by sequencing. The mutant urbs1 allele from the third mutant, UMC005, was similarly cloned using pSC3ΔStu, and the mutations were identified by DNA sequencing.
Site-Directed Mutagenesis.
The Muta-Gene phagemid in vitro mutagenesis kit (Bio-Rad) was employed. The 2.6-kb PstI/HindIII fragment of urbs1 was cloned at the PstI/HindIII sites of pTZ19U to give pAK1 (Fig. 1A). The mutagenic primers used to introduce amino acid changes were: 5′-ACTTCGACTCCGCTTTGGCTAAGAGCCCCCGAT for amino acid Arg-350 to leucine and 5′-AATTGCCAGACGACCACCACCCCACTGTGGCTACGCGACGAAGACGGCAACAATATCTGCAA for Arg-494 to leucine. Mutations were confirmed by DNA sequencing.
Construction of Epitope-Tagged Mutant urbs1 Alleles.
Plasmid pAN15 contains the wild-type urbs1 allele tagged at its C terminus with the triple influenza hemagglutinin (HA) epitope (9). Tagged mutant alleles were obtained by replacing the 2-kb StuI fragment in pAN15 with the StuI fragment from the respective mutant alleles recovered by gap repair.
Preparation of U. maydis Cellular Extracts, SDS/PAGE of Protein, and Western Blotting.
U. maydis cells, grown for 3 days at 28°C in 250 ml of low iron medium with 10 μM FeSO4 and 300 μg/ml hygromycin B to select for plasmids bearing hygromycin B resistance, were harvested and washed with Hepes buffer (50 mM Hepes, pH 7.5/100 mM KCl/1 mM EDTA/1 mM DTT/10 mM phenylmethylsulfonyl flouride). Protein extracts were prepared and analyzed by SDS/PAGE and Western blotting as described (9). The signal obtained on autoradiograms of Western blots was quantitated by scanning image analysis using Molecular Dynamics Personal Densitometer SI. Experimental determinations were repeated multiple times.
DNA-Binding Assays.
Electrophoretic mobility shift analysis was done with a 0.3-kb DNA fragment containing the two distal GATAs in the sid1 promoter region (−2594 to −2288 bp) as described (9). The ratio of the bound over total probe was determined by scanning the resulting autoradiogram with Molecular Dynamics Personal Densitometer SI. All experimental determinations were repeated multiple times.
RESULTS
Characterization of urbs1 Mutants.
A gap-repair procedure (42) was used to identify the mutations in three NTG urbs1 mutants (8). Initially, the mutations in UMC002 (Urbs1–1) and UMC007 (Urbs1–3) were localized by testing for restoration of the white colony, wild-type phenotype on siderophore detection medium following the introduction of either of two gapped plasmids, pSC3ΔAsu or pSC3ΔStu (Fig. 1A), into these strains. The plasmid pSC3 containing the wild-type copy of urbs1, was included as a control. Whereas one of every six transformants into which pSC3ΔAsu displayed the wild-type phenotype in UMC002 (urbs1–1) and UMC007 (urbs1–3), no complementation was observed when introducing pSC3ΔStu into the strains. These results suggest that the mutations in both mutants reside outside the 0.7-kb AsuII fragment, but within the 2-kb StuI fragment (Fig. 1). The urbs1 mutant alleles from these strains were subsequently isolated by recovering the gap-repaired pSC3ΔStu plasmids. The StuI fragments were then sequenced to determine the nature of the mutations. Both mutant alleles had a single nucleotide change resulting in one altered amino acid. In urbs1–1, glycine (G) at position 507 of the urbs1 ORF was mutated to an aspartic acid (D) (GGT to GAT) and in urbs1–3, proline (P) at position 491 was mutated to leucine (L) (CCA to CTA) (Fig. 1B). Both amino acids fall within the C-terminal zinc-finger domain of Urbs1 and are among the most highly conserved amino acid residues of finger domains of the GATA family of transcription factors. On the basis of the findings in UMC002 (urbs1–1) and UMC007 (urbs1–3), it was hypothesized that the mutation(s) in UMC005 (urbs1–2) might also be within the 2-kb StuI region. This was confirmed when the UMC005 urbs1 StuI fragment was recovered using the same gap-repair procedure. The sequence of this fragment revealed a one nucleotide mutation (CCC to TCC) at position 518 resulting in a proline (P) to a serine (S) substitution (Fig. 1B). This mutation is also located at a consensus amino acid within the C-terminal finger domain of Urbs1.
