

Abstract
This study examines genomic duplications, deletions, and rearrangements that have happened at scales ranging from a single base to complete chromosomes by comparing the mouse and human genomes. From whole-genome sequence alignments, 344 large (>100-kb) blocks of conserved synteny are evident, but these are further fragmented by smaller-scale evolutionary events. Excluding transposon insertions, on average in each megabase of genomic alignment we observe two inversions, 17 duplications (five tandem or nearly tandem), seven transpositions, and 200 deletions of 100 bases or more. This includes 160 inversions and 75 duplications or transpositions of length >100 kb. The frequencies of these smaller events are not substantially higher in finished portions in the assembly. Many of the smaller transpositions are processed pseudogenes; we define a “syntenic” subset of the alignments that excludes these and other small-scale transpositions. These alignments provide evidence that ≈2% of the genes in the human/mouse common ancestor have been deleted or partially deleted in the mouse. There also appears to be slightly less nontransposon-induced genome duplication in the mouse than in the human lineage. Although some of the events we detect are possibly due to misassemblies or missing data in the current genome sequence or to the limitations of our methods, most are likely to represent genuine evolutionary events. To make these observations, we developed new alignment techniques that can handle large gaps in a robust fashion and discriminate between orthologous and paralogous alignments.
Keywords: comparative genomics, cross-species alignments, synteny, chromosomal inversion, breakpoints
Evolution creates new forms and functions from the interplay of reproduction, variation, and selection. There are many types of variation; the most common and well studied is the substitution of one base for another. Small insertions and deletions are also quite common. Large-scale insertions usually involve the duplication of part of the genome. These duplications can be the starting point for the development of a new gene with a new function. The evolution of nonduplicated genes generally is quite constrained by selection, because the existing function of the gene must be maintained. After duplication, one copy is free to lose its original function and possibly assume a new function (1, 2). Deletion and rearrangement also play important roles in the long-term evolution of genomes.
This study examines patterns of variation observed at all scales by comparing the human and mouse genomes to each other. Human and mouse are at an excellent distance for studying all types of variation. The genomes are still similar enough that it is possible to align the majority of orthologous sequence at the DNA level (3) yet distant enough that a great deal of variation has had the opportunity to accumulate.
Chromosomal rearrangements of ≥1 megabase can be observed by comparing genetic maps between organisms (4) and by chromosome painting (5). Approximately 200 conserved blocks of synteny between human and mouse were discovered by gene order comparisons before the genome sequences became available, with recent estimates ranging from 98 (6) to 529 blocks (7), depending on details of definition and method. The length distribution of synteny blocks was found to be consistent with the theory of random breakage introduced by Nadeau and Taylor (8, 9) before significant gene order data became available. In recent comparisons of the human and mouse genomes, rearrangements of ≥100,000 bases were studied by comparing 558,000 highly conserved short sequence alignments (average length 340 bp) within 300-kb windows. An estimated 217 blocks of conserved synteny were found, formed from 342 conserved segments, with length distribution roughly consistent with the random breakage model (3). Subsequent analysis of these data found 281 conserved synteny blocks of size at least 1 megabase, with a few thousand further “microrearrangements” within these blocks, about one per megabase (10).
The most common variations are single-base transitions, that is C/T and G/A substitutions (11, 12). Single-base insertions and deletions are also quite common, although they are rapidly selected out of coding regions. Substitutions and small (<20-base) insertions and deletions can be studied in traditional nucleotide alignments of homologous genomic sequences. A traditional pairwise alignment consists of two segments of genomic DNA with gap characters put in to maximize the number of matching bases. A simple example is
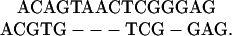 |
If the two sequences are derived from a common ancestor, then a mismatch can result from a substitution in either sequence relative to their common ancestor. Similarly, an alignment gap could be caused either by an insertion in one sequence or a deletion in the other.
