

Abstract
Approximately 3–10% of people have specific difficulties in reading, despite adequate intelligence, education, and social environment. We report here the characterization of a gene, DYX1C1 near the DYX1 locus in chromosome 15q21, that is disrupted by a translocation t(2;15)(q11;q21) segregating coincidentally with dyslexia. Two sequence changes in DYX1C1, one involving the translation initiation sequence and an Elk-1 transcription factor binding site (–3G → A) and a codon (1249G → T), introducing a premature stop codon and truncating the predicted protein by 4 aa, associate alone and in combination with dyslexia. DYX1C1 encodes a 420-aa protein with three tetratricopeptide repeat (TPR) domains, thought to be protein interaction modules, but otherwise with no homology to known proteins. The mouse Dyx1c1 protein is 78% identical to the human protein, and the nonhuman primates differ at 0.5–1.4% of residues. DYX1C1 is expressed in several tissues, including the brain, and the protein resides in the nucleus. In human brain, DYX1C1 protein localizes to a fraction of cortical neurons and white matter glial cells. We conclude that DYX1C1 should be regarded as a candidate gene for developmental dyslexia. Detailed study of its function may open a path to understanding a complex process of development and maturation of the human brain.
Dyslexia, or specific reading disability, is the most common childhood learning disorder. Available evidence suggests that dyslexia is a neurological disorder with a genetic basis. Functional brain imaging studies have illustrated that dyslexia has universal neurobiological correlates (1). Linkage and association studies have pinpointed at least six loci for dyslexia (DYX1 to DYX6, www.ncbi.nlm.nih.gov/omim). DYX1 in chromosome 15q21 was linked to dyslexia, and the results have been replicated in at least three independent studies thereafter (2–5). Despite numerous molecular genetic studies, no candidate genes for dyslexia are known so far.
In this study, we report a gene, DYX1C1, disrupted by translocation that cosegregates with dyslexia in one family, and additional genetic evidence for its possible role in dyslexia susceptibility. In two sample sets tested, the –3A allele associated significantly with dyslexia and gave an overall odds ratio of 3.2, whereas the 1249T allele associated in one but not the other sample set; however, the overall odds ratio was 2.3. A haplotype of the two markers yielded a significant transmission disequilibrium test and associated significantly with dyslexia. DYX1C1 protein appears to be rapidly up-regulated and translocated after brain ischemia. Our results suggest a candidate gene for detailed molecular and epidemiological studies in dyslexia.
Materials and Methods
Ascertainment of Patients and Psychological Assessment. A family segregating t(2;15)(q11;q21) and dyslexia (5) was studied to identify the translocation breakpoint in chromosome 15q21 as a putative marker involving a gene for dyslexia (Fig. 1A). The phenotypes of the family members have been described in detail (5). In brief, the father had a history of profound reading and writing difficulties in school but is employed in business. The two daughters with translocations have been examined and diagnosed with dyslexia at Helsinki University Hospital after being referred there at ages 7 and 9, respectively, because of reading problems in school. Their brother was referred to a neuropediatric unit at age 6 and was diagnosed with specific difficulty in reading-related skills; however, his overall performance was also slightly below normal. He has attended special education class.
Fig. 1.

(A) Pedigree of the family with t(2;15)(q11;q21). Black fill denotes translocation; gray fill, dyslexia. The boy with translocation has low overall performance and therefore his phenotype for dyslexia is unknown. (B) Fluorescent in situ hybridization with BAC clone 178D12 as a probe, showing hybridization signals in chromosomes 15, der(15), and der(2). (C) Southern hybridization with a probe derived from 178D12 shows genomic rearrangement with six restriction enzymes in DNA from a translocation patient (T) compared with the control sample (C). (D) Physical map of the breakpoint region, including DYX1C1 exons (bars) and an intronic pseudogene (bar with arrow). Black triangle illustrates the Southern hybridization probe position, gray denotes the breakpoint interval. (E) Predicted secondary structure of a 370-bp A+T-rich region within the breakpoint interval.
For association studies, 58 dyslexic and 61 nondyslexic individuals from 20 unrelated families were first recruited from the Department of Pediatric Neurology at the Hospital for Children and Adolescents, University of Helsinki (Helsinki, Finland). In addition, 3 families and 33 unrelated dyslexic-nondyslexic couples were recruited by the Child Research Centre, Jyväskylä, Finland. Additional population controls consisted of 100 anonymous blood donors. The study was approved by the appropriate Ethical Review Boards and informed consent was obtained from the participants. The diagnosis and degree of dyslexia were determined by Finnish reading and spelling tests designed for children (6) and adults (7). Intelligence was estimated by Wechsler tests for adults (WAIS-R; ref. 8) or for children (WISC-R; ref. 9). Eight subtests covering verbal and visual skills were used: Information, Digit Span, Vocabulary, Similarities, Picture Completion, Picture Arrangement, Block Design, and Coding. In addition, reading-related neurocognitive skills (phonological awareness, rapid automatized naming, and verbal short-term memory) were assessed by neuropsychological tests (10–13). The diagnostic criteria for dyslexia included normal performance intelligence quotient (PIQ >85) and remarkable deviation (depending on age, at least 2 years) in reading skills.