Functional Analysis of the Three Independent urbs1 Mutations.
To determine whether these single amino acid changes were sufficient to produce the urbs1–minus phenotype, the urbs1 gene disruption mutant UMC015 was transformed with the mutant urbs1 alleles recovered by gap repair. None of the three mutant alleles complemented the orange phenotype of UMC015 on siderophore detection medium (data not shown). To further confirm that the sequenced mutation was causal to the phenotype, the 2-kb StuI fragments from mutant alleles were cloned in place of the wild-type StuI fragment in a wild-type urbs1 gene. To avoid possible copy number effects, the phleomycin cassette-containing urbs1 allele of UMC015 was replaced with the mutant urbs1 genes by gene replacement. Double crossover events resulted in phleomycin-sensitive transformants. The presence of mutations at the urbs1 locus in the genome was confirmed by Southern hybridization analysis and sequencing of a PCR-generated StuI fragment of each allele. Regulation of siderophore production by iron in the resultant U. maydis strains (UMC002–1, UMC005–1, and UMC007–1) was tested (Table 2). The single mutations in both UMC002–1 and UMC007–1 were sufficient to give the urbs1–minus phenotype, as indicated by the low iron/high iron ratio of siderophore production (Table 2). To our surprise, the mutation in UMC005–1 was not sufficient to give a urbs1–minus phenotype; the strain carrying this allele had a wild-type phenotype. This latter result suggested that an additional mutation(s) must be present in UMC005 outside of the StuI fragment but within the urbs1 locus recovered by the gap-repair procedure. DNA recovered via gap repair may include sequences adjacent to the region represented by the plasmid gap (42). To locate another mutation(s) in this allele, regions flanking the StuI fragment were sequenced in the gap-repaired plasmid. An insertion of a cytosine at nucleotide position 2,795 of the ORF was found. This frame-shift mutation resulted in the complete change of the amino acid sequence after residue 932, adding 122 novel amino acids at the C-terminus of Urbs1 (data not shown). The presence of both mutations in the genome of UMC005 was confirmed by PCR amplification and sequencing of the relevant regions of the urbs1 ORF.
Table 2.
Siderophore production by wild-type 518, urbs1 gene disruption mutant UMC015, and urbs1 NTG mutants carrying a single amino acid mutation in the C-terminal finger domain under low and high iron conditions
| Strain | Low iron | High iron | LI/HI |
|---|---|---|---|
| 518 | 0.12 ± 0.08* | 0.01 ± 0.005 | 12 |
| UMC015 | 0.16 ± 0.02 | 0.05 ± 0.01 | 3.2 |
| UMC002-1 (urbs1-1) | 0.14 ± 0.08 | 0.06 ± 0.02 | 2.3 |
| UMC005-1 (urbs1-2) | 0.16 ± 0.01 | 0.01 ± 0.00 | 16 |
| UMC007-1 (urbs1-3) | 0.14 ± 0.02 | 0.04 ± 0.01 | 3.5 |
Cells were grown for 48 h at 28°C in low iron medium with or without 10 μM FeSO4. Siderophore production was determined by the ferric perchlorate assay as described. Equal volumes of cell free supernatant and assay solution were mixed and the adsorbance read at 495 nm. Data were normalized to cell growth using an adsorbance at 600 nm equal to 1.0. LI, low iron; HI, high iron.
Means of three replications ± SE.
Epitope Tagging of Mutant urbs1 Alleles and Detection of Mutant Urbs1 Protein in U. maydis.
