

Abstract
RNA silencing, found broadly throughout the eukaryotes, posttranscriptionally suppresses the expression of “aberrant” genes including those of many viruses and transposons. Similar to the specific immune system of vertebrates, RNA silencing works by generating specific responses against foreign elements and rapidly amplifying these responses to clear or otherwise inactivate the threat. Also like the vertebrate immune system, RNA-silencing systems risk making mistakes and mounting undesirable responses against the self. We develop a set of mathematical models of RNA silencing. We show that current models of RNA silencing do little to explain what prevents mistaken reactions from silencing vital organismal genes. We extend the basic models to show that the presumed unidirectional nature of the amplification process (namely, unidirectional RNA-directed RNA polymerase-mediated synthesis of secondary double-stranded RNA as observed in Caenorhabditis elegans) serves as a “safety mechanism” that safeguards against accidental generation of damaging self-directed reactions.
RNA silencing, also known as RNA interference, posttranscriptional gene silencing, or quelling, is broadly conserved across the eukaryotes (1) and implicated in functions ranging from transcriptional silencing (2–4) to developmental regulation (5, 6) to self-/non-self-discrimination (7, 8). In many species, RNA silencing acts to suppress the expression of transcripts corresponding to “non-self-genes,” potentially harmful elements such as those encoded in viruses or transposons (9–12). This process parallels the operation of the vertebrate-specific immune system: Similar to the immune system, RNA silencing guards against exploitive parasitic elements by (i) identifying non-selfelements, (ii) generating target-specific responses against these foreign elements, and (iii) rapidly amplifying these responses to clear or otherwise inactivate the threat.
Thus in organisms with RNA silencing, each cell has a miniature “immune system” able to generate and amplify specific responses to a variety of gene transcripts (7, 8). To function properly, this system needs to be able to discriminate self from non-self with great specificity. But, no matter how accurate the discrimination may be, errors are inevitable: Such a system will generate inappropriate self-directed responses in numerous ways. Accidental production of antisense transcripts can lead to the formation of double-stranded RNAs (dsRNAs) corresponding to self-genes, which in turn can trigger silencing (13). Alternatively, self-reactive responses may arise due to identity or cross-reactive similarity of invading viral sequences with endogenous mRNA. Consequently, we might expect to see evolved mechanisms that serve to limit the damage caused by any self-directed reactions that do occur.
In this article we develop simple mathematical models of the dynamics of RNA silencing. We use these models to explore how the RNA-silencing system tackles the central challenge faced by any immune mechanism: the need to rapidly generate specific responses to foreign pathogens while guarding against responses against self. In A Basic Model of RNA Silencing we describe a simple mathematical model that captures the basic intracellular features of the RNA silencing. We show that although this model can generate specific responses rapidly, these responses persist as long as expression of the targeted gene continues, and thus this model does not allow the cell to shut down responses that arise by mistake. Instead, even a small accidental response to self will be amplified until the expression of self-transcripts is greatly reduced or even eliminated. In Unidirectional Amplification, we extend the model to incorporate the 5′ to 3′ unidirectionality of RNA polymerization during the amplification step and show that this unidirectionality provides a safety mechanism against continuation of self-reactive responses.
Our goal throughout is to elaborate the logical consequences of schematic models of the silencing process and to make testable predictions about the nature and components of RNA-silencing pathways. To do so, we make a series of simplifications and idealizations so as to get at the essential features of a diverse and intricate set of biological processes. In particular, we concentrate on the common elements of the RNA-silencing system in nematodes, plants, Dictyostelium, and Neurospora crassa: cleavage of dsRNA, sequence-specific mRNA degradation, and primer-based RNA amplification. Even among this group the RNA-silencing pathways exhibit important differences. In Discussion we consider the consequences for our model of the apparent bidirectional amplification that occurs in plants (14) and of systemic silencing as seen in plants and nematodes (15, 16). Our model does not address the incomplete RNA-silencing systems observed in humans and Drosophila. These species have lost the RNA-directed RNA polymerase (RdRp) involved in amplification and seem to lack any alternative amplification pathways (ref. 17; but see ref. 18). Consequently, amplification of self-directed RNA silencing is unlikely to be a problem for these organisms.