Fluorescent in Situ Hybridization (FISH) and Southern Blotting. RPCI-11 bacterial artificial chromosome (BAC) clone 178D12 (GenBank accession no. AC013355), the yeast artificial chromosome (YAC) clones 770D11, 794F1, 967G2, 757D11, 952C2, and 965E5 from the Centre d'Étude du Polymorphisme Humain (CEPH), and the P1 134E18 (Genome Systems, St. Louis) were used as probes in FISH. The methods for FISH have been previously described (5). Samples (15 μg) of total genomic DNA from an individual carrying the translocation and from an unrelated control person were digested with BamHI, EcoRI, HindIII, BsaAI, PstI, or SphI, subjected to electrophoresis in a 0.7% agarose gel, and transferred to Hybond N+ membrane (Amersham Pharmacia Biotech) with alkaline blotting. PCR fragments derived from human genomic DNA were cloned into pCR2.1 TOPO-TA vector (Invitrogen), and the insert was removed with EcoRI digestion and purified by gel electrophoresis (Qiagen, Venlo, The Netherlands). α-32P-labeled insert was used as a probe in Southern hybridization. Hybridization was performed overnight at 65°C in 0.5 M NaHPO4/1 mM EDTA/7% SDS/1% BSA, and the filter was washed in 2× SSC/0.05% SDS at 65°C for 1 h. Filters were autoradiographed with a PhosphorImager.
Cloning of DYX1C1, Sequence Analysis, and Expression Study. Novel genes in the sequence of clone 178D12 were predicted with genscan and fgenes software. Predicted genes were confirmed by sequencing RT-PCR products. Mouse Dyx1c1 was constructed from two overlapping EST sequences (GenBank accession nos. BG242087 and AK005832) and verified by comparing it to all available mouse Dyx1c1 EST sequences. cDNA sequences of Dyx1c1 and DYX1C1 were aligned with clustalx. The alignment was improved manually, and shaded with box-shade. The secondary structure of the T+A-rich region was predicted with mfold (available at www.bioinfo.rpi.edu/applications/mfold/old/dna/) with default parameters. Promoter region of DYX1C1 was predicted with the tssg and tssw software, available at http://searchlauncher.bcm.tmc.edu/seq-search/gene-search.html, and neural network promoter prediction (nnpp) software, available at www.fruitfly.org/seq_tools/promoter.html. Transcription factor binding sites were predicted by tess at www.cbil.upenn.edu/tess, matinspector at www.gsf.de/biodv/matinspector.html, and tfsearch at www.cbrc.jp/research/db/TFSEARCH.html.
The expression of DYX1C1 was analyzed by RT-PCR from CLONTECH's multiple-tissue cDNA panels 1 and 2. RT-PCR was performed in 25 μl under the following conditions: 94°C 2 min (94°C 1 min, 68°C 2 min) 30 times, 1× DyNAzyme buffer with MgCl2 (Finnzymes, Espoo, Finland), 0.2 unit of DyNAzyme II polymerase (Finnzymes), 15 pmol of forward primer GTTGACAGAATGCTGTTCCACGTCG, and 15 pmol of reverse primer CA AGCTGAGGCACGA AGAGCA ATGA. For Northern blot analysis, a cDNA probe corresponding to bases 40–630, spanning exons 2–5 of DYX1C1 coding sequence, was hybridized to multiple-tissue Northern blots I and II (CLONTECH) according to the manufacturer's instructions. The genomic sequences of nonhuman primates corresponding to all exons were determined by direct sequencing after PCR amplification with human-specific intronic primers (primer sequences available from authors on request).