To study the in vitro and in vivo activities of the mutant urbs1 alleles, the C terminus was tagged with the influenza HA antigen (YPYDVPDYA)3 as described (9). Complementation studies previously showed that the wild-type tagged Urbs1 was biologically functional even at single copy (9). The 2-kb StuI fragments from the mutant urbs1 alleles were used to replace the 2-kb StuI fragment in pAN15, which carries wild-type epitope-tagged Urbs1 (9). For UMC005, the tagged chimeric allele, pAN15–2, includes only the mutation from proline (P) to serine (S) in the C-terminal finger domain (Fig. 1B). Complementation of the urbs1 gene disruption mutation of UMC015 with pAN15–1 (urbs1–1), pAN15–2 (urbs1–2), and pAN15–3 (urbs1–3) was tested in low- and high-iron media. Plasmids pAN15–1 and pAN15–3 did not complement the urbs1− mutation, whereas plasmid pAN15–2 did (data not shown). Production of the mutant Urbs1 protein was determined by Western blotting using the anti-HA antibody as shown in Fig. 2. Total protein extract from the urbs1 mutant UMC015 or this mutant restored with the wild-type urbs1 (pSC3) did not react with the anti-HA antibody. When UMC015 was transformed with the epitope-tagged wild-type urbs1 allele (pAN15) and the three tagged mutant alleles (pAN15–1, pAN15–2, and pAN15–3), a protein with the predicted molecular weight of the epitope-tagged Urbs1 (110 kDa), was detected (Fig. 2). Even though the same amount of total protein was loaded in each lane, stronger signals were detected for the tagged protein in extracts from strains containing the mutant alleles. Scanning-image analysis showed that at least 4–5 times as much Urbs1 accumulated in strains carrying the mutant alleles as those carrying the wild-type allele.
Figure 2.
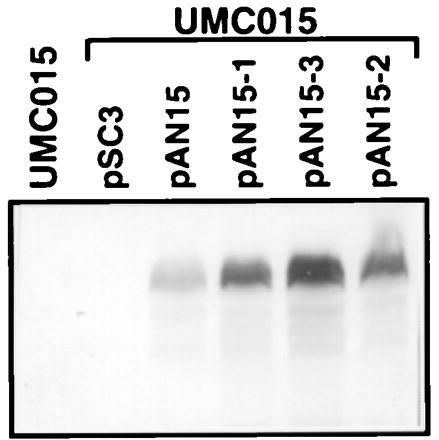
Detection of epitope-tagged wild-type (pAN15) and mutant (pAN15–1, pAN15–2, and pAN15–3) Urbs1 by Western blotting. UMC015 is a urbs1 disruption mutant. pSC3 carries the wild-type urbs1 gene. pAN15 carries the urbs1-tag gene. pAN15–1, pAN15–2, and pAN15–3 carry the tagged mutant urbs1 alleles from UMC002 (urbs1–1), UMC005 (urbs1–2), and UMC007 (urbs1–3), respectively. Assay conditions are as previously described.
Binding of the Three Mutant Urbs1 Proteins to the Distal GATA Motifs in the sid1 Promoter Region.
Urbs1 encodes a transcriptional repressor that interacts directly with a GATA sequence in the promoter region of sid1 (8, 9). To test the DNA-binding activity of the three mutant Urbs1 proteins, an in vitro gel shift assay was conducted. As shown in Fig. 3, total protein extract from the urbs1 disruption mutant UMC015 did not shift the target DNA, whereas extracts from strains expressing wild-type urbs1 (UMC015/pSC3) and the wild-type urbs1 with the tag (UMC015/pAN15) shifted the GATA-containing probe. Extracts from UMC015/pAN15–2 expressing the urbs1 mutant allele containing the P to S mutation from UMC005 shifted the target DNA to the same extent as extracts expressing the wild-type Urbs1 (Fig. 3). The gel shifted bands obtained from extracts of strains UMC015/pAN15–1 and UMC015/pAN15–3, expressing the tagged mutant alleles from UMC002 and UMC007, were even weaker (Fig. 3). We estimate, after adjusting for the elevated amounts of Urbs1 in the mutant extracts (Fig. 2), that the mutant Urbs1 bound 5 (UMC005) to 10 times (UMC002 and UMC007) less probe than the wild type.
Figure 3.
