

Abstract
Efforts to delineate the advent of many enzymes essential to protein translation are often limited by the fact that the modern genetic code evolved before divergence of the tree of life. Glutaminyl-tRNA synthetase (GlnRS) is one noteworthy exception to the universality of the translation apparatus. In eukaryotes and some bacteria, this enzyme is essential for the biosynthesis of Gln-tRNAGln, an obligate intermediate in translation. GlnRS is absent, however, in archaea, and most bacteria, organelles, and chloroplasts. Phylogenetic analyses predict that GlnRS arose from glutamyl-tRNA synthetase (GluRS), via gene duplication with subsequent evolution of specificity. A pertinent question to ask is whether, in the advent of GlnRS, a transient GluRS-like intermediate could have been retained in an extant organism. Here, we report the discovery of an essential GluRS-like enzyme (GluRS2), which coexists with another GluRS (GluRS1) in Helicobacter pylori. We show that GluRS2's primary role is to generate Glu-tRNAGln, not Glu-tRNAGlu. Thus, GluRS2 appears to be a transient GluRS-like ancestor of GlnRS and can be defined as a GluGlnRS.
Gene duplication events are often proposed as key turning points in adaptive evolution and in the acquisition of novel function (1–3). For a variety of reasons, strict redundancy in gene function is rare (2, 3). It is now well recognized that duplicated genes diverge over time to encode proteins that differ enough in function or specificity to offer a selective advantage to an organism. The timing and nature of critical ancestral gene duplication events are frequently approximated by phylogenetic analyses (4). Now, by combining genomics with direct experimental comparisons of paralogous proteins, the ability to elucidate ancestral evolutionary pathways is becoming an achievable goal.
The protein translation apparatus is perhaps one of the most ancient systems conserved in modern life and examples of apparent gene duplications have been reported (4–11). In fact, it has been argued that the 20 aminoacyl-tRNA synthetases (AARSs), enzymes that are critical to protein translation, evolved to their modern forms via multiple, sequential gene duplications and subsequent divergence (7). This hypothesis stems from the division of the AARSs into two classes (class I and class II), based on high degrees of sequence and structure homologies (12). Thus, it has been proposed that each class can be traced back to a single ancestral AARS progenitor (7). In this work, we asked whether direct evidence of gene duplication of AARS genes could have been retained in an extant organism.
Among the AARSs, the glutamyl- and glutaminyl-tRNA synthetases (GluRS and GlnRS, respectively) are related in sequence and evolutionarily (9, 13–15). In eukarya and some bacteria, GlnRS and a discriminating GluRS (GluRS-D) each catalyze a highly specific tRNA aminoacylation reaction (Eqs. 1 and 2, respectively). GluRS-D does not misacylate tRNAGln with Glu, and GlnRS does not misacylate tRNAGlu with Gln.
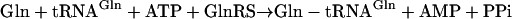 |
[1] |
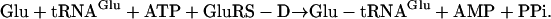 |
[2] |
In contrast, the archaea and most bacteria do not encode a functional GlnRS and Gln-tRNAGln is biosynthesized indirectly (16–18). First, tRNAGln is misacylated by a nondiscriminating GluRS (GluRS-ND), to form Glu-tRNAGln (Eq. 3) (19). [GluRS-ND still catalyzes its cognate reaction, to generate Glu-tRNAGlu, as in Eq. 2.] Next, the misacylated Glu-tRNAGln intermediate is transamidatively modified by the glutamine-dependent Glu-tRNAGln amidotransferase, designated Glu-Adt (Eq. 4) (17).
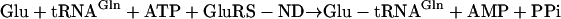 |
[3] |
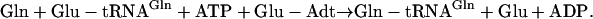 |
[4] |
In this way, the fidelity of the genetic code is accurately maintained, despite the absence of a cognate GlnRS.
A close ancestral relationship between GluRS and GlnRS has been well established (9, 13–15, 19, 20), with GlnRS predicted to be a direct descendant of GluRS (9, 13–15, 21), arising from an ancestral gene duplication event. The unifying theme of these proposals (Fig. 1) is that the two modern discriminating enzymes (GluRS-D, encoded by gltX-D, and GlnRS, encoded by glnS) arose from a common ancestral nondiscriminating GluRS (gltX-ND). This ancient GluRS-ND would be the direct ancestor of extant GluRS-NDs (Fig. 1a). A past gltX-ND gene duplication (Fig. 1b) would have enabled the evolution of more stringent tRNA and amino acid specificities. Ultimately, duplication of gltX-ND enabled the concomitant refinement of GluRS-ND into a GluRS-D and a GlnRS; the timing and nature of this divergence, however, remain unknown. It has been proposed that GlnRS emerged in eukaryotes and were horizontally transferred to a few bacteria (e.g., Escherichia coli) (9, 15).