Polymorphism Detection and Association Analysis. All 10 exons of DYX1C1 were directly sequenced after PCR with intronic primers flanking exons from 20 dyslexic individuals (all primer sequences available from authors on request). Polymorphisms –164C → T, 4C → T, and 572G → A introduced novel Tsp45I, MnlI, and MboII restriction sites, respectively. Because –3G → A, –2G → A, 270G → A, and 1249G → T changed no restriction sites, a mutation was introduced into the primer sequence to create a restriction site specific for one of the single-nucleotide polymorphism (SNP) alleles: MspI for –3G → A and –2G → A, Bstf5I for 270G → A, and HpyCH4 IV for 1249G → T. In addition, a tail containing the SNP-specific restriction site to act as an internal control for digestion was added to the primer. 1259C → G was directly sequenced. PCR products were digested with the appropriate enzyme and electrophoresed on agarose gels. 572G → A was run on a polyacrylamide gel and visualized by silver staining. SNPs –3G → A and –2G → A were in the same restriction site and, therefore, individuals showing the A allele after restriction were sequenced to verify which sequence change was present. The χ2 test or Fisher's exact test was used to evaluate the statistical significance. Bonferroni correction was applied where specified. The transmission disequilibrium test (TDT) was used to assess risk haplotype transmission.
Cellular Localization of DYX1C1 Protein. Full-length DYX1C1 cDNA was cloned into pcDNA3.1/V5–6xHis expression vector (Invitrogen). The African green monkey kidney COS-1 cell line was transfected with 3 μg of the construct, with FuGENE 6 (Roche molecular Biochemicals) as a transfection reagent, according to manufacturer's protocols. Cells were stained with mouse anti-V5 antibody (Invitrogen) and FITC-conjugated goat anti-mouse IgG (Sigma–Aldrich). Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). The specificity of anti-V5 antibody was tested with standard Western blotting methods.
Immunohistochemical Study of Brain. To investigate whether DYX1C1 is expressed in mature human brain, brain tissue from six deceased individuals was stained with anti-DYX1C1 antiserum raised in rabbits against the peptide CATEAKAAAKREDQK (antibody production purchased from Sigma-Genosys). The patients had died of cardiac arrest or ischemic stroke. In the five individuals with stroke, the postischemic time before death varied from 15 to 60 h, and brain samples were obtained at rapid autopsies with postmortem delays varying from 10 to 40 h. Tissue blocks with cortical and some subcortical tissue were obtained from the core or an area close to the core of the infarction and control samples were dissected from homologous contralateral locations for comparison. Tissues were fixed in formalin and embedded in paraffin, and were used for research by permission of the appropriate Ethical Review Board of the Helsinki University Central Hospital. Immunohistochemical methods and use of this postmortem autopsy material for studies on other proteins have been described (14). The dilutions of antiserum used were 1:100–1:200, and all stained sections were compared with adjacent tissue sections incubated with the preimmune serum in identical conditions and dilutions. No antigen-retrieval methods were used. Light microscopy of tissue sections was performed with Leitz Laborlux D microscope (Leitz, Wetzlar, Germany) equipped with Nikon Coolpix 995 digital camera (Nikon).
Results
Cloning of DYX1C1. We used fluorescent in situ hybridization to refine the translocation breakpoint within the BAC clone RP-11-178D12 (AC013355; Fig. 1B). Clones 967G2, 757D11, 952C2, and 965E5 were proximal and 134E18 distal to the breakpoint, as expected. The BAC clone contained two known genes, cell-cycle restoration protein 8 (CPR8) and complementation class B phosphoinositol glycan (PIGB) in addition to the gene described here. To identify the breakpoint, we used PCR-amplified nonrepetitive genomic DNA fragments from the BAC clone RPCI-11-178D12 as probes in Southern hybridization. A probe corresponding to nucleotides 102317–102837 of the complete sequence of 178D12 revealed a genomic rearrangement with six different restriction enzymes (Fig. 1C). Thus, we could pinpoint the breakpoint to a region of 3,229 bp, limited by the restriction sites for PstI and HindIII (Fig. 1D). This interval includes exons 8 and 9 of a novel gene, DYX1C1, and there is also a 301-bp A+T-rich region with an almost complete 72-bp inverted repeat (Fig. 1E), suggesting a repeat-induced mechanism for the translocation. A+T-rich repeats are known to occur at many chromosomal rearrangement sites (15).
We predicted the coding sequence of DYX1C1 from the genomic sequence of BAC clones RP-11-178D12 and CTD-2137J4, and confirmed the exon–intron boundaries by RT-PCR. The length of DYX1C1 mRNA, obtained by RT-PCR, is 1,993 bp, and it encodes a predicted protein of 420 aa. The human DYX1C1 protein has three C-terminal TPR domains, corresponding to amino acids 290–323, 324–357, and 366–399. Beyond weak homologies to similar domains, there are no significant homologies to known proteins. DYX1C1 consists of 10 exons spanning ≈78 kb of genomic DNA (Fig. 1D). Three promoter prediction programs identified a promoter precisely before the 5′ end of the cDNA obtained by RT-PCR, and the preceding sequence in exons 1 and 2 contains five in-frame termination codons, suggesting that the cDNA is nearly complete. The putative promoter of DYX1C1 has a TATA box (TATAAAT) at position –31. The start codon is located in exon 2, 369 bp from the predicted transcription initiation site. DYX1C1 mRNA appears to exist in several different splice forms: exons 2 and 9 can be omitted, and there is an alternative acceptor splice site in intron 2. All these arrangements, however, alter the reading frame, leading to truncated protein products.