At the heart of the pairwise alignment process is a scoring function that assigns positive values to matching nucleotides and negative values to mismatches and gaps. Most modern programs use what is called “affine” gap scoring, where the first gap character in a gap incurs a substantial “gap opening” cost, and each subsequent gap character incurs a somewhat lesser “gap extension” cost. Because gaps are frequently more than a single base long, affine scoring schemes model the underlying biological processes much better than fixed gap scoring systems. Affine gap scores generally work fairly well for protein alignments, where gaps are rare and tend to be short but do not represent the frequency of longer gaps as well (13–15). Nucleotide alignments, particularly outside of coding regions, tend to require many more gaps than protein alignments, and some of the gaps can be too large to be found by traditional pairwise alignment programs (16). Furthermore, in traditional pairwise alignment programs, at any given location a gap can occur only on one sequence. Independent deletion events in each species that delete some but not all of the same ancestral sequence cannot be represented, even though these are quite common. In these instances, the alignment program will either break the alignment into two or force nonhomologous bases to align. Traditional programs are also not able to accommodate inversions, translocations, or duplications. They can align only shorter segments of genomic DNA in which none of these events has occurred. Therefore, whereas considerable analysis of variation at the single base scale is available from traditional sequence alignments to complement the analysis of large-scale rearrangements, analyzing variation at the middle scales is still an interesting challenge. The role of segmental duplication of several thousand to several million bases in our own evolution (17) is one indication of the importance of variation at the middle scales.
In this article, we describe automated methods for linking together traditional alignments into larger structures, chains and nets, that can effectively bridge the gap between chromosome painting and sequence alignment (Fig. 1). These methods can accommodate inversions, translocations, duplications, large-scale deletions, and overlapping deletions. We apply these tools to blastz-generated alignments (18) to investigate the patterns of variation that have occurred at all scales since the divergence of the mouse and human lineages.
Fig. 1.

Mouse/human alignments at Actinin α-3 before and after chaining and netting, as displayed at the genome browser at http://genome.ucsc.edu. The RefSeq genes track shows the exon/intron structure of this human gene, which has an ortholog as well as several paralogs and pseudogenes in the mouse. The all blastz Mouse track shows blastz alignments colored by mouse chromosome. The orthologous gene is on mouse chromosome 19, which is colored purple. Although blastz finds the homology in a very sensitive manner, it is fragmented. The chained blastz track shows the alignments after chaining. The chaining links related fragments. The orthologous genes and paralogs are each in a single piece. The chaining also merges some redundant alignments and eliminates a few very low-scoring isolated alignments. The Mouse/Human Alignment Net track is designed to show only the orthologous alignments. In this case, there has been no rearrangement other than moderate-sized insertions and deletions, so the net track is quite simple. Clicking on a chain or net track allows the user to open a new browser on the corresponding region in the other species.
Methods
The November 2002 freeze of the human genome and the February 2002 freeze of the mouse genome were taken from http://genome.ucsc.edu. The two genomes were aligned by the blastz program as described in ref. 18, except that in addition to masking out transposon repeats [identified by repeatmasker (19)], simple repeats of period 12 or less found by tandem repeat finder (20) were masked out, and the artifact-prone “chrUn” mouse sequence, a sequence that could not be assembled into sizeable contigs or effectively mapped to the mouse chromosomes, was excluded. The blastz alignments were converted to axt format (see ref. 18) by using the program lavtoaxt.
By using the same nucleotide scoring matrix as blastz but a novel piecewise linear gap scoring scheme, a new program, axtchain, formed maximally scoring chained alignments out of the gapless subsections of the input alignments (Fig. 1). A chained alignment (or “chain”) between two species consists of an ordered sequence of traditional pairwise nucleotide alignments (“blocks”) separated by larger gaps, some of which may be simultaneous gaps in both species. The order of blocks within the chain must be consistent with the genomic sequence order in both species. Thus, a chain cannot have local inversions, translocations, or duplications among the parts of the DNA that it aligns. However, a chain is allowed to skip over segments of DNA in either or both species. In particular, intervening DNA in one species that does not align with the other because it is locally inverted or has been inserted in by lineage-specific translocation or duplication is skipped over during construction of the chain. Thus, the chain can represent widely scattered pieces of genomic DNA in the two contemporary species that are in fact descended from a single genomic segment in the common ancestor without rearrangement.