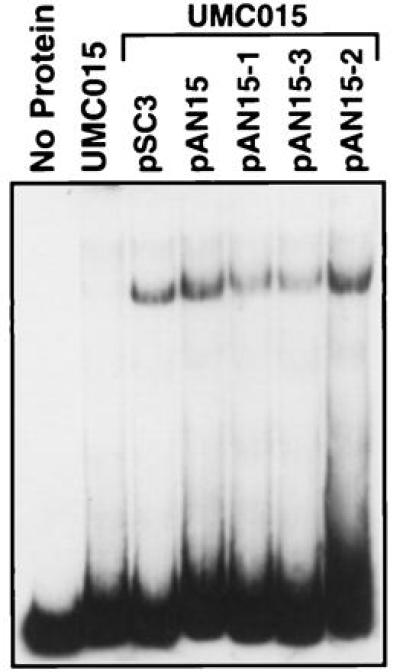
DNA-binding assay of the 0.3-kb DNA fragment with whole cell protein extracts of UMC015, UMC015/pSC3, UMC015/pAN15, UMC015/pAN15–1, UMC015/pAN15–2, and UMC015/pAN15–3. Strain UMC015 and various plasmids are described in Fig. 1 and in the text. Assay conditions are as previously described.
Function of the N-Terminal Finger Domain of Urbs1 as Tested by Site-Directed Mutagenesis.
To determine whether the N-terminal finger domain also is involved in the function of Urbs1, a urbs1 allele with an arginine to leucine mutation at Arg-350 in the N-terminal finger domain was constructed by site-directed mutagenesis (pAKR350L, Fig. 1B). As a control, an arginine to leucine mutation was generated at Arg-494 in the C-terminal finger domain (pAKR494L, Fig. 1B). The effects of these mutations on Urbs1 function were tested by attempting complementation of the urbs1 gene disruption in UMC015 with pAKR350L and pAKR494L. The Arg-350 to leucine mutation in the N-terminal finger domain did not affect Urbs1 function in siderophore production as UMC015 was restored to the wild-type phenotype by pAKR350L (Table 3). By contrast, the analogous Arg-494 to leucine mutation in the C-terminal finger domain abolished Urbs1 function as indicated by the constitutive siderophore synthesis in UMC015 carrying pAKR494L (Table 3). DNA-binding activity of the two mutant Urbs1 proteins was tested by gel shift assay. As shown in Fig. 4A, a total protein extract from UMC015/pAKR350L shifted the probe and the intensity of the band shift was approximately the same as with UMC015/pAN15. By contrast, the band shift obtained with the extract of UMC015/pAKR494L extract was relatively weak. As with the NTG-induced mutant alleles of Urbs1, Western blot analysis of protein extracts from UMC015/pAKR350L and UMC015/pAKR494L showed 4- to 8-fold enhanced amounts of Urbs1 relative to UMC015/pAN15 (Fig. 4B).
Table 3.
Siderophore production by urbs1 gene disruption mutant UMC015 transformed with plasmid containing epitope-tagged wild-type urbs1 (pAN15) and in vitro mutated urbs1 alleles (pAKR350L and pAKR494L) under low and high iron conditions
| Strain | Low iron | High iron | LI/HI |
|---|---|---|---|
| UMC015/pAN15 | 0.12 ± 0.04* | 0.02 ± 0.00 | 6 |
| UMC015/pAKR350L | 0.12 ± 0.01 | 0.02 ± 0.00 | 6 |
| UMC015/pAKR494L | 0.17 ± 0.02 | 0.13 ± 0.02 | 1.3 |
Cells were grown for 48 h at 28°C in low iron medium with or without 10 μM FeSO4. Siderophore production was determined by the ferric perchlorate assay as described. Values were normalized to a cell growth of OD600 = 1.0. LI, low iron; HI, high iron.
Means of three replications ± SE.
Figure 4.
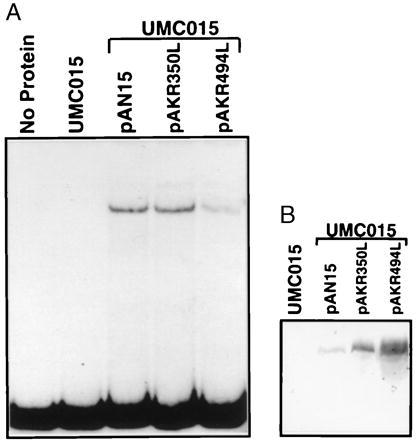
(A) DNA-binding assay of the 0.3-kb DNA fragment with whole cell protein extracts of UMC015, UMC015/pAN15, UMC015/pAKR350L, and UMC015/pAKR494L. Strain UMC015 is described in the text. pAKR350L and pAKR494L carry the Arg-350 to leucine mutation in the N-terminal finger domain and the analogous Arg-494 to leucine mutation in the C-terminal finger domain, respectively. Assay conditions are as previously described. (B) Detection of epitope-tagged wild-type (pAN15) and mutant (pAKR350L and pAKR494L) Urbs1 by Western blotting. Conditions are as in Fig. 2.