Fig. 1.

A consensus model for the evolution of GlnRS and GluRS-D from an ancestral GluRS-ND (9, 13–15, 19, 20). The dashed arrows represent evolutionary events relevant to this work (also, see Fig. 4B). The endpoints of these evolutionary paths are defined by the three modern enzymes (GluRS-ND, GluRS-D, and GlnRS), still found in extant organisms. Bs, B. subtilis; Ec, E. coli; Hs, Homo sapiens; gltX-D (GluRS-D), discriminating GluRS; gltX-ND (GluRS-ND), nondiscriminating GluRS.
To probe the inception of GlnRS, we searched for genomic evidence of gltX duplication within a single organism. Archaeal and bacterial genomes were evaluated for the presence of multiple ORFs with sequence similarity to E. coli gltX-D and/or Bacillus subtilis gltX-ND. In total, duplications were identified by us and others (22) in Helicobacter pylori, Campylobacter jejuni, Thermotoga maritima, Rickettsia conorii, Rickettsia prowazekii, Brucella melintensis, and Mesorhizobium loti. These seven bacteria all contain putative ORFs encoding for the three subunits of Glu-Adt (GatA, GatB, and GatC), suggesting that they synthesize and correct Glu-tRNAGln intermediates; additionally, none contain an ORF that is similar to E. coli glnS (Table 1). These pairs of gltX genes are attractive candidates as possible extant intermediates in the evolution of a new glnS. To evaluate this possibility, we focused on the two H. pylori gltX paralogs [gltX1 and gltX2, annotated Hp0476 and Hp0673, respectively (23)]. The gltX1 and gltX2 genes encode proteins (named GluRS1 and GluRS2, respectively) closely related in primary sequence (38% identical, 53% similar). We show herein that GluRS1 and GluRS2 have different substrate specificities. GluRS1 optimally aminoacylates the two tRNAGlu isoacceptors of H. pylori, whereas, in contrast, GluRS2 preferentially charges H. pylori tRNAGln. The implications of this discovery on the role of gene duplication events in the evolution of the AARSs will be discussed.
Table 1. Distribution of duplicate gltX genes in diverse bacteria.
| Organism | Subkingdom | GluRS1 Protein ID | GluRS2 Protein ID | Identity (%) GluRS1/GluRS2 | glnS* | Glu-Adt† |
|---|---|---|---|---|---|---|
| B. subtilis‡ | Firmicutes | NP_387973.1 | NA | NA | No | Yes |
| E. coli‡ | Proteobacteria | AAC75457.1 | NA | NA | Yes | No |
| H. pylori | Proteobacteria | NP_207274.1 | NP_207437.1 | 38.4 | No | Yes |
| C. jejuni | Proteobacteria | NP_282434.1 | NP_282006.1 | 33.8 | No | Yes |
| B. melintensis | Proteobacteria | NP_539754.1 | NP_539885.1 | 33.9 | No | Yes |
| M. loti | Proteobacteria | NP_102397.1 | NP_102536.1 | 34.3 | No | Yes |
| R. conorii | Proteobacteria | NP_360085.1 | NP_360603.1 | 27.6 | No | Yes |
| R. prowazekii | Proteobacteria | NP_220708.1 | NP_220990.1 | 33.1 | No | Yes |
| T. maritima | Thermotogales | NP_229152.1 | NP_229671.1 | 43.6 | No | Yes |
NA, not available.
The presence or absence of a putative glnS ORF was determined by tblastn analysis using the E. coli GlnRS protein sequence (GenBank accession no. P00962). Probability scores of <1.0E-30 were considered a negative response.
Glu-Adt is composed of three subunits encoded by the genes gatA, gatB, and gatC. The presence or absence of a putative Glu-Adt was determined by tblastn analysis using the B. subtilis GatA subunit (GenBank accession no. O06491).
B. subtilis and E. coli are included as representative examples of bacteria that do not contain duplicate gltX ORFs.
Materials and Methods
Bacterial Strains and Growth Conditions. E. coli DH5α was used as the host strain for cloning and the production of GluRS1 and GluRS2 recombinant proteins. Each H. pylori tRNA was overproduced in E. coli MV1184 (24). All E. coli strains were grown at 37°C on solid or liquid LB medium. Ampicillin and spectinomycin were used at a final concentration of 100 μg/ml. H. pylori strains N6 (25) (used as a recipient for isogenic mutant construction) and 26695 (23) were each cultured on horse blood (10%) agar (Oxoid, Basingstoke, U.K.), as described by Chevalier et al. (26).