To build chains efficiently, axtchain uses a variation of the k-dimensional tree (kd-tree) based algorithm described in ref. 21. To detect cases of overlapping deletions in both species, scoring is defined such that the alignment program will typically open a simultaneous gap rather than forcing alignments of nonhomologous regions, because penalties incurred by a run of mismatches exceed the simultaneous gap penalty. This strategy for allowing simultaneous gaps also helps cope with local inversions and missing sequence (blocks of Ns) in unfinished genomes. The resulting chains often span multiple megabases.
For many purposes, one wants to select only a single best alignment for every region of the human genome. Previously (18), we developed a program, axtbest, for this purpose. However, the chains in many ways represent a better substrate for picking the “best” alignment than single blastz alignments. We developed a new program, chainnet, to improve on this process. In this program, all bases in all chromosomes are initially marked as unused. The chains are then put into a list sorted with the highest-scoring chain first. The program goes into a loop, at each iteration taking the next chain off of the list, throwing out the parts of the chain that intersect with bases already covered by previously taken chains, and then marking the bases that are left in the chain as covered. The program uses red–black trees to keep track of which areas of a chromosome are already covered. If a chain covers bases that are in a gap in a previously taken chain, it is marked as a child of the previous chain. In this way, a hierarchy of chains is formed that we call a net. The chainnet program also keeps track of which bases are covered by more than one chain to help distinguish duplicated from nonduplicated regions.
The resulting net files are further annotated by the program netsynteny, which notes which chains in the net are inverted relative to their parents, displaced, or on different chromosomes. Additional annotations on repeats, including lineage-specific repeats and repeats that predate the mouse/human split, are added by the netclass program. Short chains embedded within a longer chain that come from regions distinct from that of the longer chain, as well as short chains between long chains at the top level of the net, are often processed pseudogenes. Because these can confound many types of analysis, we also prepared a subset of the chains that are judged to be “syntenic.” To be considered syntenic, a chain has to either have a very high score itself or be embedded in a larger chain, on the same chromosome, and come from the same region as the larger chain. Thus, inversions and tandem duplications are considered syntenic. The syntenic subset of the alignments was created with the netfilter program by using the–syn flag. Although netfilter does not eliminate pseudogenes that arise from tandem duplication, it does eliminate processed pseudogenes, which are much more plentiful.
Further details of these algorithms are described in the source code, which, along with Linux executables for lavtoaxt, axtchain, chainnet, netsynteny, netclass, and netfilter, are available at www.soe.ucsc.edu/~kent. The chains and nets are displayed alongside other annotations at the genome browser at http://genome.ucsc.edu (22) and may also be downloaded in bulk from that site.
Results
The initial blastz mouse alignments cover 35.9% of the human genome. This is less than the 39.9% reported in ref. 3, due to masking of the tandem repeats of period 12 and less and removal of the artifact-prone mouse sequence in “ChrUn.” The construction of longer chains from the initial blastz alignments resulted in fewer and substantially longer alignments (Fig. 1). Chained alignment length can be measured in two different ways. We define the (human) span of a chain to be the distance in bases in the human genome from the first to the last human base in the chain, including gaps, and we define the size of the chain as the number of aligning bases in it, not including gaps. Both of these showed substantial increases due to chain construction, with the average human span increasing from 608 to 22,830 bp and the average size increasing from 574 to 7,062 bp. Because many smaller blastz alignments were often merged into a single longer chain, there was also a substantial reduction in the total number of alignments involved in the human–mouse comparison, from ≈8.5 million to ≈150,000. In some cases, small low-scoring blastz alignments are discarded after chaining as well. This results in a decrease from 35.9% to 34.6% of the bases in human genome being aligned to mouse. These and other comparative statistics are listed in Table 1.