DYX1C1 Polymorphisms and Association to Dyslexia. To study the possible role of DYX1C1 in other individuals with dyslexia, we screened the DYX1C1 cDNA for polymorphism in 20 unrelated dyslexic individuals by direct sequencing. We found eight SNPs. Four of the SNPs (4C → T, 270G → A, 572G → A, and 1259C → G) were in the coding region and resulted in amino acid substitutions, whereas three (–164C → T, –3G → A, and –2G → A) resided in the 5′ untranslated region (Table 1). The eighth SNP is a G-to-T transversion at position 1249 of the DYX1C1 mRNA, which results in the substitution of a glutamic acid for an ochre stop codon at amino acid position 417 and the deletion of the C-terminal tetrapeptide Glu-Leu-Lys-Ser.
Table 1. Frequency of SNPs in dyslexic subjects and controls.
| First sample set
|
Replication set
|
Combined
|
|||||
|---|---|---|---|---|---|---|---|
| SNP | Coding change | Dyslexia % (n) | Control % (n) | Dyslexia % (n) | Control % (n) | P | OR (CI) |
| -164C → T | NC | 6.4 (55) | 3.2 (111) | 1.0 (51) | 1.8 (82) | 0.3827 | 1.5 (0.6-3.9) |
| -3G → A | NC | 8.3 (54) | 3.1 (113) | 8.7 (52) | 2.5 (81) | 0.0020 | 3.2 (1.5-6.9) |
| -2G → A | NC | 0.9 (54) | 0 (113) | 0 (49) | 1.3 (78) | 0.7150 | 1.0 (0.1-10.7) |
| 4G → T | P2S | 0.9 (54) | 0.5 (103) | 0 (34) | 2.3 (65) | 0.8748 | 0.5 (0.1-4.4) |
| 270G → A | V90I | 1.9 (54) | 5.3 (113) | 8.2 (49) | 5.0 (80) | 0.9248 | 1.0 (0.4-2.1) |
| 572G → A | G191E | 51 (46) | 47 (98) | 44 (16) | 44 (18) | 0.6447 | 1.1 (0.7-1.7) |
| 1249G → T | E417X | 13.2 (53) | 5.8 (113) | 10.2 (54) | 5.0 (80) | 0.006 | 2.3 (1.2-4.2) |
| 1259C → G | S420C | 9.3 (43) | 7.1 (106) | 10.0 (35) | 2.0 (25) | 0.1610 | 1.7 (0.8-3.5) |
The percentages are allele frequencies; n, number of subjects. Comparisons with P values <0.05 are in boldface. NC, noncoding change; OR, odds ratio; CI, 95% confidence interval.
All SNPs were then genotyped in 35 additional dyslexic subjects and 113 controls (including both family-based and population controls; no significant differences were observed between them in allele frequencies). SNPs –3A and 1249T showed significant association with dyslexia (P = 0.006 and 0.02, respectively; Table 1). Because the first set included subjects from only 20 families, we recruited a replication set with 54 dyslexic and 82 control individuals. Their genotyping yielded P values of 0.02 and 0.1 for –3G → A and 1249 G → T, respectively. Combining all data, the –3A allele frequency in dyslexic subjects was 0.085 (18/212 chromosomes) and 0.028 in controls (11/388 chromosomes), yielding an odds ratio of 3.2 (95% confidence interval 1.5–6.9, P = 0.002). The 1249T allele frequency in dyslexic subjects was 0.117 (25/214 chromosomes) and 0.054 (21/386 chromosomes) in controls, yielding an odds ratio of 2.3 (95% confidence interval 1.2–4.2, P = 0.006). Association tests for –3G → A and 1249G → T remained significant after Bonferroni correction (P = 0.016 and 0.048, respectively). The other SNPs did not show significant differences (Table 1).
A common haplotype of –3A and 1249T was seen in 14 dyslexic subjects from eight families but only in 4 normal readers from three families and six population controls. The –3A/1249T haplotype frequency in dyslexic subjects was 0.13 (14/106 cases) and 0.05 (10/192) in controls, yielding an odds ratio of 2.8 (95% confidence interval 1.2–6.5, P = 0.015). We performed also TDT in informative trios (n = 9). There were five transmissions and zero nontransmissions of the risk haplotype and zero transmissions and five nontransmissions of other haplotypes (P = 0.025).