Genome Analyses and Alignments. The tblastn program (27) was used to search all finished bacterial genomes at the National Center for Biotechnology Institute (www.ncbi.nlm.nih.gov/BLAST). Putative proteins showing similarity to either E. coli GluRS-D (GenBank accession no. NP_416899) or B. subtilis GluRS-ND (GenBank accession no. P22250) were further examined. Innumerable bacteria were identified that contained two gltX-like ORFs; however, in nearly all cases, the second putative gltX ORF did not encode a full-length GluRS and was therefore eliminated from consideration. Only the coexistence of two full-length copies of the gltX gene (encoding proteins >400 aa long) were considered as possible candidates for gltX duplication. Each pair of duplicates shared at least 25% sequence identity with each other and with either E. coli GluRS-D or B. subtilis GluRS-ND.
The complete sequences of all GluRSs studied herein were directly aligned with clustalw (28). (An alignment of representative GluRSs, including the two H. pylori GluRSs, is shown in Fig. 5, which is published as supporting information on the PNAS web site, www.pnas.org). Care was taken to ensure that all conserved motifs were properly aligned. The alignments generated by clustalw were edited to remove gaps and used for phylogenetic studies.
Phylogenetic Calculations. Initial analyses were performed with all available GluRS sequences, and the results obtained were confirmed with a smaller set of bacterial sequences (see Fig. 4A). Maximum parsimony and neighbor-joining analyses were done by using the phylip 3.57c software package (29). The numbers and lengths of maximum parsimony minimal trees were estimated from 1,000 replicate random heuristic searches, whereas confidence limits of branch points were estimated by 1,000 bootstrap replications. The neighbor-joining phylogeny was based on pairwise distances between amino acid sequences by using the programs neighbor and protdist. The “Dayhoff” program option was invoked in the protdist program, which estimates the expected amino acid replacements per position by using a replacement model based on the Dayhoff 120 matrix. The programs seqboot and consense were used to estimate the confidence limits of branching points from 1,000 bootstrap replications.
Fig. 4.

Evolution of bacterial GluRSs. (A) Distance tree of bacterial GluRS, GluRS1, and GluRS2 sequences. An ancient gene duplication event (*) separates GluRS1 (dark gray) and GluRS2 (light gray) sequences in the proteobacteria. The genomes of organisms labeled with bullets (•) contain a glnS ORF. (B) Model for the evolution of GluRS in the proteobacteria. Dashed arrows indicate possible future events in evolution of tRNA specificities. See Materials and Methods for tree derivations.
Cloning and Purification of GluRS1 and GluRS2. The gltX1 and gltX2 ORFs were amplified from H. pylori genomic DNA [strain 26695 (23)]; primers were designed based on published sequences for the two genes (Table 2, which is published as supporting information on the PNAS web site) (23). Each gene was cloned into the BamHI and SmaI sites of the pQE-80 vector (Qiagen, Chatsworth, CA; Apr), for overexpression with the addition of an N-terminal six-histidine tag. The resulting plasmids are pSS001 and pSS002, carrying gltX1 and gltX2, respectively.
GluRS1 and GluRS2 were overproduced and purified by Ni-NTA affinity chromatography (Qiagen). Overexpression of gltX1 was induced with 1 mM isopropyl β-d-thiogalactoside (IPTG) at an OD600 of 0.4–0.6, for 4 h. Because GluRS2 overproduction is toxic to E. coli, 1 mM IPTG was added at an OD600 of 0.9–1.0, and the induction time was reduced to 30 min (from 3–4 h). Cells were immediately pelleted and frozen (at –80°C) until ready for use. Moreover, because of the toxicity of gltX2, the plasmid was carefully monitored for inadvertent point mutations that would lead to a less toxic mutant GluRS. After each growth, plasmids were therefore isolated and resequenced to verify that both gltX genes were unmodified.
Overexpression and Purification of H. pylori tRNAGlu1, tRNAGlu2 and tRNAGln. Each H. pylori tRNA gene was amplified and cloned into the EcoRI and BamHI sites of pES300 (24) (Table 2). This vector enables isopropyl β-d-thiogalactoside-inducible overexpression of tRNA genes in E. coli.