Table 1. Comparison of blastz alignments/chains before and after processing with axtchain and after building the human net.
| blastz | axtchain | |
|---|---|---|
| Number of alignments/chains | 8,560,148 | 147,445 |
| Longest human span, bp | 63,780 | 115,044,604 |
| Average human span, bp | 608 | 22,830 |
| Most aligning bases, bp | 59,559 | 27,056,473 |
| Average aligning bases, bp | 574 | 7,062 |
| Bases aligned in human genome, % | 35.9 | 34.6 |
The genomewide collection of these chains was used to study the length distribution of gaps induced by indels of all sizes since the divergence of these species, from single base to multikilo-base-size indels. The affine gap score model conventionally used in sequence alignment programs corresponds to a statistical model where indel sizes follow a geometric distribution, in which the number of gaps of size N + 1 would be a constant fraction of the number of gaps of size N for all N. The length distribution we observed violates this model. Fig. 2 shows a histogram of gap sizes observed in the set of chains we constructed, with frequency of occurrence for various indel lengths plotted on a logarithmic scale. A geometric distribution would appear as a straight line on such a plot. In actuality, the plot is fairly complex. In general, there are more short and long gaps than we would see in a geometric distribution, and there are sharp spikes in the numbers of gaps observed in the human sequence at ≈300 bases corresponding to ALU insertions (19).
Fig. 2.

(a) Small insertions and deletions. (b) Large insertions and deletions and transposons. Counts of genomewide insertions and deletions plotted vs. their size. The blue line shows a combination of human insertions and mouse deletions, whereas the red line shows mouse insertions and human deletions. The vertical scale is the natural logarithm of the number of insertions/deletions of that size. In b, the counts are grouped in bins of 25. The green line shows the percentage of bases in mouse gaps of that size that are covered by human-specific transposons. The peaks in the insertion/deletion graphs appear to be due to transposons.
The chains also included a substantial number of simultaneous gaps in both species (Fig. 3). For smaller gap sizes, simultaneous gaps are quite rare, but the phenomenon becomes increasingly important with increasing gap size. The frequency of large simultaneous gaps is roughly consistent with a model in which they arise from two independent indel events, one in each species (Fig. 3c).
Fig. 3.

(a) Log histogram of gap frequencies for gaps <50 bases long. (b) Log histogram of gap frequencies for gaps up to 1,500 bases long. (c) Log histogram of gap frequencies for gaps up to 50,000 bases long. Relative frequency of simultaneous and single gaps are shown in both sequences. The horizontal axis is used for gaps in the mouse sequence, which represent either insertions in human or deletions in mouse. The vertical axis is used for gaps in human. The log of the number of simultaneous gaps of a particular size range is converted into a level of gray to create a 2D histogram. The horizontal and vertical axes are not drawn in; the dark lines where the axis would be reflect the log frequencies of gaps that are in only one sequence. (a) Gaps of <50 bases. Gaps that are purely in mouse or purely in human are especially prominent here. (b) Gaps of 10–1,500 bases. The transposon-induced effects in Fig. 1 can also be seen here. Note also the relative concentration near the diagonal for inserts of <200 bases. This occurrence is mostly due to small inversions and locally divergent sequence. (c) Gaps of 1,000–50,000 bases. In this range, the log frequency of simultaneous gaps of a given combined (human and mouse) gap size differs roughly by a constant from the sum of the log frequencies of the individual one-sided gap sizes in each species. In this sense, these longer simultaneous gaps act as if they arise from independent gaps in each individual species.