DISCUSSION
By applying the gap-repair procedure developed for Saccharomyces cerevisiae (42) and an easily scorable plate assay for siderophore production, mutations in three independent, NTG-generated urbs1 mutant alleles have been mapped and cloned. This procedure has proven to be very efficient in U. maydis as reported by Ferguson and Holloman (43).
The StuI fragment within the urbs1 region was determined to contain the mutations responsible for the phenotype of UMC002 and UMC007. The urbs1 allele from the remaining NTG mutant, UMC005, was also recovered. As expected, the gap-repaired plasmid would not complement the mutation in UMC005, and a base pair substitution was discovered within the StuI fragment. This mutation, however, was not sufficient to produce the urbs1–minus phenotype as determined by introducing the recovered StuI fragment into an otherwise wild-type urbs1 gene. Additional sequencing uncovered a second mutation near the 3′ end of the gene resulting in a frame shift and the addition of 122 novel amino acids to the C terminus of Urbs1. Evidently, the gap-repair procedure resulted not only in the recovery of the gapped DNA, but also DNA adjacent to the gapped region. Either this second frame-shift mutation or the combination of both mutations is responsible for the urbs1–minus phenotype in UMC005. Since only one gap-repaired plasmid was recovered in the analysis of UMC005, this finding indicates that multiple overlapping gapped constructs should be used to map the mutation prior to further analysis.
Variability has been observed in the quantity of siderophore secreted by urbs1 mutant strains grown in the presence of iron (refs. 8 and 9 and this study). This can be attributed in part to inherent differences seen in siderophore production from experiment to experiment and by different approaches to normalize siderophore production (ref. 9 and this study). At first view, the observation of reduced levels of siderophore produced in high iron medium by urbs1 mutants suggests that some iron regulation exists in these mutants (Tables 2 and 3). However, primer extension, Northern hybridization, and β-glucoronidase reporter analyses have demonstrated a nearly absolute regulation of siderophore gene expression by Urbs1 and iron (refs 6, 7, and 9, and W.M.Y. and S.A.L., unpublished results). The orange colony phenotype of urbs1 mutants suggests that siderophores are hyperaccumulated within cells. Extraction of wild-type and urbs1 cells for siderophores has revealed abnormally high levels of cell-associated siderophores in urbs1 mutant UMC015 grown in the presence of iron (A. D. Budde and S.A.L., unpublished results) and low levels in the wild type consistent with those previously reported (5). The quantities of cell-associated siderophore found in the urbs1 mutant were sufficient to account for the decreased amounts found extracellularly in high iron medium.
The fact that single base pair substitutions in the C-terminal finger domain of Urbs1 (Fig. 1B) can be sufficient for a urbs1–minus phenotype demonstrates that the C-terminal finger domain of Urbs1 is required for the iron-mediated regulation of siderophore production. Sequence alignment with the consensus sequence of GATA family transcription factors indicated that the single base pair substitutions occur at conserved amino acids in the finger domains of GATA family transcription factors (8). Identical mutations in corresponding conserved residues in the single-finger domain of AreA lead to similar effects on DNA binding as reported here. For example, the G507D mutant of Urbs1 corresponds to the loss-of-function areAr217 mutant (12). The P518S mutant of Urbs1 corresponds to the areA120/1203 (P709S) mutant, which does not result in loss-of-function (44).
