

Abstract
Ribonucleases H from the thermophilic bacterium Thermus thermophilus and the mesophile Escherichia coli demonstrate a dramatic and surprising difference in their change in heat capacity upon unfolding (ΔCp°). The lower ΔCp° of the thermophilic protein directly contributes to its higher thermal denaturation temperature (Tm). We propose that this ΔCp° difference originates from residual structure in the unfolded state of the thermophilic protein; we verify this hypothesis by using a mutagenic approach. Residual structure in the unfolded state may provide a mechanism for balancing a high Tm with the optimal thermodynamic stability for a protein's function. Structure in the unfolded state is shown to differentially affect the thermodynamic profiles of thermophilic and mesophilic proteins.
Thermophilic organisms thrive at temperatures where proteins from mesophilic organisms are often completely unfolded and nonfunctional. Understanding the mechanisms by which proteins function at such high temperatures will help to optimize and design thermostable functional proteins for a variety of biotechnological applications. To learn how proteins from thermophilic organisms (thermophilic proteins) function at such elevated temperatures, we need to understand what makes these proteins different from their mesophilic homologs. This difference clearly does not reside in the overall structure of the native conformation; structures of numerous pairs of homologous proteins show that the thermophilic and mesophilic proteins invariably adopt the same fold (1). Examining the differences between individual amino acids and their specific interactions has led to the conclusion that the rules are extremely complex (2, 3).
Thermodynamic comparisons of thermophilic and mesophilic pairs of proteins can provide a rational framework for understanding the functional differences between thermophilic and mesophilic proteins. A protein's thermodynamic stability is defined as the difference between the free energies of the native and the unfolded states (ΔGunf = GU – GN). The manner in which protein stability depends on temperature is illustrated by the so-called “protein stability curve,” which is defined by the Gibbs–Helmholtz equation (4, 5) (for an example, see Fig. 4). The temperature at which the ΔGunf° equals zero is the thermal denaturation midpoint (Tm), and the curvature of the stability curve, determined under standard conditions, is given by the heat capacity change upon unfolding (ΔCp°). There are several ways in which a protein stability curve can be altered to result in a larger Tm. An increase in the number of enthalpic interactions will raise the curve and make the protein more stable at every temperature. Alternatively, a lowering of the ΔCp° produces a “flatter” curve, which results in a higher Tm for the same stability maximum. The question then becomes, how do thermophilic proteins alter these protein stability curves and how are they encoded in the structure and sequence?
Fig. 4.

Stability curves of core T. thermophilus I53D (a), T. thermophilus I53A (b), E. coli I53D (c), and E. coli I53A (d) variants in comparison to WT E. coli (lower solid trace) and T. thermophilus (upper solid trace) RNase H stability curves. Symbols represent results of a two-state fit to thermal and guanidine-induced denaturation, and the line connecting them is the fit to the Gibbs–Helmholtz equation.
Thermus thermophilus and Escherichia coli RNase H are homologous proteins with the same folds (Fig. 1) (6, 7). As expected, based on the different optimal host growth temperatures (68°C for T. thermophilus and 37°C for E. coli), the energetic profiles of the two proteins are very different (8). The thermophilic protein undergoes a more reversible thermal denaturation and has a higher Tm (86°C for the thermophile vs. 66°C for the mesophile), and therefore remains folded at higher temperatures. The stability curve for T. thermophilus RNase H is above that of E. coli RNase H, reflecting the fact that the thermophilic protein is more stable at all temperatures (see Results). Surprisingly, however, the thermophilic RNase H has a significantly lower ΔCp° than E. coli RNase H (1.8 ± 0.1 kcal·mol–1·K–1 for T. thermophilus vs. 2.7 ± 0.2 kcal·mol–1·K–1 for E. coli RNase H), resulting in a broader stability curve. Both the raising and broadening of the thermophilic stability curve contribute to the higher Tm. Interestingly, the two RNases H have comparable stabilities at their optimum living temperatures, suggesting a balance between stability and flexibility that might be needed for a protein's optimal function (8, 9).
Fig. 1.