Cultures of each tRNA-encoding plasmid were grown in 1–2 liters of LB, supplemented with 100 μg/ml ampicillin. Induction was initiated at an OD600 of 0.4–0.6, with 1 mM isopropyl β-d-thiogalactoside. The cells were pelleted and frozen at –80°C until ready for use. In each case, a heterogeneous mixture of tRNAs, enriched with the given H. pylori tRNA, was purified by Nucleobond column purification (Clontech), as per the manufacturer's instructions. This kit uses a KCl gradient to elute different types of RNAs (e.g., mRNA, tRNA, etc.) in a stepwise fashion. After purification, each tRNA was visualized on a denaturing urea gel to verify that other contaminating RNAs had been removed (data not shown). Because it is possible that a given tRNA purification would yield a heterogeneous mixture of modified and hypomodified tRNAs, GluRS1 and GluRS2 were always assayed concomitantly, using the same tRNA preparation. The concentration of each crude tRNA preparation was first estimated by UV absorbance (OD260). The concentration of each heterologous H. pylori tRNA was then calculated by performing charging assays with l-[3,4-3H]glutamate (NEN, typical assays were diluted ≈300–500 dpm/pmol) and an excess of GluRS (500 nM) or GluRS2 (400 nM) (see below), depending on the substrate propensities of each GluRS. The tRNA concentrations used in all subsequent assays were based on these determinations.
Aminoacylation Assays and Kinetics. Glutamylation assays were performed with minor modifications of a previously described procedure (30). All assays were run in triplicate. Each tRNA was prefolded by incubation at 75°C for 3 min, followed by the addition of 2 mM MgCl2, once the solution had cooled to 65°C. The samples were cooled to room temperature. For the determination of kinetic constants, each assay contained 50 mM Hepes-KOH (pH 7.5), 4 mM ATP, 20 mM MgCl2, 25 μM glutamate (l-[3,4-3H]-Glu at 195 dpm/pmol), 8 μM tRNA, 12.5 nM GluRS1, or 25 nM GluRS2 (30). One unit of GluRS1 aminoacylates tRNAGlu1 at a rate of 0.1 pmol/s. One unit of GluRS2 aminoacylates tRNAGln at a rate of 0.1 pmol/s. Time points were quenched in trichloroacetic acid (5%) and quantified as described (30).
Glutaminylation of tRNAGln by GluRS2 was assayed as described above, with 200 nM GluRS2, 2 μM tRNAGln, and 100 μM glutamine (l-[3,4-3H]-Gln at 230 dpm/pmol) or 100 μM glutamate (l-[3,4-3H]-Glu at 220 dpm/pmol).
Acid Gel Electrophoresis and Northern Blot Analyses. Each tRNA was aminoacylated for 90–120 min at 37°C under the following conditions: 8 μM tRNA, 50 mM Hepes (pH 7.5), 20 mM MgCl2, 4 mM ATP, 50 μM glutamate (no radiolabel), and 500 nM GluRS1 or GluRS2. NaOAc was added to the reactions (0.3 M final concentration; pH 4.5), to stabilize the aminoacyl-tRNA bonds; the tRNAs were then extracted with phenol (pH 4.5) and ethanol-precipitated. Transfer RNA pellets were redissolved in buffer containing 10 mM NaOAc (pH 4.5) and 1 mM EDTA.
Acid PAGE and Northern Blot analyses were performed essentially as described (31). Hybridization oligonucleotide sequences are given in Table 2. These tRNA-specific oligonucleotides were radiolabeled with 40 μCi of [γ-32P]ATP (NEN), using 10 units of T4 polynucleotide kinase (NEB, Beverly, MA). Hybridization was visualized with a PhosphorImager (Molecular Dynamics).
Construction of Plasmids Carrying Disrupted Copies of gltX1 or gltX2. The gltX1 and gltX2 genes were amplified and cloned into the pILL570Δ vector (Spr). Plasmid pILL570Δ is a deleted version (from the HindIII site to the AvaI site) of the pILL570 plasmid (32). The two resultant plasmids, carrying gltX1 or gltX2, were then used for a second round of PCR amplification. For this step, oligonucleotides (24 bp) were synthesized to amplify (i) intragenic sequences located ≈300 bp downstream of the 5′ end (primers gltX1–5′ and gltX2–5′, introducing a 5′ BamHI restriction site; Table 2) and ≈300 bp upstream of the 3′ end (primers gltX1–3′ and gltX2–3′, introducing a 5′ KpnI restriction site; Table 2) of both gltX genes, and (ii) the whole vector sequence. After PCR, the template DNA was digested with DpnI. In each case, the PCR product and a promotorless nonpolar kanamycin cassette (33) were digested by KpnI and BamHI and ligated together. In the resultant plasmids, the kanamycin resistance cassette is flanked by 300-bp regions of either gltX1 or gltX2, to direct allelic exchange. The two recombinant plasmids were then purified and independently introduced by natural transformation into H. pylori competent cells (26). Transformation experiments were repeated in triplicate on selective plates (20 μg/ml kanamycin) at 37°C, under microaerobic conditions; plates were examined for colonies after 5 and 10 days.