The nested “net” structure of chains produced by the chainnet program was used to examine the rearrangements that have occurred since the divergence of human and mouse in the form of inversion (Fig. 4), deletion, transposition, retrotransposition, tandem duplication, and interspersed duplication. Because of various types of duplications, 52% of coding regions and 3.3% of the human genome as a whole are covered by more than one blastz mouse alignment (3) even after excluding transposons. This can make it difficult to separate ortholog from paralog and gene from pseudogene. The net structure helps to disambiguate these. The net structure is not symmetric between species: the human net is constructed by allowing only the single best aligning DNA from the mouse genome to align to any single place in the human genome and the mouse net is constructed in the opposite manner. This one-sided “best-in-genome” requirement is not as strong as the requirement of “reciprocal best” matching, where only alignments that are best-in-genome for both species are allowed. The latter prohibits the investigation of lineage-specific duplications by cross-species alignments, because it allows at most one copy of the duplicated region to align to the other species, and often not even one copy can be fully aligned. With human and mouse, this results in a substantial reduction of the total amount of genomic DNA covered by cross-species alignments. We found that requiring reciprocal-best alignment reduces the coverage in the human net by 11%, and in the mouse net by 9%. These numbers are quite close, suggesting that a similar level of duplication has been occurring in both genomes since the common ancestor. In what follows, we will explore the human net further, constructed as described in Methods, without the reciprocal best requirement.
Fig. 4.

A 15,000-base inversion containing two transcripts and showing chr7:2077222–2497100 in the November 2002 assembly of the human genome. Numerous smaller rearrangements are also visible in the net track in this picture. In some cases, the smaller ones simply represent paralogous mouse regions filling in when the orthologous mouse region is not yet sequenced.
On the basis of analysis of the human net, the frequency of various rearrangements on the draft mouse sequence as a whole and on the 48-million-base (2%) subset of the sequence that is finished is shown in Table 2. Overall rearrangement patterns were very similar in the finished subset of the mouse genome as in the genome as a whole. The same held for the finished subset of the human genome (data not shown). There were significantly more local duplications in the finished subset, likely reflecting the collapse of local duplications, a common artifact of the whole-genome shotgun assembly techniques used in the mouse. In general, the most common type of rearrangement is a section of the genome being duplicated and inserted in a different chromosome (nonsyntenic duplication). Most of these appear to be processed pseudogenes. Nonsyntenic apparent transpositions were also surprisingly common.
Table 2. Rearrangement statistics of the mouse genome relative to human.
| Genomewide frequency (events per megabase) | Finished frequency (events per megabase) | Genome median size | Finished median size | |
|---|---|---|---|---|
| Inversion | 2.0 | 1.8 | 814 | 762 |
| Inversion + local duplication | 0.5 | 1.0 | 275 | 302 |
| Inversion + local part duplication | 0.7 | 0.8 | 517 | 1235 |
| Local move | 0.8 | 1.0 | 204 | 246 |
| Local duplication | 1.9 | 4.0 | 211 | 351 |
| Local part duplication | 0.9 | 1.2 | 343 | 388 |
| Syntenic move | 0.8 | 1.6 | 223 | 322 |
| Syntenic duplication | 1.3 | 1.2 | 283 | 286 |
| Syntenic part duplication | 0.7 | 0.8 | 474 | 946 |
| Nonsyntenic move | 5.0 | 5.2 | 104 | 109 |
| Nonsyntenic duplication | 11.9 | 11.6 | 235 | 228 |
| Nonsyntenic part duplication | 4.6 | 4.6 | 282 | 256 |
| Mouse 1 base gaps | 1,461.8 | 1,513.4 | 1 | 1 |
| Mouse 10 base gaps | 39.7 | 46.4 | 10 | 10 |
| Mouse gaps ≥ 100 | 68.8 | 80.8 | 207 | 201 |
| Double gaps ≥ 100 | 398.6 | 419.9 | 444 | 411 |
| H likely deletion ≥ 100 | 230.0 | 223.5 | 685 | 633 |
The finished columns show rearrangements within the 96.3 megabases of mouse sequence that were finished in this assembly. The genome columns refer to the entire 2.47-gigabase mouse assembly. A move or inversion involves no duplicated sequence. “Duplication” means at least 80% of the aligning bases of the rearranged chain align to multiple places in the human genome. “Part duplication” means some sequence, but <80% is duplicated. The moves and duplications of <100,000 bases are considered local. Syntenic moves and duplications are on the same chromosome but >100,000 bases away and may be inverted as well. Nonsyntenic moves and duplications fill gaps in a chain with sequence from another chromosome. The “single gaps ≥100” row shows gaps of 100 or more bases in mouse and 0 bases in human. The “double gaps ≥ 100” row shows gaps of 100 or more in mouse and >0 bases in human. The “h likely deletion ≥100” row counts gaps in the mouse genome that are not the result of human lineage-specific transposons or Ns, and that are at least 100 bases.