Ribbon diagrams of crystal structures of E. coli RNase H (A) (7) and T. thermophilus RNase H (B) (6). The folding cores (residues 43–122) are shown in purple, and the peripheries are shown in gold. Side chains of the mutated residues, Ile-53 in the core and Leu-26 in the periphery, are shown in red.
The dramatic difference in ΔCp° values between the two homologous RNases H is surprising. A major component of ΔCp° originates from the difference between solvent interactions with the folded and unfolded state, which has been found to correlate with the change in the solvent-accessible surface area as a protein unfolds (17–19). Because the proteins are homologous and have the same three-dimensional folds (6), the solvent-accessible surface areas in the folded conformation are similar. Normally, this implies similar ΔCp° values (19). The difference in observed ΔCp° values would suggest a change of ≈6,000 Å2 buried surface area (19), and small variations in the native states not captured by the crystal structures (such as small regions of disorder) are unable to account for such a large disparity in the ΔCp° values. The lack of obvious differences in the native conformations has led us to propose that residual structure in the unfolded state of the thermophilic protein might account for the important changes in ΔCp° [because ΔCp° is the difference between absolute heat capacities of the unfolded and the native states (ΔCp° = Cp°unfolded – Cp°native)].
Previous studies on chimeric versions of RNase H suggest the core region of the protein is responsible for the abnormally low ΔCp° of the thermophilic protein (11). The stability profiles of the two chimeras, constructed by swapping the folding core of the thermophilic protein with that of the mesophilic protein and vice versa, showed that the unusually low ΔCp° tracks with the thermophilic core. This suggested that the residual structure in the unfolded state of T. thermophilus RNase H may reside within the core region of the protein (Fig. 1). Experimental verification of such residual structure and its nature would lend insight into a potentially important general mechanism of thermostabilization. Here, using a novel mutagenic approach, we provide evidence for the presence of residual structure in the unfolded state of the thermophilic protein. This residual structure directly contributes to the difference in melting temperatures (Tm) between the thermophilic and mesophilic RNases H.
Methods
Construction and Purification of Variant RNases H. The RNase H variants were constructed by using either Kunkel or QuikChange (Stratagene) mutagenesis starting with pSM101 (plasmid encoding cysteine-free E. coli RNase H) or pJH109 (encoding the cysteine-free T. thermophilus RNase H). The constructs were sequenced and transformed into BL21 pLys S E. coli cells. The variants EcL26A, EcL26D, and EcI53D were purified from inclusion bodies as described (10). The remaining variants were purified from the soluble fraction as described (11).
Circular Dichroism (CD) and Stability Curves. CD spectra of the variants were collected on an Aviv 62DS spectropolarimeter (Aviv Associates, Lakewood, NJ) in a 1 cm-pathlength cuvette at 25°C. The spectra were taken in 5 mM NaOAc (50 mM KCl, pH 5.5). Data points were recorded from 300 to 200 nm, at 0.5-nm intervals. Each data point was averaged for 3 sec. Thermal and guanidinium chloride-induced denaturation profiles were monitored by CD at 222 nm. All experiments were performed in 1-cm-pathlength cuvettes, using 50 μg/ml protein in 20 mM NaOAc/50 mM KCl (pH 5.5). For thermal denaturation, data were gathered every 3°C, with a 3-min equilibration time, and each data point was averaged for 1 min. To test the reversibility of thermal denaturation a CD spectrum was taken at room temperature after thermal denaturation and compared with the spectrum taken before denaturation. Reversibility was defined as preservation of >95% of the CD signal between 220 and 225 nm. For guanidinium chloride-induced denaturation, individual samples with increasing concentrations of denaturant were prepared and equilibrated at the defined temperature. The CD signal of each sample was averaged for 1 min. Denaturant concentrations were verified by using a refractometer (12).
To generate stability curves, guanidinium chloride-induced denaturation experiments (see above) were performed at different temperatures, ranging from 4 to 45°C. Guanidinium chloride (GdmCl) was chosen as the denaturant for comparison with previous studies on the parent proteins (8). Samples were equilibrated at appropriate temperatures (in a refrigerated water bath) between 4 and 24 h before CD measurements (longer at lower temperatures). Each sample was equilibrated for 2 more min, after it was placed in the CD sample holder with a Peltier temperature regulator.