Results and Discussion
The gltX1 and gltX2 Genes Are both Essential for H. pylori Survival. To test the physiological relevance of the two H. pylori gltX genes, plasmids were constructed to individually disrupt, by insertion of a nonpolar kanamycin cassette, the chromosomal copy of each gltX ORF. H. pylori N6 strain competent cells were separately transformed with each of the resultant plasmids and then tested for viability. These experiments were repeated three times (see Materials and Methods for details). In each case, no viability was observed, demonstrating that both gltX1 and gltX2 are essential. These results argue that the corresponding gene products, GluRS1 and GluRS2, are functionally nonredundant and are implicated in essential aspects of H. pylori metabolism.
Expression of gltX2 Is Toxic to E. coli. Each gltX ORF was amplified from H. pylori genomic DNA and cloned for overexpression in E. coli. Overexpression of nondiscriminating GluRS orthologs in E. coli has been shown previously to induce a toxic growth phenotype, caused by misacylation of E. coli tRNAGln1 by the foreign GluRS-ND (19). E. coli does not encode Glu-Adt and therefore cannot correct the misacylated Glu-tRNAGln. Overexpression of H. pylori GluRS2, but not of GluRS1, resulted in toxicity (Fig. 2A). Acid gel electrophoresis and Northern blot hybridization can be used to separate and identify aminoacyl-tRNAs from nonacylated tRNAs (31) by careful design of specific hybridization oligonucleotides. In these gels, deacylated tRNAs migrate more quickly than their aminoacylated counterparts. As seen in Fig. 2B, GluRS2 (and not GluRS1) does indeed misacylate E. coli tRNAGln1 (consistent with its deleterious effect on E. coli growth).
Fig. 2.

Overexpression of GluRS2 induces a toxic growth phenotype in E. coli, caused by misacylation of E. coli tRNAGln1. (A) E. coli DH5α was transformed with pSS001 (GluRS1, □), pSS002 (GluRS2, •), or unmodified pQE-80 (○). Cultures were grown in LB, supplemented with 100 μg/ml ampicillin. Growth was monitored at 600 nm. After each growth, pSS001 and pSS002 were purified and each insert was resequenced to verify that random mutations did not occur. Similar results were obtained by using E. coli K-12 (data not shown). (B) Acid gel and Northern blot analysis of E. coli tRNAGln1 misacylation by H. pylori GluRS2. Hybridization was conducted with an E. coli tRNAGln1-specific oligonucleotide (Table 2); the oligonucleotide was kinased with [γ-32P]ATP before hybridization.
GluRS1 and GluRS2 Have Divergent Aminoacylation Specificities. To determine the tRNA aminoacylation specificities of H. pylori GluRS1 and GluRS2, both enzymes were purified to homogeneity (>95% by SDS/PAGE) by Ni-NTA affinity chromatography. Two different techniques were used to evaluate the tRNA substrate specificities of GluRS1 and GluRS2. First, each enzyme was assayed in vitro for glutamylation activity by using crude E. coli tRNA preparations that were enriched with a specific H. pylori tRNA isoacceptor (either H. pylori tRNAGlu1, tRNAGlu2, or tRNAGln). Each H. pylori tRNA isoacceptor was overtranscribed in E. coli (rather than prepared by in vitro transcription) to allow 2-thiouridine modification at position 34 in each tRNA's anticodon. In E. coli, 2-thiouridine is the only posttranscriptional modification essential for aminoacylation of tRNAGlu and tRNAGln (30, 34, 35); the 2-thiouridine modification machinery is conserved in H. pylori (23), suggesting a similar requirement.
The relative aminoacylation preferences for GluRS1 and GluRS2 are shown in Fig. 3A. Interestingly, H. pylori GluRS1 contains a semiconserved arginine (Arg-350). In the Thermus thermophilus GluRS-D crystal structure (36), the corresponding arginine contributes to specific recognition of tRNAGlu and discrimination against tRNAGln, suggesting that H. pylori GluRS1 may be or resemble a GluRS-D. As shown in Fig. 3A, GluRS1 activity is optimal with tRNA preparations that are enriched with either of the two H. pylori tRNAGlu isoacceptors, consistent with the previously proposed role of Arg-350. In contrast, GluRS2 activity is optimal only when the enzyme is confronted with a tRNA mixture that has been enriched with H. pylori tRNAGln (Fig. 3A). H. pylori GluRS2 is therefore an example of an AARS that preferentially aminoacylates a noncognate tRNA.
Fig. 3.