A Basic Model of RNA Silencing
In this section we work out the mathematical consequences of the primer-based amplification model (18–21), commonly used as a conceptual framework for the silencing process (1, 7, 8, 22, 23). Fig. 1 provides a schematic outline of the basic steps that comprise this model.
Fig. 1.

Schematic diagram of the basic RNA-silencing model (see A Basic Model of RNA Silencing).
The system uses the presence of dsRNA as a signal of possible danger; the presence of long dsRNA is a good indicator of non-self-genetic material in that dsRNA is generally not found in an uninfected and properly functioning eukaryotic cell. Provided that the dsRNA is sufficiently long, the Dicer enzyme cleaves it into fragments ≈22 nt in length (24).
The RNA fragments formed in the previous step become associated with a multienzyme complex and are reduced to a single-stranded form. Stabilized by the multienzyme complex, these 22-nt single-stranded short RNAs serve as templates in the RNA-induced silencing complex (RISC) units.
The RISC units are targeted to mRNA having the same sequence as the dsRNA that triggers this process, binding with this mRNA to form the RISC–mRNA complex (simply “complex” for short).
Once RISC binds to complementary mRNA, one of two reactions proceeds: (a) the targeted mRNA may be degraded in a sequence-specific manner, or (b) dsRNA complementary in sequence to the mRNA and thus similar to the dsRNA that initiated the entire process may be synthesized by an RdRp.
We can model the steps described above by using the system of differential equations described in Appendix A.
Because of our concern with the problem of avoiding self-reactivity, the initial conditions in our model correspond to a situation in which silencing is directed toward mRNA corresponding to a self-gene. These mRNAs are transcribed at a constant rate and degraded at a fixed per-molecule rate such that, before silencing, mRNA concentration attains an equilibrium value. We then introduce a small or large amount of dsRNA specific to the gene and follow the silencing reaction. This situation is also similar to that of in vitro laboratory models where the experimenter adds exogenous dsRNA to silence a native gene.
In this model, the silencing reaction never takes off in the absence of an initial inoculum of dsRNA; instead, mRNA levels remain constant, and no dsRNA, complex, or short interfering RNA (siRNA) specific to the gene are observed. When initiated by a small amount of dsRNA, the silencing reaction takes off and ultimately reaches a new steady state with considerably reduced levels of mRNA and an ongoing silencing reaction (Fig. 2). The model successfully captures the following features of RNA-silencing dynamics from empirical studies (14, 15, 18, 21, 25–27):
Fig. 2.

Dynamics of RNA silencing after the introduction of a small (a) or large (b) dose of dsRNA as predicted by the basic model. In both cases we observe identical long-term silencing. Parameters are as described in Appendix A.
Detection and degradation of the initial dose of dsRNA, as evinced in Fig. 2, by the strong initial drop in dsRNA concentration.
Rapid generation of sequence-specific siRNAs, as seen in Fig. 2, by the rapid rise of RISC in the early stages of the reaction.
Amplification of the response, producing “secondary” dsRNA and siRNA molecules, as seen in Fig. 2, by the increase in dsRNA concentration subsequent to the initial decline and by the maintenance of a high RISC level despite continued RISC turnover.
Silencing of mRNA corresponding to the dsRNA that set off the reaction, as evinced in Fig. 2, by the sharp decline and prolonged reduction of the mRNA concentration.
Although successful in these ways, the model also makes two qualitative predictions that seem inconsistent with some of the more subtle aspects of the empirical data and that moreover seem troubling when one considers the possibility of accidental self-directed silencing.
Any starting dose of dsRNA will lead to the same ultimate level of silencing as long as the reaction is able to take off at all. We see this in Fig. 2, in which the same degree of silencing is obtained for both low and high initial dsRNA concentrations.
Once initiated, silencing is self-perpetuating even in the absence of further dsRNA input. Fig. 2 reveals no decline in silencing over time despite the absence of further dsRNA input.
The first prediction is troubling because it suggests that RNA-silencing systems should not exhibit “dosage dependence,” i.e., the strength of the reaction should not be influenced by the size of the initial dose of dsRNA. Experimental studies (13, 18, 21, 28) suggest otherwise: Larger initial doses of dsRNA engender larger silencing reactions or induce silencing more effectively. Moreover, we might expect dosage dependence to be useful in avoiding self-directed reactions. Because greater quantities of dsRNA typically will be more reliable indicators of the presence of non-self, dosage dependence could reduce the impact of mistaken reactions to self-derived genetic material.