Denaturation free energies (ΔGunf°) were determined from GdmCl-induced denaturation experiments at different temperatures, assuming a two-state model and a linear dependence of ΔGunf° on the concentration of GdmCl (13). Thermal melts were fit to a two-state model to determine the Tm of each protein and the ΔGunf° in the transition range of the thermal denaturation profile. The free energies of unfolding, obtained from both GdmCl and thermal denaturation experiments, were plotted as a function of temperature. Each point on the stability curve is the average of at least two experiments. The stability curve data were fit to the Gibbs–Helmholtz equation:
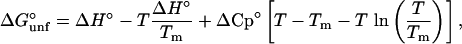 |
[1] |
where ΔH° is the change in the enthalpy with respect to the reference point Tm, and ΔCp° is the change in heat capacity upon unfolding (4). We assumed that ΔCp° is constant in the experimental temperature range. All curve fitting was done with sigmaplot (Jandel, San Rafael, CA).
Clearly, the ΔCp° values reported in this work reflect the curvature of ΔG° vs. T profiles obtained from guanidine-denaturation experiments using the linear extrapolation method. Experimental work (14, 15) supports that the guanidine-concentration dependence of unfolding ΔG° is linear over an extended concentration range, and that significant deviations from linearity only occur at low guanidine concentrations (below 1 M), mainly as a result of the screening of charge–charge interactions. Thses type of deviations are not expected to change strongly with temperature and, therefore, they should not affect significantly the curvature of the ΔG° vs. T profile (14). In fact, the ΔCp° value for T. thermophilus RNase H unfolding derived from the analysis of the guanidine-denaturation ΔG° vs. T profile (8) is in excellent agreement with the value obtained from differential scanning calorimetry (DSC) in the absence of denaturants (see legend to Fig. 2).
Fig. 2.

(a) DSC thermogram for T. thermophilus RNase H in 20 mM NaOAc/50 mM KCl (pH 5.5). The symbols are the experimental heat capacity values, and the line is the best fit of the two-state model (20). The profile shown was obtained with a protein concentration of 0.82 mg/ml, but five DSC experiments were carried out under the same buffer conditions with different protein concentrations within the range 0.17–0.82 mg/ml. The averages of the thermodynamic parameters derived from the two-state fittings are Tm = 357.9 ± 0.1 K, ΔH (at the Tm) = 131.5 ± 1.4 kcal/mol, and ΔCp (at the Tm) = 1.8 ± 0.2 kcal·K–1·mol–1. These values are in excellent agreement with those calculated in ref. 8 on the basis of the stability curve derived from guanidine denaturation experiments. (b) Absolute heat capacities for the native states of T. thermophilus and E. coli RNases H. These values were determined from the protein-concentration dependence of the apparent heat capacities (20). The small temperature dependencies shown are similar to those found for the native states of other proteins (see, for instance, ref. 27).
Activity Assays. A UV-based spectrophotometric RNase H assay was used to test the activity of the variants (partially based on ref. 16). The assay measures the loss of hypochromic effect, resulting from the cleavage of the RNA moiety in DNA–RNA hybrids. Reactions were initiated by addition of 5 nM of protein to a solution containing 25 μg/ml of an RNA/DNA hybrid (polyrA/polydT) in the presence of 10 mM MgCl2/50 mM Tris (pH 8.0) at 25°C. The loss of hypochromic effect was measured by monitoring the increase of absorbance at 260 nm. Activity was determined from the slope of the initial linear phase of the kinetic profile.