Transfer RNA specificities of GluRS1 and GluRS2. (A) Glu-tRNAAA biosynthesis by GluRS1 (black) versus GluRS2 (red). (Top) tRNAGlu1.(Middle) tRNAGlu2. (Bottom) tRNAGln. Assays shown are the average of triplicate experiments (see Materials and Methods). (B) Acid gel and Northern blot analysis of H. pylori tRNAGlu1 (Top), tRNAGlu2 (Middle), and tRNAGln (Bottom). The black line in the middle of the bottom two gels represents empty gel lanes that were removed for clarity. Each tRNA/GluRS combination was evaluated individually (lanes 2 and 3) and as a mixture of tRNAs (lanes 4 and 5). For comparison, lane 1 shows each tRNA preparation in the deacylated form, before incubation with either GluRS1 or GluRS2. (C) Glutamine is not a substrate for GluRS2. A standard aminoacylation assay was performed (see Materials and Methods) with GluRS2, 2 μM tRNAGln, and 100 μM glutamate (□) or 100 μM glutamine (▪). The graph represents averaged data from experiments run in triplicate.
The in vitro aminoacylation assays reflect enzyme activities in the absence of competing tRNAs and may not accurately represent the true ability of each GluRS to select and aminoacylate a tRNA from a pool of competing tRNAs, as would be found in vivo. Assays are further complicated by the presence of contaminating and highly variable levels of total E. coli tRNA. As shown in Fig. 2, H. pylori GluRS2 misacylates E. coli tRNAGln1. H. pylori GluRS1 aminoacylates E. coli tRNAGlu isoacceptors in a similar manner (data not shown). It is impossible to accurately quantify the contribution of these activities (which vary between different tRNA preparations) to the rates shown in Fig. 3A. These two challenges can be overcome by evaluating each enzyme's tRNA propensities by acid gel electrophoresis and Northern blot hybridization (31). By designing hybridization oligonucleotides that are rigorously specific for only one H. pylori tRNA isoacceptor (e.g., H. pylori tRNAGlu1, tRNAGlu2, or tRNAGln), these experiments eliminate complications arising from misdetection of E. coli tRNA aminoacylation. Furthermore, aminoacylation of each tRNA can also be determined in the presence of competing tRNAs, to more accurately mimic in vivo conditions.
Standard aminoacylation reactions (30) were incubated for extended periods of time to ensure that each reaction was complete. Each tRNA/GluRS pair was evaluated independently and in the presence of an equimolar mixture of the three tRNAs preparations (H. pylori tRNAGlu1, tRNAGlu2, and tRNAGln) relevant to this study. Fig. 3B shows the results of our acid gel and Northern blot analyses. As in Fig. 2B, glutamylation of each tRNA is detected by an upward band shift, in comparison to deacylated tRNA (which is shown as a control in lane 1 of each gel). Under these assay conditions, GluRS1 optimally and totally aminoacylates both tRNAGlu1 (Fig. 3B Top, lanes 2 and 4) and tRNAGlu2 (Fig. 3B Middle, lanes 2 and 4), but does not misacylate tRNAGln (Fig. 3B Bottom, lanes 2 and 4), at least at a detectable limit. In sharp contrast, GluRS2 only misacylates tRNAGln (Fig. 3B Bottom, lanes 3 and 5), regardless of the presence of competing tRNAs. Measurable levels of Glu-tRNAGlu1 and Glu-tRNAGlu2 were not evident when these tRNAs were incubated with GluRS2 (Fig. 3B Top and Middle, respectively, lanes 3 and 5). It should be noted that tRNAGlu2 migrates as two distinct bands by acid gel. This duplication may reflect posttranscriptional hypomodification. However, both populations of tRNAGlu2 are aminoacylated by GluRS1 and not by GluRS2. Therefore, the presence of two bands does not affect the interpretation of these gels.
GluRS2 Is a Noncognate GluRS and Not a GlnRS. The acid gel and Northern blot data shown in Fig. 3B delineate the dichotomy of H. pylori GluRS1 and GluRS2. GluRS1 is a GluRS-D whose predominant activity is to aminoacylate only its cognate tRNAGlu isoacceptors. In contrast, GluRS2 is not a GluRS-ND. This enzyme is specific only for the noncognate tRNAGln (generating misacylated Glu-tRNAGln) and has little to no activity with the two cognate tRNAGlu isoacceptors. Thus, H. pylori GluRS2 has the hallmark of a GluRS that is evolving into a GlnRS. For this reason, we asked whether this enzyme had begun to develop the ability to aminoacylate tRNAGln directly with glutamine, in addition to glutamate. Even in the presence of stoichiometric levels of GluRS2 and elevated concentrations of glutamine, direct GluRS2-catalyzed formation of Gln-tRNAGln was never observed (Fig. 3C). Thus, although GluRS2 preferentially aminoacylates tRNAGln, it is not yet a GlnRS.