Similar structure–function correlations can be made with the N- and C-terminal fingers of Urbs1, GATA-1, and GATA-2. Site-directed mutation of the highly conserved Arg-494 in the C-terminal finger of Urbs1 impaired Urbs1 function in DNA binding. This residue is critical to both the structure of the C-terminal finger domain of GATA-1 and its interaction with the major groove of DNA (45). The finding that the corresponding Arg-350 finger mutation of Urbs1 had only a moderate effect on DNA binding and no visible effect on siderophore gene regulation mirrors findings reported for GATA-1 and GATA-2. Visvader et al. (30) found that the C-terminal finger domain of human GATA-1 or GATA-2 is sufficient to induce megakaryocytic differentiation of an early myeloid cell line, whereas the N-terminal finger plays a role in the attenuation of differentiation. In the mouse and chicken erythroid factor GATA-1, the C-terminal finger is essential for DNA binding, whereas the N-terminal finger can help to achieve binding specificity and stability (25, 26). Finally, stable binding by GATA-1 to high-affinity palindromic GATA sites in the mouse and chicken GATA-1 promoters is dependent on both finger domains (29). The target binding sites of Urbs1 in the sid1 promoter also contain palindromic GATA sequences (9). DNA footprinting and off-rate analysis of the wild-type and Arg-350-Leu Urbs1 proteins with these palindromic GATAs may shed further light on the function of the N-terminal finger in DNA binding. Phenotypic analysis of the Arg-350-Leu urbs1 at single copy in place of wild-type urbs1 may reveal a phenotype missed in this study; the elevated amounts of Arg-350-Leu mutant protein produced from the multicopy plasmid may account for our failure to detect an effect on siderophore gene regulation.
In a study on the DNA-binding specificity of GATA proteins, Merika and Orkin (28) constructed chimeric proteins between the body of GATA-1 and the finger domain of AreA. They found that the AreA finger or carboxyl finger of GATA-1 alone were slightly more restricted in sequence specificity than those of some two-finger GATA proteins. On the basis of these results, they speculated that the addition of a second finger might permit a wider DNA-binding specificity (28). Consistent with this hypothesis is the finding that the AreA-300 mutant, which contains two finger motifs, gains the ability to regulate new genes while losing the ability to regulate others (32). Interestingly, the single fingers of several fungal GATA proteins (12–16) are more closely related to the carboxyl finger than the amino finger of the vertebrate members. We previously reported that the sequence identity of the C-terminal finger motif of Urbs1 and the single-finger motif of Nit-2 was 64% over 50 amino acids (8). By extending the analysis to include sequences upstream and downstream of these fingers, we found additional sequence identity, albeit lower. Comparison of residues 328–734 of Urbs1 with residues 666–1,020 of Nit-2 using the Global Alignment Program of the Genetics Computer Group gave 49.3% similarity and 29.8% identity. These comparisons further support the critical role of the C-terminal finger domain of Urbs1 and suggest a possible common evolutionary origin for these two proteins.
The two fingers in Urbs1 are separated by 94 amino acids (8), whereas those of other two-finger GATA family members directly follow one another (8, 16, 19–24). The two exceptions are gaf2 and SreP, in which the two fingers are separated by 110 (10) and 92 amino acids (11), respectively. Interestingly, the region between the two-finger domains of gaf2 and SreP also shows considerable sequence conservation with that of Urbs1. The unique organization of these fingers suggests that Urbs1, gaf2, and SreP may belong to a new subgroup of transcription factors within the GATA family.
Epitope tagging and Western blot analysis of the Urbs1 mutant alleles indicated that not only were the mutant proteins expressed, but they were expressed in greater quantities; that is, when compared with accumulation of wild-type Urbs1, at least four times more mutant protein accumulated in cells carrying the mutant alleles. These results suggested that expression of Urbs1 in the cell is autoregulated. DNA sequence analysis of the 1.1-kb urbs1 promoter region has identified 10 GATA boxes upstream of the two major transcription sites of urbs1 (GenBank accession L54058L54058). Some of these GATA sites could serve as DNA-binding sites for Urbs1. The autoregulation model predicts that Urbs1 acts as a transcriptional repressor of its own promoter by binding to the GATA(s) in the promoter region of its gene. Voisard et al. (8) found similar levels of urbs1 transcript in low and high iron grown cells of U. maydis, indicating that autoregulation by Urbs1 is independent of iron. Moreover, we found no evidence for different levels of Urbs1 in cells grown in the absence or presence of iron. Consistent with the autoregulation of urbs1, we found elevated levels of mRNA for urbs1 in all mutant cell extracts surveyed when compared with extracts for wild-type urbs1. However, these extracts were prepared from cells expressing Urbs1 from a multicopy plasmid. Southern hybridization analysis indicated that the copy number of urbs1 in the wild-type cells was lower than that in cells carrying the mutant urbs1 alleles. This indicates that the level of RNA and protein are likely related to copy number and not to the presence or absence of mutations in urbs1, thus making an autoregulatory mechanism unlikely.