The second prediction is troubling with regard to the problem of self-/non-self-discrimination. Imagine a small mistaken response to self such as generation of RISC with specificity for self-mRNA. According to the model, once a self-directed reaction is initiated, the cell will be unable to correct the mistake and attenuate the response. Taken together with the prediction of no dosage dependence, this is a major problem: Even a small mistake will generate a full-scale and permanent silencing reaction. As described in more detail in Discussion, in certain organisms such a silencing response could even spread systemically with severely detrimental consequences.
Unidirectional Amplification
In this section we take a more detailed look at RNA silencing systems in an effort to answer the questions above. In doing so, we will highlight what we hypothesize to be a crucial mechanism for limiting accidental reactions against self-RNAs.
A closer look at the molecular biology of the RdRp suggests that the long dsRNAs it produces after binding of RISC to the mRNA differ from the dsRNAs that initiated the silencing process. In many RNA-silencing systems that feature RdRp-mediated amplification, amplification is unidirectional. For example, Sijen et al. (21) show that in Caenorhabditis elegans, secondary siRNA production occurs only for segments upstream of the original siRNA primer. This unidirectionality is thought to occur because RdRp operates only in the 3′ direction when synthesizing dsRNA from a RISC–mRNA complex. We illustrate this process in Fig. 3 and its consequences in Fig. 4 (for simplicity, both figures ignore RISC-mediated cleavage of mRNA). A mathematical model of the process is developed in Appendix B. We find that:
Fig. 3.

The amplification process in RNA silencing. Each letter (A–E) represents a 22-nt segment of the gene in question. The reaction is initiated by a RISC-associated siRNA corresponding to segment D. RdRp mediates polymerization in the 3′ direction along the nascent strand, generating a dsRNA covering regions A–D. Dicer cleaves this dsRNA, yielding new double-stranded siRNAs corresponding to segments A–D. Polymerization does not occur on segment E, and thus no antisense siRNA segment E′ is produced.
Fig. 4.

Distribution of siRNAs after successive rounds of amplification. The fraction of upstream segments increases with each round. For illustrative clarity, no siRNA turnover is shown; turnover only strengthens the general pattern seen here.
The silencing reaction initially takes off, amplifying the target-specific siRNAs to levels many-fold higher than their initial prevalence.
The composition of the siRNA population changes over the course of the silencing reaction. As the reaction proceeds, upstream siRNAs increasingly make up the population of siRNA molecules as shown in Fig. 4.
Because these upstream siRNAs are unable to prime further RNA polymerization, the silencing reaction eventually dies out of its own accord.
Fig. 5 illustrates these results, showing the expected size of the siRNA population after a reaction is triggered by a single type E siRNA. The size of the siRNA population initially expands at a high rate but soon crashes back down toward zero. A full analysis of the branching process reveals that the median siRNA population will return to zero even more quickly. That is, the vast majority of silencing reactions will cease before the mean reaction does so, because the mean reaction is prolonged by inclusion of few rare cases where by chance no siRNA molecules are degraded or lost early on.
Fig. 5.

Expected size of the siRNA population after initiation of a silencing reaction by a single siRNA of type E as predicted by the branching process model with d = 0.2, p = 0, and q = 0(Appendix B). Individual siRNA types are shown by the curves that range from green (D) to blue (A). The total ensemble of siRNAs is shown by the red curve (Total). For comparison, the black line (Exponential) shows the size of an exponentially growing siRNA population that increases 5-fold each time unit, i.e., the siRNA population size that would be observed if each segment A–D had equal reproduction potential to that of segment E.
To understand better how the change in the distribution of the siRNA population affects the overall dynamics of silencing, we can expand the differential equation model described in A Basic Model of RNA Silencing to include the multiple siRNA types in the branching process described above. The dynamics of this extended model are shown in Fig. 6 and described in Appendix C. Two principal results emerge. First, silencing can persist indefinitely only with the continual input of dsRNA. Otherwise, only a transient silencing reaction is induced. Second, the magnitude of a silencing reaction now depends on the initial dose of dsRNA, as has been observed in the empirical studies (13, 18, 21). Although many experimental studies and applications of RNA silencing rely on the production of self-directed reactions, this is achievable only with high and/or sustained input of dsRNA.