The distribution of the spans of the chains from the human net (Fig. 5) shows a roughly bimodal distribution for the chains that span <100,000 bases (“short chains”) and a long flat tail consisting of 579 chains that span between 100,000 and ≈115 million bases (“long chains,” average length 983 kb). The long chains combined span 90.9% of the human genome (excluding gaps <100,000 bases in individual chains) and their aligned bases cover 32.9% of the bases in the human genome. In contrast, all chains together (including arbitrarily large gaps in individual chains) span 96.3% of the human genome and align to 34.6% of it. Thus the long chains alone (without their long gaps) span 94.4% of the bases spanned by all chains and include 95.1% of all aligned bases.
Fig. 5.

Distribution of the spans of all 147,445 chains in the human net. The distribution consists of a bimodal portion for short chains of span <105 and a flat tail for 579 long chains of size between 105 and ≈108.
Of the 579 long chains, 344 appear at the top level of the net and form large primary units of synteny with the mouse. The locations of these chains are largely in agreement with the regions of synteny with mouse identified in earlier studies (see Discussion). The remaining 235 long chains appear at lower levels of the net because they are embedded within the gaps of these primary synteny chains. Of these, 160 represent inversions, 29 represent duplications or translocations of nearby regions on the same chromosome, and 46 represent distant duplications or translocations at great distance on the same chromosome or between chromosomes. Some of these were also detected in earlier studies.
In addition to the 344 long chains at the top level of the net, there are 19,800 short chains at the top level and many more at lower levels. The short chains at the top level often appear in long runs in regions where no significant synteny between the human and mouse genomes was previously found and constitute what appear to be hot spots for rearrangements or duplications. In these regions, the best alignment in the other species shifts rapidly from one chromosome to another. Many of these regions contain clusters of genes from families that have undergone recent lineage-specific expansions. They include many genes involved in the immune system, olfactory receptors (23), and Krüppel-associated box C2H2-type zinc fingers, which are known to have been highly duplicated and mobile in mammalian evolution (24). For a table of such regions, see Table 3, which is published as supporting information on the PNAS web site, www.pnas.org.
Because the 344 large chains that are at the top level of the net are similar to the regions of synteny identified in earlier studies, the distribution of their spans agrees roughly with the simple random breakage model that has worked well in previous synteny (8). However, the presence of large numbers of small chains suggests that other processes may be at work not only within but also between the synteny blocks defined by the long chains. These intervening short chains cannot be explained as additional blocks of synteny along with the longer blocks by using the parameters of a simple random breakage model.
Finally, as described in Methods, we also derived a “syntenic” subset of the human net. The complete net covers 96.7% of bases in RefSeq (www.ncbi.nlm.nih.gov/RefSeq; ref. 25) coding regions, whereas the syntenic subset covers 93.0%. The complete net covers 71% of bases in pseudogenes on chromosome 22 annotated by the Sanger Centre (26), whereas the syntenic subset covers 13%. Thus, the syntenic subset should be useful in locating pseudogenes and in gene analysis applications where specificity is important, such as described in refs. 27 and 28.