Mouse Dyx1c1 and Primate Genes. We could predict the mouse Dyx1c1 by connecting overlapping mouse EST clones. The Dyx1c1 mRNA encodes a 421-residue protein that is 78% identical with human DYX1C1 (Fig. 2A). The nonhuman primates chimpanzee, pygmy chimpanzee, gorilla, and orangutan were sequenced for the genomic segments corresponding to human exons, and the predicted proteins differed by 3, 2, 5, and 6 amino acids (0.7%, 0.5%, 1.2%, and 1.4% of residues), respectively (Table 2; GenBank accession nos. AY178583–AY178618 and AH012450–AH012453).
Fig. 2.

(A) Comparison of the protein sequences of human (h) DYX1C1 and mouse (m) Dyx1c1. The SNPs found in this study are marked with arrowheads, and the three TPR domains are marked with asterisks. SNPs in the 5′-UTR are also shown. (B) RT-PCR from human multiple-tissue cDNA panels I and II (Clontech). Lanes: 1, phage λ/φX174 DMA size marker; 2, heart; 3, brain; 4, placenta; 5, lung; 6, liver; 7, skeletal muscle; 8, kidney; 9, pancreas; 10, spleen; 11, thymus; 12, prostate; 13, testis; 14, ovary; 15, small intestine; 16, colon; and 17, leukocytes. (C) DYX1C1 Northern blot showing a band of ≈2 kb. Lanes: 1, spleen; 2, thymus; 3, prostate; 4, testis; 5, ovary; 6, small intestine; 7, colon; 8, leukocytes; 9, brain; and 10, placenta. (D) Cellular localization of DYX1C1 protein. COS-1 cells transfected with DYX1C1-V5 fusion construct were stained with monoclonal mouse anti-V5 antibody and FITC-conjugated anti-mouse-IgG (green). 4′,6-Diamidino-2-phenylindole (DAPI)-stained nuclei are shown blue.
Table 2. Comparison of DYX1C1 cDNA between human and four nonhuman primates.
| Exon | Nucleic acid change | Amino acid change | Chimpanzee | Pygmy chimpanzee | Gorilla | Orangutan |
|---|---|---|---|---|---|---|
| 1 | None | |||||
| 2 | 6T → C | + | + | + | + | |
| 2 | 47C → T | A16V | + | |||
| 2 | 48G → C | + | ||||
| 2 | 107C → T | T36M | + | |||
| 3 | None | |||||
| 4 | 284T → C | M95T | + | |||
| 4 | 384C → T | + | + | + | + | |
| 5 | 473C → A | A158E | + | + | + | + |
| 5 | 516A → C | Q172H | + | |||
| 5 | 520A → G | K174E | + | |||
| 5 | 540A → G | + | ||||
| 5 | 572G → A | G191E | + | + | + | + |
| 5 | 583A → T | I195L | + | + | ||
| 5 | 591T → C | + | ||||
| 5 | 611T → C | L204S | + | |||
| 5 | 624T → C | + | ||||
| 6 | 639G → A | + | ||||
| 7 | 789A → G | + | ||||
| 8 | 909G → A | + | + | + | + | |
| 9 | None | |||||
| 10 | None |
Nucleic acid and amino acid changes are shown for each exon of DYX1C1 in comparison to the human sequence; + indicates the presence of a change in a nonhuman species.
Expression of DYX1C1 and Cellular Localization of DYX1C1 Protein. We detected DYX1C1 mRNA in several adult human tissues by RT-PCR. It is most abundantly expressed in the brain, lung, kidney, and testis (Fig. 2B). Northern hybridization revealed an ≈2-kb transcript, corresponding to the predicted size of DYX1C1 mRNA in all tissues studied (Fig. 2C). In addition, ≈1-kb and 5-kb transcripts were seen in skeletal muscle but not in any of the other tissues studied (data not shown).
We studied the cellular localization of DYX1C1 protein in transiently transfected monkey kidney COS-1 cells by using immunofluorescence. The full-length DYX1C1 cDNA was cloned into a mammalian expression vector containing a C-terminal V5 epitope and a polyhistidine tail. The fusion protein showed a staining pattern similar to 4′,6-diamidino-2-phenylindole staining, suggesting that DYX1C1 is a nuclear protein (Fig. 2D). The result was not different with a construct including a deletion of the last four amino acids (data not shown).