Fig. 6.

Dynamics of the extended model for low starting dsRNA concentration with no further input (a), high starting dsRNA concentration with no further input (b), and low starting dsRNA concentration with continued low-level (1/10th the rate of mRNA production) dsRNA input (c). Parameters are as described in Appendix C.
The RNA-silencing system modeled here exhibits the remarkable property of amplification while maintaining sensitivity: The system massively amplifies an initial signal (dsRNA) yet requires continued low-level input of additional signal if amplification is to persist. This is a crucial property for certain information-processing circuits. Rapid and large-scale responses require signal amplification, but that amplification can easily swamp later inputs, drastically reducing the sensitivity of the system once it has been activated.
In this section we have shown that unidirectional amplification offers a solution to the basic problem of limiting mistaken responses to self-mRNA. Other aspects of the silencing pathway may contribute as well. For example, competition among siRNAs for the RISC-associated proteins and/or competition among RISCs for mRNA-binding sites would limit reaction priming by the under-represented downstream segments of siRNA, and thus further limit the expansion of a response in the absence of continued dsRNA input.
Discussion
The RNA-silencing system acts as a miniature immune system within each somatic cell. In this article we have shown how unidirectional amplification can prevent the analogue of autoimmune reactions: silencing responses directed toward self-genes. Should a self-directed silencing reaction occur, it is likely to impact an individual in two ways. First, there will be detrimental consequences within the cell in which the self-direction reaction begins; second, in many species a self-directed silencing reaction can spread throughout the organism.
At the single-cell level, silencing decreases gene expression with consequences that depend on gene function and the normal expression level. Because our model predicts that silencing will only be sustained for mRNA with sufficiently high rates of generation and low rates of clearance (as determined by equation Eq. 1), we predict silencing responses to be more vigorous against more highly expressed mRNAs, with siRNA production roughly proportional to mRNA expression level.
If a critical gene is targeted by an accidental silencing reaction, cell death could ensue. This may not be disastrous for multicellular organisms; we expect that most multicellulars could easily be able to absorb the cost of losing one or a few individual cells to mistakes in RNA silencing. After all, the cytotoxic T cell branch of the vertebrate immune response controls pathogens by directly killing infected cells, which are either expendable or can be regenerated. In organisms with systemic silencing, however, a mistaken silencing reaction may not be a one-cell problem. Instead, silencing could spread from one part of the organism to another and have more serious consequences. How is this spread prevented or controlled?
By way of answering this question, we have shown that the unidirectional nature of RNA amplification limits the extent to which a single copy of dsRNA can be amplified. Hence unidirectional amplification restricts the size and duration of most accidental responses directed against cellular mRNA but not those responses directed to viruses or transposons that continually generate dsRNA.
We do not expect unidirectional amplification to be the sole mechanism by which self-reactive responses are controlled. As with most potentially damaging processes, we expect multiple and redundant safeguards. Plants must rely on some alternative mechanism; they exhibit extensive systemic silencing but nonetheless feature an amplification cycle involving bidirectional RNA amplification. Bidirectional amplification does not exhibit the automatic self-limiting properties observed for unidirectional amplification; extending the unidirectional amplification model to incorporate bidirectional amplification yields a persistent silencing reaction (data not shown).
Other possible mechanisms include gene silencing, cooperativity, and localized enzymatic modification of dsRNA; we will address these briefly in turn. Because the intercellular transmission of self-reactive RNA silencing is probably the major problem for multicellulars, sufficient protection against self-directed responses may be conferred by mechanisms that merely reduce transmission without necessarily reducing the frequency or magnitude of mistaken reactions within the initial cell. Chromosome-level silencing, in which elements of the RNA-silencing system often play a role (2, 3, 29), is one such mechanism. Because the amplification cycle depends on the level of mRNA circulating with the cell, chromosome-level suppression will cause the mRNA level to fall below a threshold (such that R0 < 1; see Eq. 3. in Appendix A), and in turn will cause the silencing reaction to die out. Although in this case the gene will be silenced within the initial cell, intercellular spread would presumably be prevented. Indeed, the coupling of the RNA-silencing pathways to the transcriptional silencing process might have evolved first as a safeguard against accidental self-directed silencing reactions rather than as a defense against transposons as suggested previously (2, 3, 29).