We examined in detail the coding regions covered by the complete net but not the syntenic net on chromosome 22. This includes part or all of 40 of the 450 RefSeq genes mapped to chromosome 22 in the November 2002 assembly at http://genome.ucsc.edu. Eighteen of these genes were in areas that seem to be hot spots for rearrangement and duplication, as discussed above. Twelve of the genes with parts present in the full net but not the syntenic net are caused by gaps in the mouse draft genome. In the complete net, paralogous regions are filled in for the missing orthologous regions. Eight genes appeared to be deleted or partially deleted in the mouse. Extrapolating this to the complete genome would imply that the mouse has deleted or partially deleted ≈2% of genes present in the common ancestor. Two of the RefSeq gene mappings missing from the syntenic net turned out, on closer examination, to be mappings to processed pseudogenes. As the human and mouse genomes become more fully finished, the syntenic subnet will be increasingly useful for identifying processed pseudogenes and studying gene evolution in the entire genome.
Discussion
The chaining and netting technique presented here is very useful in tracing the evolution of the genome. It has some major advantages over methods such as GRIMM (4) for studying genomic rearrangments. It can accommodate duplication and deletion as well as transposition and inversion, and it provides resolution down to the base level. However, the netting technique assumes that each rearrangement is independent, not overlapping other rearrangements. It can detect and correctly classify translocations only if they insert in the middle of a chromosome rather than at chromosome ends. GRIMM can deal with overlapping rearrangements and translocations at the ends of chromosomes. When we restrict our attention to blocks of 100,000 bases or more, we find 160 inversions, which is similar to the 149 inversions of 1 million bases or more reported with GRIMM. The level of transpositions <100,000 bases is lower than observed by GRIMM due to the limitations of our technique. The breakpoints we observe between large syntenic areas do correlate well with those observed with GRIMM and with windowing-based techniques such as reported in ref. 3. Comparisons of the chains from the human net and blocks of synteny found by other methods can be viewed as side-by-side tracks on the genome browser by means of the link at http://genome.ucsc.edu/cauldron/syntenies.html.
It is clear that inversions and other rearrangements happen at all scales, and indeed that there are more inversions <1,000 bases that there are inversions <1,000 bases. Due to limits in our alignment techniques, we may not currently be observing many of the smallest rearrangements, particularly those <50 bases. The most common rearrangement we observe (excluding transposon insertion) is nonsyntenic duplication, most of which appears to be due to processed pseudogenes created by transposon machinery. However, there is a surprising amount of nonsyntenic nonduplicating transposition of small blocks. Some of these, as well as other events we record, may be artifacts due to the incompleteness of the mouse genome: second-best matches to the mouse genome are occasionally used in the net where the best match is missing because it falls in an unsequenced region in the mouse genome. However, this does not account for the majority of the events. We currently do not know of a mechanism that would generate movement of small pieces of the genome like this.
Finally, this work lets us define orthologous mouse and human genes more clearly than we could in the past and has the potential to help eliminate false annotation of pseudogenes as true genes.
Supplementary Material
Acknowledgments
We thank the International Human Genome Sequencing Consortium and the Mouse Genome Sequencing Consortium for providing the data. W.J.K., R.B., A.H., and D.H. were supported by National Human Genome Research Institute Grant 1P41HG-02371. D.H. was also supported by the Howard Hughes Medical Institute. W.M. is supported by National Human Genome Research Institute Grant HG-02238.
This paper was submitted directly (Track II) to the PNAS office.
See commentary on page 11188.
References
- 1.Haldane, J. B. S. (1932) The Causes of Evolution (Longmans and Green, London).
- 2.Graur, D. & Li, W. H. (2000) Fundamentals of Molecular Evolution (Sinauer, Sunderland, MA).