Acknowledgments
We thank Dr. James Kronstad for supplying pUble20 and Allen Budde and Paige Taylor for excellent technical support. This work was supported by U.S. Department of Agriculture, University of Wisconsin Graduate School, and Public Health Service (GM33716) Grants to S.A.L.
ABBREVIATIONS
- NTG
N′-methyl-N′-nitro-N-nitrosoguanidine
- ORF
open reading frame
- HA
hemagglutinin
Footnotes
References
- 1.Crichton R, Ward R J. Biochemistry. 1992;25:11255–11264. doi: 10.1021/bi00161a001. [DOI] [PubMed] [Google Scholar]
- 2.DeSilva D M, Askwith C C, Kaplan J. Physiol Rev. 1996;76:31–47. doi: 10.1152/physrev.1996.76.1.31. [DOI] [PubMed] [Google Scholar]
- 3.Guerinot M L. Annu Rev Microbiol. 1994;48:743–772. doi: 10.1146/annurev.mi.48.100194.003523. [DOI] [PubMed] [Google Scholar]
- 4.Halliwell B, Gutteridge J M C. Biochem J. 1984;219:1–14. doi: 10.1042/bj2190001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Budde A D, Leong S A. Mycopathologia. 1989;108:125–133. doi: 10.1007/BF00436063. [DOI] [PubMed] [Google Scholar]
- 6.Mei B, Leong S A. In: Metal Ions in Fungi. Winkelmann G, Winge D R, editors. New York: Dekker; 1994. pp. 117–148. [Google Scholar]
- 7.Mei B, Budde A D, Leong S A. Proc Natl Acad Sci USA. 1993;90:903–907. doi: 10.1073/pnas.90.3.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Voisard C, Wang J, McEvoy J L, Xu P, Leong S A. Mol Cell Biol. 1993;13:7091–7100. doi: 10.1128/mcb.13.11.7091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.An Z, Mei B, Yuan W M, Leong S A. EMBO J. 1997;16:1742–1750. doi: 10.1093/emboj/16.7.1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hoe K-L, Won M-S, Yoo O-J, Yoo H-S. Biochem Mol Biol Int. 1996;39:127–135. doi: 10.1080/15216549600201131. [DOI] [PubMed] [Google Scholar]
- 11.Haas H, Angermayr K, Stöffler G. Gene. 1997;184:33–37. doi: 10.1016/s0378-1119(96)00570-7. [DOI] [PubMed] [Google Scholar]
- 12.Kudla B, Caddick M X, Langdon T, Martinez-Rossi N M, Bennett C F, Sibley S, Davies R W, Arst H N., Jr EMBO J. 1990;9:1355–1364. doi: 10.1002/j.1460-2075.1990.tb08250.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cunningham T S, Cooper T G. Mol Cell Biol. 1991;11:6205–6215. doi: 10.1128/mcb.11.12.6205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Minehart P L, Magasanik B. Mol Cell Biol. 1991;11:6216–6228. doi: 10.1128/mcb.11.12.6216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fu Y, Marzluf G A. Mol Cell Biol. 1990;10:1056–1065. doi: 10.1128/mcb.10.3.1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ballario P, Vittorioso P, Magrelli A, Talora C, Cabibbo A, Macino G. EMBO J. 1996;15:1650–1657. [PMC free article] [PubMed] [Google Scholar]
- 17.Sil A, Herskowitz I. Cell. 1996;84:711–722. doi: 10.1016/s0092-8674(00)81049-1. [DOI] [PubMed] [Google Scholar]
- 18.Zon L I, Tsai S, Burgess S, Matsudaira P, Bruns G A P, Orkin S H. Proc Natl Acad Sci USA. 1990;87:668–672. doi: 10.1073/pnas.87.2.668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tsai S, Martin D I K, Zon L I, D’Andrea A D, Wong G G, Orkin S H. Nature (London) 1989;339:446–451. doi: 10.1038/339446a0. [DOI] [PubMed] [Google Scholar]
- 20.Evans T, Felsenfeld G. Cell. 1989;58:877–885. doi: 10.1016/0092-8674(89)90940-9. [DOI] [PubMed] [Google Scholar]