Results and Discussion
Implication of a Role for the Unfolded State in Thermophilic RNase H. DSC experiments (20) implicate that the low ΔCp° of T. thermophilus RNase H originates in the unfolded state. Although a complete calorimetric comparison between the two proteins was not possible because of aggregation of E. coli RNase H under the experimental conditions, it was possible to obtain reliable measurements of the absolute heat capacities of the native states for both proteins (aggregation only becomes a problem at the higher temperatures used for measurement of the unfolded state). There is only a small difference (≈0.2 kcal·K–1·mol–1) between the native-state heat capacities of the E. coli and T. thermophilus RNases H (Fig. 2). In addition, it is the thermophilic protein that shows the lower Cp°native value, a fact that, because ΔCp° = Cp°unfolded – Cp°native, could only contribute to a (slightly) larger ΔCp° for the thermophilic protein. It is clear, therefore, that the origin of the experimental ΔCp° value for T. thermophilus RNase H (lower that that for E. coli RNase H) must be sought in the unfolded state. Thus, both the stability curves and the DSC results, coupled with the fact that the proteins have the same native conformations, implicate a novel role for the unfolded state in contributing to the thermostability of a protein. Although we were not able to characterize the unfolded state structurally under native conditions, CD spectra of thermally denatured RNases H support this hypothesis: T. thermophilus RNase H shows more structure than the thermally denatured E. coli RNase H (data not shown).
Using Point Mutations to Probe the Unfolded State. The difficulties in detecting and populating the unfolded state motivated the development of a new mutational strategy to probe the residual structure in the unfolded state of T. thermophilus RNase H. The idea behind this approach is that introduction of a mutation that perturbs this putative residual structure in the unfolded state should give rise to a change in the ΔCp°, which we can monitor with protein stability curves. Disrupting residual structure in the unfolded state of T. thermophilus RNase H should result in an increase in its ΔCp°, making it more “mesophile-like.” Because no residual structure is expected in the unfolded state of the mesophilic protein, similar mutations in the E. coli RNase H should not affect the ΔCp°. Moreover, these effects should be limited to regions of the protein involved in the residual structure.
We tested this hypothesis by replacing buried hydrophobic residues (one in the folding core and one in the periphery) with polar, ionizable amino acids in an attempt to perturb the residual structure in the unfolded state. Position 53 (normally an Ile in both proteins) resides in the folding core, and position 26 (a Leu in both proteins) is outside the region suspected to be structured in the unfolded state (Fig. 1). In addition to substitution with the ionizable amino acid aspartate, we also evaluated a more conservative substitution, alanine. Based on our hypothesis, only the more dramatic polar substitution, when placed in the core region, should result in a loss of residual structure.
All proteins were overexpressed in E. coli and purified either from the soluble fraction or from inclusion bodies (see Methods). Both CD spectra (Fig. 3) and a UV-based activity assay (13) suggest that the native conformations of these proteins are not significantly perturbed by the point mutations.
Fig. 3.

Circular dichroism spectra of WT and variant RNases H in 20 mM NaOAc/50 mM KCl (pH 5.5), shown as follows. ○, WT E. coli RNase H; □, WT T. thermophilus RNase H; ▴, EcI53A; ♦, EcI53D; •, Ec L26A; ▾, TthI53A; ▴, TthL26D; ▾, TthI53D. CD spectra of E. coli and T. thermophilus WT RNases H differ from each other; however, the spectra of all of the variants overlay those of their respective WT parent protein.
Effects of Mutations on the ΔGunf° and ΔCp°. As expected, all of the mutations affect protein stability. Most of the mutations, however, do not result in a change in ΔCp° (Table 1). The effects on stability and ΔCp° are therefore uncoupled in these variants. Because all of the variants adopt the RNase H fold, we use the ΔCp° values as an indicator for the presence or absence of structure in the unfolded state.
Table 1. Thermodynamic parameters of WT* (cysteine-free version) and variant E. coli and T. thermophilus RNases H.