GluRS1 and GluRS2 Arose Via Gene Duplication. The additive nature of the catalytic activities of GluRS1 and GluRS2 offers experimental support of a bacterial gltX gene duplication event with subsequent divergence of function (Fig. 1). To place these results in an evolutionary context, the phylogenetic relationship between known bacterial GluRS and GluRS2 paralogs was analyzed (Fig. 4A). Cladistic trees were generated with 42 GluRS sequences, including the seven GluRS1/GluRS2 pairs (14 sequences) given in Table 1. The six proteobacterial GluRS1/GluRS2 pairs are monophyletic and clustered as paralogs (Fig. 4A). This organization suggests that these sequences are the result of the duplication of an ancestral proteobacterial gltX gene (see Fig. 4A, asterisk). Organisms whose genomes contain an ORF for glnS (the gene encoding GlnRS) are highlighted by a bullet in Fig. 4A. Importantly, the sequence of H. pylori GluRS2 is not related to eukaryotic GlnRSs (data not shown), but retains high similarity to GluRS-NDs (for example, GluRS2 and the B. subtilis GluRS-ND are 37% identical). Each genome that contains two gltX genes also contains the gatA, gatB, and gatC genes, encoding the three subunits of Glu-Adt, the Glu-tRNAGln amidotransferase. In this way, the accuracy of the genetic code is maintained despite the noncognate tRNA specificity of GluRS2. Thus, we propose that H. pylori, and presumably other proteobacteria, are in the process of evolving their own GlnRS and GluRS-D, via a path that is independent of the orthologs that have already emerged in the eukaryotes.
GluRS2, Named a GluGlnRS, May Be an Intermediate in the Evolution of a Bacterial GlnRS. Our experimental data point to GluRS1 more as a modern GluRS-D than a canonical GluRS-ND. In contrast, GluRS2 could be defined as a GluGlnRS, an intermediate between a GluRS-ND and a new bacterial GlnRS (Fig. 4B). This discovery is an example of an extant enzyme whose activity lies intermediate between an ancestral and a modern AARS. Because the proteobacteria appear to be in the process of evolving a new GlnRS/GluRS-D pair, we are now in the unique position to directly assess the evolution of specificity in tRNA aminoacylation reactions.
Supplementary Material
Acknowledgments
We thank Dr. Agnès Labigne for helpful discussions and experimental expertise, Chantal Ecobichon for technical assistance, Prof. Uttam Rajbhandary and Dr. Caroline Koehrer for acid gel electrophoresis advice, Prof. Marc Greenberg for PhosphorImager access, and Prof. Paul Schimmel for helpful comments on the manuscript. This work was supported by funding from The Johns Hopkins University and a Research Corporation Innovation Award. S.S. was supported by a 1-year fellowship from l'Association pour la Recherche sur le Cancer and a grant awarded by the Philippe Foundation. Preliminary sequence data were obtained from The Institute for Genomic Research web site (www.tigr.org).
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: GluRS, glutamyl-tRNA synthetase; GluRS-D, discriminating GluRS; GluRS-ND, nondiscriminating GluRS; GlnRS, glutaminyl-tRNA synthetase; AARS, aminoacyl-tRNA synthetase.
References
- 1.Ohno, S. (1970) Evolution by Gene Duplication (Springer, New York).