Expression of DYX1C1 Protein in Human Brain. Light microscopy of normal human brain sections revealed a strikingly nuclear expression pattern for DYX1C1 immunoreactivity, consistent with the transfection results. In both cortical neuronal cell populations and white matter glial cells, a minority of cell nuclei expressed DYX1C1 immunoreactivity (Fig. 3 A, B, and E). Characteristically, even neighboring, identical-appearing, cells had different expression (Fig. 3 C and D), which, together with the lack of staining obtained with preimmune serum (Fig. 3F), supports the specificity of immunoreactivity.
Fig. 3.
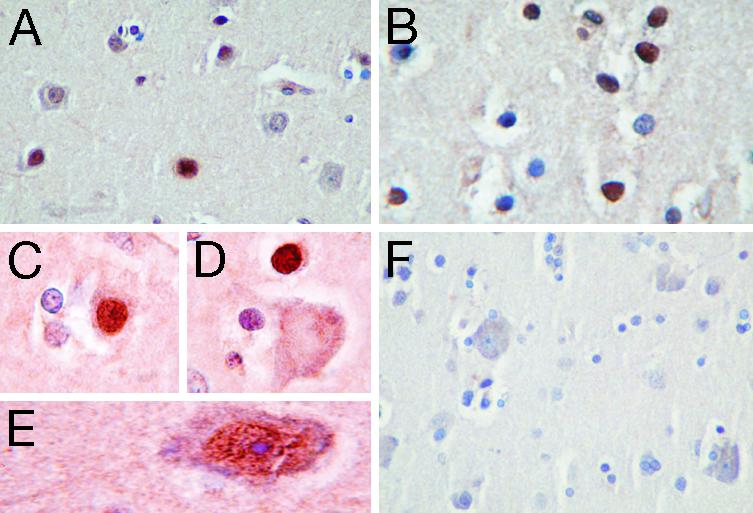
Immunostaining patterns for DYX1C1 observed in normal human brain tissue from an individual who died immediately after sudden cardiac arrest. (A) Immunoreactivity in cortical brain tissue, demonstrating variable nuclear expression in a subset of neurons. (Original magnification: ×30.) (B) Typical staining result in white matter, where also a fraction of cell nuclei are densely positive in contrast to clearly negative adjacent cell nuclei. (×40.) (C) Positive neuronal nucleus adjacent to negative glial cell nuclei. (×100.) (D) Negative neuronal cell body adjacent to neighboring small cells, probably glial cells. (×100.) (E) High-magnification illustration of a large pyramidal neuron expressing clearly intranuclear localization of DYX1C1 protein. (×100.) (F) Typical view of an adjacent tissue section stained with preimmune (control) serum, indicating the lack of nonspecific staining in neuronal and glial cell nuclei. (×30.)
We studied DYX1C1 immunoreactivity also in individuals who died soon after the onset of acute ischemic stroke (Fig. 4). In contrast to the typically nuclear expression in the normal brain, also cytoplasmic expression was observed in ischemic brain areas (Fig. 4A). In cortical areas representing early ischemic morphology, the fraction of positive cell nuclei or cytoplasms appeared increased (Fig. 4C) as compared with nonischemic brain or contralateral hemispheres. In most ischemic sections studied, also structures corresponding to neuronal processes were frequently found positive for DYX1C1 (Fig. 4E). Quantitative or statistical analysis of expression was not attempted.
Fig. 4.
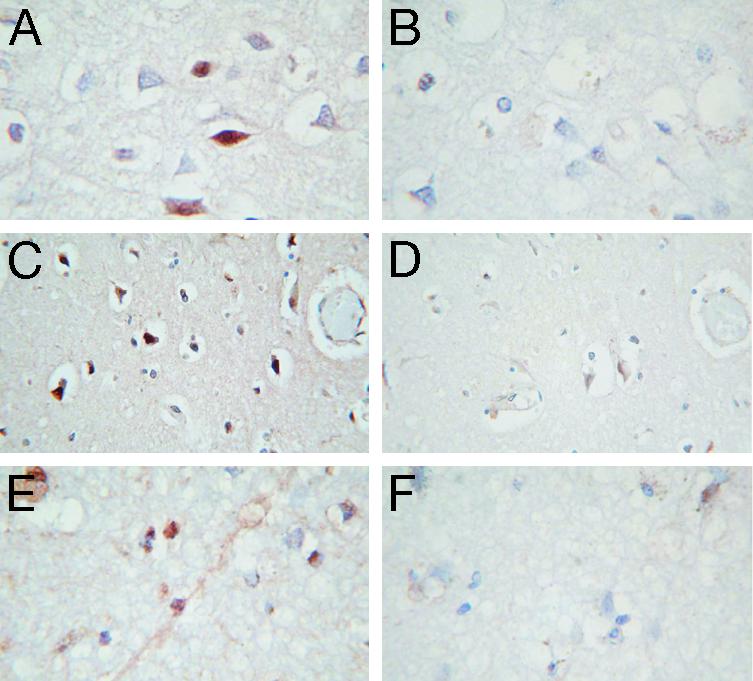
Immunostaining patterns for DYX1C1 observed in human ischemic brain tissue of three victims of acute ischemic stroke. (A) Immunostaining result in a subject deceased at 23 h after the onset of stroke symptoms. Note the cytoplasmic staining pattern present in a subset of ischemic neurons. Compare with Fig. 3 from nonischemic brain tissue, where typically nuclear expression was found. (Original magnification: ×40.) (B) Same neuronal population identified from adjacent section stained with preimmune (control) serum. (×40.) (C) Immunostaining pattern in ischemic brain tissue area of another subject deceased 26 h after the onset of stroke symptoms. Note the increased fraction of pyknotic neurons expressing dense, predominantly nuclear, DYX1C1 immunoreactivity. (×20.) (D) Same neuronal population identified from adjacent section stained with preimmune (control) serum. (×20.) (E) Immunoreactivity for DYX1C1 observed in a more advanced, vacuolized, stage of tissue ischemia, demonstrating faint expression also in neuronal processes. This subject died 60 h after the onset of stroke symptoms. (×40.) (F) Same tissue area identified from adjacent section stained with preimmune (control) serum. (×40.)