| RNase H variant | ΔGunf at 25°C, kcal·mol-1 | m at 25°C, kcal·mol-1·M-1 | ΔCp, kcal·mol-1·K-1 | Reversibility in the absence of denaturant | Tm, °C |
|---|---|---|---|---|---|
| T. thermophilus WT* | 12.1 ± 0.4 | 4.4 ± 0.2 | 1.8 ± 0.1 | + | 86 |
| T. thermophilus 153A | 8.8 ± 0.7 | 4.0 ± 0.3 | 1.6 ± 0.1 | + | 81 |
| T. thermophilus 153D | 7.0 ± 0.5 | 4.0 ± 0.3 | 2.4 ± 0.2 | - | 69† |
| T. thermophilus L26A | 10.4 ± 0.6 | 4.4 ± 0.3 | 1.5 ± 0.2 | + | 77 |
| T. thermophilus L26D | 9.5 ± 0.4 | 4.2 ± 0.2 | 1.5 ± 0.2 | + | 86 |
| E. coli WT* | 7.5 ± 0.2 | 4.9 ± 0.2 | 2.7 ± 0.2 | - | 66† |
| E. coli 153A | 6.5 ± 0.4 | 5.8 ± 0.2 | 2.8 ± 0.4 | - | 56† |
| E. coli 153D | 4.6 ± 0.2 | 5.8 ± 0.2 | 2.8 ± 0.3 | - | 48† |
| E. coli L26A | 4.3 ± 0.2 | 3.3 ± 0.2‡ | ND | - | ND |
| E. coli L26D | 4.2 ± 0.2 | 2.0 ± 0.2‡ | ND | - | ND |
ND, not determined.
The Tm is determined by fitting to the Gibbs—Helmholtz equation and not measured directly for proteins that do not undergo reversible thermal denaturation.
Low m values suggest that a partially unfolded form is significantly populated under equilibrium conditions. For these two proteins, stability curve analyses were not performed.
T. thermophilus I53D RNase H (TthI53D) shows a dramatically different ΔCp° from the parent protein (Fig. 4 and Table 1). This single replacement of a buried core hydrophobic residue with a polar amino acid results in an increase in the ΔCp° from 1.8 to 2.4 kcal·mol–1·K–1. Not only is the magnitude of this change surprising, but the direction of this change also does not follow what we know about the relationship between the ΔCp° and the burial of polar and nonpolar residues. Burial of a polar residue makes a negative contribution to the ΔCp° (18, 21), suggesting that the ΔCp° of TthI53D RNase H should be lower than that of the WT T. thermophilus RNase H, based on native state effects. The observed increase in ΔCp° is, however, in agreement with our hypothesis of residual structure in the unfolded state. We propose that the substitution TthI53D disrupts the hydrophobic clustering within the unfolded state of T. thermophilus RNase H, thus making the unfolded state more solvent-accessible and random coil-like. Importantly, the same mutation I53D has no effect on the ΔCp° of E. coli RNase H (EcI53D), consistent with the hypothesis that, unlike T. thermophilus RNase H, E. coli RNase H does not contain any significant residual structure in its unfolded state.
Despite their effects on protein stability, alanine substitutions at position 53 show no effect on ΔCp° (Fig. 4 and Table 1). Both TthI53A and EcI53A have ΔCp° values within error of WT protein, suggesting that the change from isoleucine to alanine does not perturb any residual hydrophobic clustering in the unfolded state.
Although the replacement of a core Ile with a polar amino acid (TthI53D) dramatically alters the ΔCp° for the thermophilic protein, a polar mutation outside the core region does not show the same effect. None of the mutations at position 26 affect the curvature of the protein stability curve; both E. coli and T. thermophilus variants (i.e., TthL26A, TthL26D, EcL26A, and EcL26D) have ΔCp° values similar to those of their parent proteins, suggesting that they do not perturb the change in solvent-accessible surface area (Table 1) and that the residual structure is defined by the core region of the protein.
Interestingly, the m values determined from our denaturant melts do not parallel the changes seen in the ΔCp° values (Table 1). Although this might seem surprising given that m values as well as ΔCp° values are believed to correlate with the change in solvent-accessible surface area as a protein unfolds (19), it is not inconsistent with our model of residual structure in the unfolded state under native conditions. Data to determine m values are obtained at high denaturant concentrations, and the unfolded state under these conditions is not necessarily the same as the unfolded state achieved under native conditions.
Implications for the Stability of Thermophilic Proteins. The presence of sequence-specific residual structure in the unfolded state of thermophilic RNase H explains the observed difference in the ΔCp° values between the thermophilic and the mesophilic RNase H. A reduction in ΔCp° presents an excellent mechanism for fine-tuning protein stability. A lower ΔCp° will raise the Tm without the need for an unusually high stability, which might compromise function.