- 2.Nowak, M. A., Boerlijst, M. C., Cooke, J. & Smith, J. M. (1997) Nature 388, 167–171. [DOI] [PubMed] [Google Scholar]
- 3.Prince, V. E. & Pickett, F. B. (2002) Nat. Rev. Genet. 3, 827–837. [DOI] [PubMed] [Google Scholar]
- 4.Kollman, J. M. & Doolittle, R. F. (2000) J. Mol. Evol. 51, 173–181. [DOI] [PubMed] [Google Scholar]
- 5.Laursen, R. A. & Duffy, L. (1978) FEBS Lett. 92, 200–202. [DOI] [PubMed] [Google Scholar]
- 6.Golden, B. L., Ramakrishnan, V. & White, S. W. (1993) EMBO J. 12, 4901–4908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nagel, G. M. & Doolittle, R. F. (1995) J. Mol. Evol. 40, 487–498. [DOI] [PubMed] [Google Scholar]
- 8.Cousineau, B., Leclerc, F. & Cedergren, R. (1997) J. Mol. Evol. 45, 661–670. [DOI] [PubMed] [Google Scholar]
- 9.Brown, J. R. & Doolittle, W. F. (1999) J. Mol. Evol. 49, 485–495. [DOI] [PubMed] [Google Scholar]
- 10.Peeters, N. M., Chapron, A., Giritch, A., Grandjean, O., Lancelin, D., Lhomme, T., Vivrel, A. & Small, I. (2000) J. Mol. Evol. 50, 413–423. [DOI] [PubMed] [Google Scholar]
- 11.Makarova, K. S., Ponomarev, V. A. & Koonin, E. V. (2001) Genome Biol. 2, research 0033.1–0033.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eriani, G., Delarue, M., Poch, O., Gangloff, J. & Moras, D. (1990) Nature 347, 203–206. [DOI] [PubMed] [Google Scholar]
- 13.Lamour, V., Quevillon, S., Diriong, S., N′Guyen, V. C., Lipinski, M. & Mirande, M. (1994) Proc. Natl. Acad. Sci. USA 91, 8670–8674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rogers, K. C. & Soll, D. (1995) J. Mol. Evol. 40, 476–481. [DOI] [PubMed] [Google Scholar]
- 15.Siatecka, M., Rozek, M., Barciszewski, J. & Mirande, M. (1998) Eur. J. Biochem. 256, 80–87. [DOI] [PubMed] [Google Scholar]
- 16.Wilcox, M. & Nirenberg, M. (1968) Proc. Natl. Acad. Sci. USA 61, 229–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Curnow, A. W., Hong, K., Yuan, R., Kim, S., Martins, O., Winkler, W., Henkin, T. M. & Soll, D. (1997) Proc. Natl. Acad. Sci. USA 94, 11819–11826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tumbula, D. L., Becker, H. D., Chang, W. Z. & Soll, D. (2000) Nature 407, 106–110. [DOI] [PubMed] [Google Scholar]
- 19.Lapointe, J., Duplain, L. & Proulx, M. (1986) J. Bacteriol. 165, 88–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hong, K. W., Ibba, M. & Soll, D. (1998) FEBS Lett. 434, 149–154. [DOI] [PubMed] [Google Scholar]
- 21.Rogers, K. C. & Soll, D. (1993) Biochemistry 32, 14210–14219. [DOI] [PubMed] [Google Scholar]
- 22.Woese, C. R., Olsen, G. J., Ibba, M. & Soll, D. (2000) Microbiol. Mol. Biol. Rev. 64, 202–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tomb, J. F., White, O., Kerlavage, A. R., Clayton, R. A., Sutton, G. G., Fleischmann, R. D., Ketchum, K. A., Klenk, H. P., Gill, S., Dougherty, B. A., et al. (1997) Nature 388, 539–547. [DOI] [PubMed] [Google Scholar]
- 24.Schmidt, E. (1995) Ph.D. Dissertation (Massachusetts Institute of Technology, Cambridge).
- 25.Ferrero, R. L., Cussac, V., Courcoux, P. & Labigne, A. (1992) J. Bacteriol. 174, 4212–4217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chevalier, C., Thiberge, J. M., Ferrero, R. L. & Labigne, A. (1999) Mol. Microbiol. 31, 1359–1372. [DOI] [PubMed] [Google Scholar]
- 27.Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W. & Lipman, D. J. (1997) Nucleic Acids Res. 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thompson, J. D., Higgins, D. G. & Gibson, T. J. (1994) Nucleic Acids Res. 22, 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Felsenstein, J. (1988) Annu. Rev. Genet. 22, 521–565. [DOI] [PubMed] [Google Scholar]
- 30.Madore, E., Florentz, C., Giege, R., Sekine, S., Yokoyama, S. & Lapointe, J. (1999) Eur. J. Biochem. 266, 1128–1135. [DOI] [PubMed] [Google Scholar]
- 31.Varshney, U., Lee, C. P. & RajBhandary, U. L. (1991) J. Biol. Chem. 266, 24712–24718. [PubMed] [Google Scholar]
- 32.Labigne-Roussel, A., Courcoux, P. & Tompkins, L. (1988) J. Bacteriol. 170, 1704–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Skouloubris, S., Thiberge, J. M., Labigne, A. & De Reuse, H. (1998) Infect. Immun. 66, 4517–4521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seno, T., Agris, P. F. & Soll, D. (1974) Biochim. Biophys. Acta 349, 328–338. [DOI] [PubMed] [Google Scholar]
- 35.Kambampati, R. & Lauhon, C. T. (2003) Biochemistry 42, 1109–1117. [DOI] [PubMed] [Google Scholar]
- 36.Sekine, S., Nureki, O., Shimada, A., Vassylyev, D. G. & Yokoyama, S. (2001) Nat. Struct. Biol. 8, 203–206. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.