Another hypothetical mechanism of limiting intercellular spread would involve a requirement for “cooperativity” in transmission such that multiple intercellular signals from multiple other cells are required to start a secondary silencing reaction in the recipient of the intercellular signals. Such a system would not initiate a mistaken self-reaction without simultaneous mistakes by several cells, and thus if p is the probability of a single self-reactive response per cell per unit time, and systemic spread requires n cells (in a very close proximity) to make this mistake, the probability of spread would drop to approximately pn. Because spreading viruses do infect multiple cells, such a cooperative mechanism would allow transmission of silencing directed to a spreading virus but not to self-mRNA.
Third, a localized process of dsRNA modification could prevent inadvertent silencing reactions. Knight and Bass (30) proposed that adenosine deaminases that act on RNA (ADARs) serve precisely this function. If ADARs are localized to the nucleus and Dicer is primarily in the cytoplasm, ADARs may modify any dsRNA produced accidentally within the nucleus before RNA silencing takes off (30). This would prevent self-directed reactions but would not inhibit the silencing of viral dsRNA located outside of the nucleus. Although this mechanism could prevent self-directed responses, ADARs alone would not explain the dosage dependence observed in experiments and predicted by the unidirectional model.
In this article we have drawn attention to the problem of avoiding accidental self-directed silencing reactions. Our unidirectional amplification model provides a hypothesis for a mechanism by which cells solve this problem: Unidirectional amplification ensures that a silencing reaction will persist only if dsRNA input is itself persistent. The unidirectional amplification model makes a number of additional testable predictions as well. For example, the model predicts that when a silencing reaction is initiated experimentally by adding a particular type of siRNA, the magnitude of the reaction will depend on the position of the siRNA within the silenced gene. Specifically, downstream siRNAs should initiate larger silencing responses than do upstream siRNAs. We expect superadditive scaling; moving to an siRNA twice as far downstream should more than double the magnitude of ensuing reaction. Second, the model predicts that when a silencing reaction is initiated experimentally by adding long dsRNAs corresponding to a whole gene, longer genes will experience stronger silencing reactions with all else being equal. Third, even early in a silencing reaction before target mRNA availability becomes limiting, the rate of increase in siRNA concentration will be subexponential rather than exponential as predicted by the basic model. Further empirical work will be needed to confirm or reject these predictions.
Acknowledgments
We thank Charles Laird and three anonymous reviewers for numerous helpful suggestions. E.M. is supported by a Howard Hughes Medical Institute predoctoral fellowship. R.A. acknowledges support from National Institutes of Health Grant R01 AI-48334.
Appendix
A. The Basic Model. The process depicted in Fig. 1 can be described quantitatively as a system of differential equations. We allow D(t), R(t), C(t), and M(t) to represent the concentrations of the dsRNA, RISC, RISC–mRNA complex, and mRNA at time t, respectively. The rates of changes in these quantities then are given by
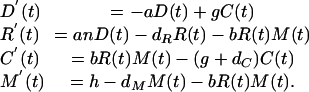 |
[1] |
We describe these terms as:
dsRNA D(t): The net rate of degradation of dsRNA by Dicer and background processes equals aD. The regeneration of dsRNA by RdRp (from the RISC–mRNA complex, C) equals gC(t).
RISC R(t): The net rate of generation of RISC (from the association of short fragments of RNA produced by Dicer to a multienzyme complex) equals anD(t), where n is the average number of complexes generated per dsRNA molecule. Two terms account for the loss of RISC: The term dRR(t) represents background loss, and the term bR(t)M(t) results from the binding of RISC to mRNA to form the complex C(t).
RISC–mRNA complex C(t): The mass action term bR(t)M(t) describes the formation of complex from RISC and mRNA. The complex is used in dsRNA synthesis at a rate gC and degraded at rate dCC for a net rate of loss (g + dC)C.
mRNA M(t): mRNA is generated at a constant rate h and is lost by nonspecific degradation at a rate dMM(t). Under mass-action kinetics, mRNA binds to RISC to form complex at a rate bR(t)M(t).