So far only a handful of mesophilic and thermophilic protein homologs have been subjected to a thorough biophysical comparison based on their stability curves. Of the five thermodynamically characterized thermophile–mesophile protein pairs, four thermophilic proteins have up-shifted and broadened stability curves compared with their mesophilic homologs (8, 22–25). It is difficult to make conclusions based on this limited data set, but it is possible that a large group of enzymes and other proteins requiring a finely tuned energy landscapes use lower ΔCp° values to balance a high melting temperature with the thermodynamic stability needed for optimal activity. Residual structure in the unfolded state may provide another benefit in that this physiologically relevant unfolded state sequesters hydrophobic residues that would normally be prone to aggregation, especially at the elevated temperatures under which these organisms grow.
Our study demonstrates that the often ignored interactions in the unfolded state are as important for thermodynamic differences between thermophilic and mesophilic proteins as are the interactions that stabilize the native state. A common working assumption is that the unfolded state always assumes a random distribution of structures (i.e., random coil) and that this distribution is not affected by the amino acid sequence. The heterogeneous nature of the unfolded state, however, makes its structural characterization extremely difficult. Recent technological advances have allowed a glimpse at the unfolded state, and a more complex picture of it is emerging. Surprisingly, NMR experiments have shown that native-like topology of staphylococcal nuclease and long-range interactions of lysozyme persist even in denaturant concentrations as high as 8 M urea (26, 27). The role of these structures under highly denaturing conditions is unclear. Our novel mutagenic and thermodynamic approach adds a new dimension to these recent developments in studies of the unfolded state. Unfolded states populated under high denaturant or at high temperatures may differ from each other and, more importantly, from the physiologically relevant unfolded state populated under native conditions (28–30). Future high-resolution structural studies on the unfolded state of T. thermophilus RNase H will provide important insight into the unfolded state and how such features are encoded in the primary sequence.
Conclusions
By using a comparative mutagenesis study we have been able to decouple effects on protein stability (ΔGunf°) and heat capacity (ΔCp°), allowing us to infer the presence and thermodynamic consequences of residual structure in the unfolded state of the thermophilic RNase H. The exact nature of this residual structure and how it is encoded in the primary sequence remain unclear. Residual structure within the unfolded state appears to be an important and novel mechanism for fine-tuning a protein's energetics and function. It may be an important feature permitting thermophilic proteins to avoid irreversible denaturation in vivo, and to balance a higher melting temperature with the thermodynamic stability required for a protein's function.
Acknowledgments
We thank Jackie Gilmore and Neda Ghaffari for help with protein purification, Dave King for mass spectrometry, and the Marqusee and Sanchez-Ruiz labs for helpful discussions and critical reading of the manuscript. This work was supported by a grant to S.M. from the National Institutes of Health.
Abbreviations: CD, circular dichroism; DSC, differential scanning calorimetry.