Discussion
Our study identifies a specific gene, DYX1C1, as a candidate susceptibility gene for developmental dyslexia, the most common childhood learning disorder, and provides a starting point for prospective population studies and further biochemical and functional research. We have three independent lines of evidence. First, a translocation breakpoint disrupting DYX1C1 is transmitted with dyslexia in one family. This breakpoint is located within a TPR-domain coding region of the gene, and thus is likely to disrupt protein function. Second, two SNPs show association to dyslexia independently and as a haplotype with a significant TDT result. Even though we performed replication in a second study set, the associations will need independent verification in larger studies. Third, the associated 5′ SNP, –3G → A, is located in the binding sequence of the transcription factors Elk-1, HSTF, and TFII-I. Elk-1 is a transcriptional activator expressed in rat brain neurons and its activation has been associated with learning in rats (16–18). The –3G → A SNP affects also the Kozak sequence near the translation initiation site. The coding SNP, 1249C → T, truncates the protein by four amino acids, suggesting a functional effect.
In a complex disorder, even a modest increase in genetic risk may be interesting. There is overwhelming evidence that dyslexia is a genetically complex condition. Linkage of dyslexia to chromosome 15 was suggested first (19), and the locus DYX1 in chromosome 15q21 has been confirmed (2–4). The second dyslexia locus, DYX2, maps to chromosome 6p21 (20), confirmed by three independent studies (21–23). There are, however, also negative results on DYX2 (24, 25). Furthermore, different studies have indicated loci in chromosomes 1p34-p36 (26, 27), 2p15-p16 (DYX3; ref. 28), 3p14.1-q13 (DYX5; ref. 29), and 18p11 (DYX6; ref. 30). Dominant inheritance has been proposed only for the dyslexia loci in chromosomes 2 and 3 (28, 29). For other dyslexia loci, including DYX1 in 15q, the gene is merely expected to increase risk for dyslexia, compatible with the common disease/common variant hypothesis. For example, a Crohn disease susceptibility allele in chromosome 5q has a population frequency of 37% and heterozygosity for this allele increases the risk for Crohn disease 2-fold (31).
There is some uncertainty about the position of the DYX1 locus (2–5). The peaks of two linkage studies map about 7 megabases or 2.2 centimorgans proximally from the breakpoint defined in our study. Given the imprecision of genetic linkage for multifactorial phenotypes, DYX1C1 might correspond to DYX1. Alternatively, there might be more than one locus for dyslexia in chromosome 15. There are previous examples of neighboring genes contributing to a single phenotype. For example, the Griscelli syndrome is caused by mutations in either MYO5A or RAB27A (32, 33). Strikingly, RAB27A resides 165 kb proximal to DYX1C1, whereas MYO5D is located close to DYX1.
The amino acid sequence offers little information about the cellular function of DYX1C1. Remarkably, the DYX1C1 protein differs from its pygmy chimpanzee and chimpanzee counterparts at two or three amino acids, but from gorilla and orangutan at five of six residues, and six of nine amino acid changes cluster in exon 5 (Table 2). For comparison, the rate of coding divergence is approximately three times higher for DYX1C1 than for FOXP2, the product of a gene implicated in a speech and language disorder (34). Thus, as suggested for FOXP2, the DYX1C1 gene may reveal important evolutionary differences related to brain functions between the primates.