Here we assumed that the proteins involved (Dicer, RdRp, and the protein components of the RISC) are present at sufficient levels so as not to limit reaction rates and that, once the RISC–mRNA complex is formed, the mRNA is cleaved before the dissociation of RISC. Although these assumptions are likely to be met early in the course of a silencing reaction, they may need to be modified for the later stages of the silencing process. We also assumed that the RISC is not reused on multiple mRNAs. Allowing RISC to be recycled after mRNA degradation only increases the magnitude of accidental self-directed responses (data not shown).
This system of equations has two steady states. At the first steady state, silencing does not occur (the levels of D, R, and C are zero) and mRNA persists at its normal level (M̂ = h/dM). In the second steady state the components of the RNA-silencing system D, R and C, are all positive and the silencing reaction controls the level of mRNA below its normal level. The values at this steady state are
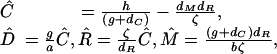 |
[2] |
where ζ = [g(n – 1) – dC]. This second state is biologically meaningful only if Ĉ, D̂, R̂, and M̂ are nonnegative, for which the condition hζ > dMdRdC is both necessary and sufficient. Interestingly, the level of mRNA present at this steady state is independent of the rate of mRNA expression h.
We can determine whether a silencing reaction will take off in this model by computing the “basic reproductive number” R0 for dsRNA, which equals the number of secondary copies of D generated after the introduction of a single copy of D:
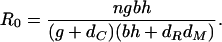 |
[3] |
If R0 < 1, the addition of a small amount of dsRNA is unable to trigger a silencing reaction; if R0 > 1, then even a small amount of dsRNA will be sufficient to trigger a silencing reaction, and this silencing reaction will stably persist with a permanent reduction in the level of the target mRNA.
Fig. 2 shows sample reaction trajectories for this model. The parameter values, intended simply as examples with which to illustrate the general qualitative features of these dynamics, are as follows.
a = 10: Rate of dsRNA degradation by Dicer (per molecule per time unit).
b = 0.001: Mass action rate constant for RISC mRNA formation [per (molecule)2 per time unit].
h = 1,000: Rate of target mRNA synthesis (per cell per time unit).
g = 1: Rate of dsRNA synthesis from RISC–mRNA complex (per complex per time unit).
dM = 1: Rate of nonspecific mRNA degradation (per molecule per time unit).
dR = 0.1: Rate of RISC dissociation (per RISC per time unit).
dC = 1: Rate at which complex is destroyed (per complex per unit time).
n = 5: Number of siRNAs produced from one secondary dsRNA (per molecule).
The initial levels of mRNA, RISC, complex, and dsRNA are M(0) = 1,000, R(0) = 0, C(0) = 0, and D(0) = 10 (Fig. 2a) or D(0) = 1,000 (Fig. 2b). Time can be thought of as measured in hours. As the explicit solutions indicate, the qualitative features of Fig. 2 are generally robust to changes in these parameter values.
B. Modeling the Kinetics of Unidirectional Amplification. In this section, we model the amplification of a gene with five 22-bp “segments” labeled A–E as in Fig. 3. We assume that a silencing reaction is initiated with a single siRNA corresponding to segment E. Generalization to longer genes and to reactions triggered by multiple or even overlapping siRNA is straightforward.
During each time-step (or “generation,” in terms of the branching process), various events may occur to each siRNA in the system.
With probability d, the siRNA is destroyed before any copying can take place.
With probability p(1 – d), nothing happens at all. (Notice that this means that p essentially scales time.)
With probability (1 – p)(1 – d), the siRNA (presumably as part of a RISC) binds to a complementary mRNA, and the RdRp starts synthesizing and manages to create at least one new 22-bp segment. With probability q, the polymerization breaks off in between two segments. Once the RdRp finishes with segment A, the reaction halts.
If v(t) is a vector giving the number of siRNAs of each type A–E at time t, the expected number of siRNAs one time step later, at time t + 1, will be v(t + 1) = v(t)M, where M is the matrix giving the expected number of siRNAs of each type.