- 3.The Mouse Sequencing Consortium (2002) Nature 420, 520–562. [DOI] [PubMed] [Google Scholar]
- 4.Tesler, G. (2002) Bioinformatics 18, 492–493. [DOI] [PubMed] [Google Scholar]
- 5.Yang, F., Alkalaeva, E. Z., Perelman, P. L., Pardini, A. T., Harrison, W. R., O'Brien, P. C., Fu, B., Graphodatsky, A. S., Ferguson-Smith, M. A. & Robinson, T. J. (2003) Proc. Natl. Acad. Sci. USA 100, 1062–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Housworth, E. A. & Postlethwait, J. (2002) Genetics 162, 441–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kumar, S., Gadagkar, S. R., Filipski, A. & Gu, X. (2001) Genetics 157, 1387–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nadeau, J. H. & Taylor, B. A. (1984) Proc. Natl. Acad. Sci. USA 81, 814–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nadeau, J. H. & Sankoff, D. (1998) Mamm. Genome 9, 491–495. [DOI] [PubMed] [Google Scholar]
- 10.Pevzner, P. & Tesler, G. (2003) Genome Res. 13, 37–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chiaromonte, F., Yap, V. B. & Miller, W. (2002) Pac. Symp. Biocomput. 115–126. [DOI] [PubMed]
- 12.Makalowski, W. & Boguski, M. S. (1998) Proc. Natl. Acad. Sci. USA 95, 9407–9412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Qian, B. & Goldstein, R. A. (2001) Proteins 45, 102–104. [DOI] [PubMed] [Google Scholar]
- 14.Ophir, R. & Graur, D. (1997) Gene 205, 191–202. [DOI] [PubMed] [Google Scholar]
- 15.Gu, X. & Li, W. H. (1995) J. Mol. Evol. 40, 464–473. [DOI] [PubMed] [Google Scholar]
- 16.Kent, W. J. & Zahler, A. M. (2000) Genome Res. 10, 1115–1125. [DOI] [PubMed] [Google Scholar]
- 17.Bailey, J. A., Gu, Z., Clark, R. A., Reinert, K., Samonte, R. V., Schwartz, S., Adams, M. D., Myers, E. W., Li, P. W. & Eichler, E. E. (2002) Science 297, 1003–1007. [DOI] [PubMed] [Google Scholar]
- 18.Schwartz, S., Kent, W. J., Smit, A., Zhang, Z., Baertsch, R., Hardison, R. C., Haussler, D. & Miller, W. (2003) Genome Res. 13, 103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smit, A. F. (1999) Curr. Opin. Genet. Dev. 9, 657–663. [DOI] [PubMed] [Google Scholar]
- 20.Benson, G. (1999) Nucleic Acids Res. 27, 573–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang, Z., Raghavachari, B., Hardison, R. C. & Miller, W. (1994) J. Comput. Biol. 1, 217–226. [DOI] [PubMed] [Google Scholar]
- 22.Kent, W. J., Sugnet, C. W., Furey, T. S., Roskin, K. M., Pringle, T. H., Zahler, A. M. & Haussler, D. (2002) Genome Res. 12, 996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Young, J. M., Friedman, C., Williams, E. M., Ross, J. A., Tonnes-Priddy, L. & Trask, B. J. (2002) Hum. Mol. Genet. 11, 1683. [DOI] [PubMed] [Google Scholar]
- 24.Looman, C., Abrink, M., Mark, C. & Hellman, L. (2002) Mol. Biol. Evol. 19, 2118–2130. [DOI] [PubMed] [Google Scholar]
- 25.Pruitt, K. D., Tatusova, T. & Maglott, D. R. (2003) Nucleic Acids Res. 31, 34–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Collins, J. E., Goward, M. E., Cole, C. G., Smink, L. J., Huckle, E. J., Knowles, S., Bye, J. M., Beare, D. M. & Dunham, I. (2003) Genome Res. 13, 27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Couronne, O., Poliakov, A., Bray, N., Ishkhanov, T., Ryaboy, D., Rubin, E., Pachter, L. & Dubchak, I. (2003) Genome Res. 13, 73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alexandersson, M., Cawley, S. & Pachter, L. (2003) Genome Res. 13, 496–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.