- 21.Joulin V, Bories D, Eleouet J, Labastie M, Chretien S, Mattei M, Romeo P H. EMBO J. 1991;10:1809–1816. doi: 10.1002/j.1460-2075.1991.tb07706.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Spieth J, Shim Y H, Lea K, Conrad R, Blumenthal T. Mol Cell Biol. 1991;11:4651–4659. doi: 10.1128/mcb.11.9.4651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ramain P, Heitzler P, Haenlin M, Simpson P. Development (Cambridge, UK) 1993;119:1277–1291. doi: 10.1242/dev.119.4.1277. [DOI] [PubMed] [Google Scholar]
- 24.Zon L I, Mather C, Burgess S, Bolce M E, Harland R M, Orkin S H. Proc Natl Acad Sci USA. 1991;88:10642–10646. doi: 10.1073/pnas.88.23.10642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martin D I K, Orkin S H. Genes Dev. 1990;4:1886–1898. doi: 10.1101/gad.4.11.1886. [DOI] [PubMed] [Google Scholar]
- 26.Yang H, Evans T. Mol Cell Biol. 1992;12:4562–4570. doi: 10.1128/mcb.12.10.4562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Whyatt D J, deBoer E, Grosveld F. EMBO J. 1993;12:4993–5005. doi: 10.1002/j.1460-2075.1993.tb06193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Merika M, Orkin S H. Mol Cell Biol. 1993;13:3999–4010. doi: 10.1128/mcb.13.7.3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Trainor C D, Omichinski J G, Vandergon T L, Gronenborn A M, Clore G M, Felsenfeld G. Mol Cell Biol. 1996;16:2238–2247. doi: 10.1128/mcb.16.5.2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Visvader J E, Crossley M, Hill J, Orkin S H, Adams J M. Mol Cell Biol. 1995;15:634–641. doi: 10.1128/mcb.15.2.634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang Z, Gu L, Romeo P L, Bories D, Motohashi H, Yamamoto M, Engel J D. Mol Cell Biol. 1994;14:2201–2212. doi: 10.1128/mcb.14.3.2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Caddick M X, Arst H N. Gene. 1990;95:123–127. doi: 10.1016/0378-1119(90)90422-n. [DOI] [PubMed] [Google Scholar]
- 33.Wang J, Budde A D, Leong S A. J Bacteriol. 1989;171:2811–2818. doi: 10.1128/jb.171.5.2811-2818.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Holliday R. In: Handbook of Genetics. King R C, editor. Vol. 1. New York: Plenum; 1974. pp. 575–595. [Google Scholar]
- 35.Atkin C L, Neilands J B, Phaff H J. J Bacteriol. 1970;103:722–733. doi: 10.1128/jb.103.3.722-733.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gold S E, Bakkeren G, Davies J E, Kronstad J W. Gene. 1994;142:225–230. doi: 10.1016/0378-1119(94)90265-8. [DOI] [PubMed] [Google Scholar]
- 37.Wang J, Holden D W, Leong S A. Proc Natl Acad Sci USA. 1988;85:865–869. doi: 10.1073/pnas.85.3.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Holmes D S, Quigley M. Anal Biochem. 1981;114:193–197. doi: 10.1016/0003-2697(81)90473-5. [DOI] [PubMed] [Google Scholar]
- 39.Lee S, Rasheed S. BioTechniques. 1990;9:676–679. [PubMed] [Google Scholar]
- 40.Elder R T, Loh E Y, Davis R W. Proc Natl Acad Sci USA. 1983;80:2432–2436. doi: 10.1073/pnas.80.9.2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 42.Rothstein R. In: Guide to Yeast Genetics and Molecular Boliogy. Guthrie C, Fink G R, editors. San Diego: Academic; 1991. pp. 281–301. [Google Scholar]
- 43.Ferguson D O, Holloman W K. Proc Natl Acad Sci USA. 1996;93:5419–5424. doi: 10.1073/pnas.93.11.5419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Platt A, Langdon T, Arst H N, Jr, Kirk D, Tollervey D, Mates Sanchez J M, Caddick M X. EMBO J. 1996;15:2791–2801. [PMC free article] [PubMed] [Google Scholar]
- 45.Omichinski J G, Clore G M, Schaad O, Felsenfeld G, Trainor C, Appella E, Stahl S J, Gronenborn A M. Science. 1993;261:438–446. doi: 10.1126/science.8332909. [DOI] [PubMed] [Google Scholar]