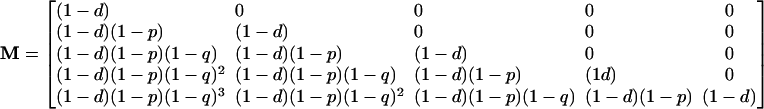 |
[4] |
This matrix tells us a great deal about the behavior of the multitype branching process (31). The maximal eigenvalue λ of this matrix is (1 – d), which gives us the asymptotic net rate of growth of the system. Because this rate is below unity, we are dealing with a subcritical branching process that will inevitably go to extinction. From this we see that the level of siRNA in the system will never increase on the long term. The reaction might proceed for a long time and reach a very large size, but it will eventually and inevitably die down. The left eigenvector of M (v̄ such that v̄M = λv̄) gives the limiting distribution of siRNA types. This eigenvector is (1, 0, 0, 0, 0), which implies that in the limit, the population of siRNAs is dominated by the terminal element A. Finally, the right eigenvector of M gives the “reproductive values” of each type; this eigenvector is (0, 0, 0, 0, 1), which means that, in the long run, only the progeny of element E make up an appreciable part of the siRNA population.
This analysis pertains to the mean trajectory of the system; the theory of branching processes would also allow us to compute the probabilities of reaching some threshold, probabilities of extinction after time t, and so forth (31). In this way, we could compute the probability that a reaction started by k siRNAs reaches an ultimate level of j siRNAs or that it continues after time t*.
C. Extending the Differential Equation Model. Here we adapt the basic model (Eq. 2) to a target mRNA composed of n successive 22-bp segments. We assume nonoverlapping siRNA frames such that the gene in question is composed of n distinct 22-base siRNAs: one corresponding to segment 1 (R1), one corresponding to segment 2 (R2), etc. Similarly, we consider only n distinct siRNA–mRNA complexes (Ci), one corresponding to each siRNA. We assume that once RdRp is in place, polymerization continues to the end of the mRNA target. Taken together with the assumption that Dicer quickly degrades dsRNA completely into its component 22-base segments, this allows us to track only n classes of dsRNA: one spanning from segment 1 through segment n (we call this Dn), one spanning segments 1 through n – 1 (Dn–1), one from segment i to n – i (Dn–i), etc., and finally one that is composed of only segment 1 (D1). We assume that polymerization occurs with sufficient rapidity that we can safely ignore differences in synthesis times for the different dsRNA types.
These assumptions lead us to a system of 3n + 1 differential equations. Where i = 1... n.
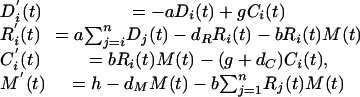 |
[5] |
Fig. 6 shows sample reaction trajectories for this model directed toward a gene with five segments of 22 bases in length each labeled A–E, respectively. The parameters and initial values are as given in Appendix A. In Fig. 6c, dsRNA is added continually at a rate r = 100 molecules per cell per time unit, 1/10th the rate of mRNA production.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: dsRNA, double-stranded RNA; RdRp, RNA-directed RNA polymerase; RISC, RNA-induced silencing complex; siRNA, short interfering RNA.