References
- 1.Szilagyi, A. & Zavodszky, P. (2000) Struct. Fold. Des. 8, 493–504. [DOI] [PubMed] [Google Scholar]
- 2.Xiao, L. & Honig, B. (1999) J. Mol. Biol. 289, 1435–1444. [DOI] [PubMed] [Google Scholar]
- 3.Kumar, S., Tsai, C.-J. & Nussinov, R. (2001) Biochemistry 40, 14152–14165. [DOI] [PubMed] [Google Scholar]
- 4.Becktel, W. J. & Schellman, J. A. (1987) Biopolymers 26, 1859–1877. [DOI] [PubMed] [Google Scholar]
- 5.Pace, C. N. & Laurents, D. V. (1989) Biochemistry 28, 2520–2525. [DOI] [PubMed] [Google Scholar]
- 6.Ishikawa, K., Okumura, M., Katayanagi, K., Kimura, S., Kanaya, S., Nakamura, H. & Morikawa, K. (1993) J. Mol. Biol. 230, 529–542. [DOI] [PubMed] [Google Scholar]
- 7.Goedken, E. R., Keck, J. L., Berger, J. M. & Marqusee, S. (2000) Protein Sci. 9, 1914–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hollien, J. & Marqusee, S. (1999) Biochemistry 38, 3831–3836. [DOI] [PubMed] [Google Scholar]
- 9.Rees, D. C. & Robertson, A. D. (2001) Protein Sci. 10, 1187–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Spudich, G. & Marqusee, S. (2000) Biochemistry 39, 11677–83. [DOI] [PubMed] [Google Scholar]
- 11.Robic, S., Berger, J. M. & Marqusee, S. (2002) Protein Sci. 11, 381–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pace, C. N. (1986) Methods Enzymol. 131, 266–280. [DOI] [PubMed] [Google Scholar]
- 13.Santoro, M. M. & Bolen, D. W. (1988) Biochemistry 27, 8063–8068. [DOI] [PubMed] [Google Scholar]
- 14.Ibarra-Molero, B. & Sanchez-Ruiz, J. M. (1996) Biochemistry 35, 14689–14702. [DOI] [PubMed] [Google Scholar]
- 15.Ibarra-Molero, B., Loladze, V. V., Makhatadze, G. I. & Sanchez-Ruiz, J. M. (1999) Biochemistry 38, 8138–8149. [DOI] [PubMed] [Google Scholar]
- 16.Black, C. B. & Cowan, J. A. (1994) Inorg. Chem. 33, 5805–5808. [Google Scholar]
- 17.Sturtevant, J. M. (1977) Proc. Natl. Acad. Sci. USA 74, 2236–2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Makhatadze, G. I. & Privalov, P. L. (1990) J. Mol. Biol. 213, 375–384. [DOI] [PubMed] [Google Scholar]
- 19.Myers, J. K., Pace, C. N. & Scholtz, J. M. (1995) Protein Sci. 4, 2138–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guzman-Casado, M., Parody-Morreale, A., Robic, S., Marqusee, S. & SanchezRuiz, J. M. (2003) J. Mol. Biol. 329, 731–743. [DOI] [PubMed] [Google Scholar]
- 21.Loladze Vakhtang, V., Ermolenko Dmitri, N. & Makhatadze George, I. (2001) Protein Sci. 10, 1343–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li, W. T., Grayling, R. A., Sandman, K., Edmondson, S., Shriver, J. W. & Reeve, J. N. (1998) Biochemistry 37, 10563–10572. [DOI] [PubMed] [Google Scholar]
- 23.Shiraki, K., Nishikori, S., Fujiwara, S., Hashimoto, H., Kai, Y., Takagi, M. & Imanaka, T. (2001) Eur. J. Biochem. 268, 4144–4150. [DOI] [PubMed] [Google Scholar]
- 24.Deutschman, W. A. & Dahlquist, F. W. (2001) Biochemistry 40, 13107–13113. [DOI] [PubMed] [Google Scholar]
- 25.Wassenberg, D., Welker, C. & Jaenicke, R. (1999) J. Mol. Biol. 289, 187–193. [DOI] [PubMed] [Google Scholar]
- 26.Shortle, D. & Ackerman, M. S. (2001) Science 293, 487–489. [DOI] [PubMed] [Google Scholar]
- 27.Klein-Seetharaman, J., Oikawa, M., Grimshaw, S. B., Wirmer, J., Duchardt, E., Ueda, T., Imoto, T., Smith, L. J., Dobson, C. M. & Schwalbe, H. (2002) Science 295, 1719–1722. [DOI] [PubMed] [Google Scholar]
- 28.Dill, K. A. & Shortle, D. (1991) Annu. Rev. Biochem. 60, 795–825. [DOI] [PubMed] [Google Scholar]
- 29.Hammarstrom, P. & Carlsson, U. (2000) Biochem. Biophys. Res. Commun. 276, 393–398. [DOI] [PubMed] [Google Scholar]
- 30.Makhatadze, G. I. & Privalov, P. L. (1992) J. Mol. Biol. 226, 491–505. [DOI] [PubMed] [Google Scholar]
- 31.Gupta, R., Yadav, S. & Ahmad, F. (1996) Biochemistry 35, 11925–11930. [DOI] [PubMed] [Google Scholar]