References
- 1.Hannon, G. J. (2002) Nature 418, 244–251. [DOI] [PubMed] [Google Scholar]
- 2.Mette, M. F., Aufsatz, W., van der Winden, J., Matzke, M. A. & Matzke, A. J. (2000) EMBO J. 19, 5194–5201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jones, L., Ratcliff, F. & Baulcombe. D. D. (2001) Curr. Biol. 11, 747–757. [DOI] [PubMed] [Google Scholar]
- 4.Volpe, T. A., Kidner, C., Hall, I. M., Teng, G., Grewal, S. I. S. & Martienssen, R. A. (2002) Science 297, 1833–1837. [DOI] [PubMed] [Google Scholar]
- 5.Reinhart, B. J., Weinstein, E. G., Rhoades, M. W., Bartel, B. & Bartel, D. P. (2002) Genes Dev. 16, 1616–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kasschau, K. D., Xie, Z., Allen, E., Llave, C., Chapman, E. J., Krizan, K. A. & Carrington, J. C. (2003) Dev. Cell 4, 205–217. [DOI] [PubMed] [Google Scholar]
- 7.Waterhouse, P. M., Wang, M.-B. & Lough, T. (2001) Nature 411, 834–842. [DOI] [PubMed] [Google Scholar]
- 8.Plasterk, R. H. A. (2002) Science 296, 1263–1265. [DOI] [PubMed] [Google Scholar]
- 9.Ketting, R. F., Haverkamp, T. H., van Luenen, H. G. & Plasterk, R. H. (1999) Cell 99, 133–141. [DOI] [PubMed] [Google Scholar]
- 10.Tabara, H., Sarkissian, M., Kelly, W. G., Fleenor, J., Grishok, A., Timmons, L., Fire, A. & Mello, C. C. (1999) Cell 99, 123–132. [DOI] [PubMed] [Google Scholar]
- 11.Hamilton, A., Voinnet, O., Chappell, L. & Baulcombe, D. (2002) EMBO J. 21, 4671–4679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li, H., Li, W. X. & Ding, S. W. (2002) Science 296, 1319–1321. [DOI] [PubMed] [Google Scholar]
- 13.Giordano, E., Rendina, R., Peluso, I. & Furia, M. (2002) Genetics 160, 637–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klahre, U., Crété, P., Leuenberger, S. A., Iglesias, V. A. & Meins, F., Jr. (2002) Proc. Natl. Acad. Sci. USA 99, 11981–11986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fire, A., Xu, S., Mongomery, M. K., Kostas, S. A., Driver, S. E. & Mello, C. C. (1998) Nature 391, 806–811. [DOI] [PubMed] [Google Scholar]
- 16.Jorgensen, R. A. (2002) Proc. Natl. Acad. Sci. USA 99, 11561–11563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schwarz, D. S., Hutvágner, G., Haley, B. & Zamore, P. D. (2002) Mol. Cell 10, 537–548. [DOI] [PubMed] [Google Scholar]
- 18.Lipardi, C., Wei, Q. & Patterson, B. M. (2001) Cell 107, 297–307. [DOI] [PubMed] [Google Scholar]
- 19.Sijen, T. & Kooter, J. M. (2000) BioEssays 22, 520–531. [DOI] [PubMed] [Google Scholar]
- 20.Dalmay, T., Hamilton, A., Rudd, S., Angell, S. & Baulcombe, D. C. (2000) Cell 101, 543–553. [DOI] [PubMed] [Google Scholar]
- 21.Sijen, T., Fleenor, J., Simmer, F., Thijssen, K. L., Parrish, S., Timmons, L., Plasterk, R. H. A. & Fire, A. (2001) Cell 107, 465–476. [DOI] [PubMed] [Google Scholar]
- 22.Ahlquist, P. (2002) Science 296, 1270–1273. [DOI] [PubMed] [Google Scholar]
- 23.Zamore, P. D. (2002) Science 296, 1265–1269. [DOI] [PubMed] [Google Scholar]
- 24.Bernstein, E., Caudy, A. A., Hammond, S. M. & Hannon, G. J. (2001) Nature 409, 363–366. [DOI] [PubMed] [Google Scholar]
- 25.Hammond, S. M., Bernstein, E., Beach, D. & Hannon, G. J. (2000) Nature 404, 293–296. [DOI] [PubMed] [Google Scholar]
- 26.Zamore, P. D., Tuschl, T., Sharp, P. A. & Bartel, D. P. (2000) Cell 101, 25–33. [DOI] [PubMed] [Google Scholar]
- 27.Nykänen, A., Haley, B. & Zamore, P. D. (2001) Cell 107, 309–321. [DOI] [PubMed] [Google Scholar]
- 28.Parrish, S., Fleenor, J., Xu, S., Mello, C. & Fire, A. (2000) Mol. Cell 6, 1077–1087. [DOI] [PubMed] [Google Scholar]
- 29.Allshire, R. (2002) Science 297, 1818–1819. [DOI] [PubMed] [Google Scholar]
- 30.Knight, S. W. & Bass, B. L. (2002) Mol. Cell 10, 809–817. [DOI] [PubMed] [Google Scholar]
- 31.Athreya, K. B. & Ney, P. E. (1972) Branching Processes (Springer, Berlin).