Based on the human variation, we hypothesize that the Elk-1 binding site and possibly the C-terminal part of the DYX1C1 protein are functionally important. TPR motifs are found in a wide variety of proteins in many different phyla; they are general protein–protein interaction modules, thought to be of ancient origin. Computational analysis of the human genome has revealed a total of 72 genes encoding proteins with at least one TPR motif (35). Most of the TPR-domain-containing proteins are associated with multiprotein complexes (36). Among known proteins, TPR motifs are present in kinesin light chain, a subunit of kinesin I molecule involved in axonal cargo transport (37), and receptor-associated protein at synapse (rapsyn), which is essential for clustering of postsynaptic nicotine acetylcholine receptors (38).
Observations from both normal and ischemic human brains demonstrated that DYX1C1 protein is expressed in a subset of human glial and neuronal cells. Because only some of the neighboring neurons expressed DYX1C1 immunoreactivity, we suggest that DYX1C1 is not a housekeeping gene. Instead, it may relate to the functional state of the cells. Examination of DYX1C1 expression in ischemic brain tissue suggested that it is involved dynamically in the functional cell state, changing in the face of metabolic challenge. Similar changes in protein distribution in ischemia have been observed for the Elk-1 transcription factor (39). These results from human cerebral ischemia warrant more systematic studies on the role of DYX1C1 in cell stress and ischemia as well as dyslexia.
Acknowledgments
Riitta Lehtinen is acknowledged for skillful assistance in the laboratory. We thank the families involved in this study for their cooperation. The research was supported by the Finnish National Technology Agency, the Sigrid Jusélius Foundation, the Helsingin Sanomat Centenary Foundation, the Arvo and Lea Ylppö Foundation, Helsinki University Central Hospital research funds, and the Academy of Finland.
Abbreviations: BAC, bacterial artificial chromosome; SNP, single-nucleotide polymorphism; TDT, transmission disequilibrium test; TPR, tetratricopeptide repeat.
Data deposition: The sequences reported in this paper have been deposited in the GenBank database (accession nos. AF337549 for the DYX1C1 cDNA sequence, AC013355 and AC013355 for the genomic sequence of the BAC clones 178D12 and RP-11-178D12, BG242087 and AK005832 for the mouse Dyx1c1 cDNA sequence, and AY178583–AY178618 and AH012450–AH012453 for the primate DYX1C1 sequences).
See commentary on page 11190.
References
- 1.Paulesu, E., Demonet, J. F., Fazio, F., McCrory, E., Chanoine, V., Brunswick, N., Cappa, S. F., Cossu, G., Habib, M., Frith, C. D., et al. (2001) Science 291, 2165–2167. [DOI] [PubMed] [Google Scholar]
- 2.Grigorenko, E. L., Wood, F. B., Meyer, M. S., Hart, L. A., Speed, W. C., Shuster, A. & Pauls, D. L. (1997) Am. J. Hum. Genet. 60, 27–39. [PMC free article] [PubMed] [Google Scholar]
- 3.Schulte-Körne, G., Grimm, T., Nothen, M. M., Müller-Myhsok, B., Cichon, S., Vogt, I. R., Propping, P. & Remschmidt, H. (1998) Am. J. Hum. Genet. 63, 279–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morris, D. W., Robinson, L., Turic, D., Duke, M., Webb, V., Milham, C., Hopkin, E., Pound, K., Fernando, S., Easton, M., et al. (2000) Hum. Mol. Genet. 9, 843–848. [DOI] [PubMed] [Google Scholar]
- 5.Nopola-Hemmi, J., Taipale, M., Haltia, T., Lehesjoki, A. E., Voutilainen, A. & Kere, J. (2000) J. Med. Genet. 37, 771–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Häyrinen, T., Serenius-Sirve, S. & Korkman, M. (1999) Reading and Writing Test Designed for and Normalized in Finnish Elementary School (Psykologien Kustannus, Helsinki) (Finnish).
- 7.Leinonen, S., Müller, K., Leppänen, P., Aro, M., Ahonen, T. & Lyytinen, H. (2001) Read. Writ. Interdiscip. J. 14, 265–296. [Google Scholar]
- 8.Wechsler, D. (1992) Wechsler Adult Intelligence Scale Revised (WAIS-R) (Psykologien Kustannus, Helsinki, and Psychological Corp., San Antonio, TX).
- 9.Wechsler, D. (1984) Wechsler Intelligence Scale for Children Revised (WISC-R) (Psykologien Kustannus, Helsinki, and Psychological Corp., San Antonio, TX).
- 10.Korkman, M., Kirk, U. & Kemp, S. (1997) NEPSY: Neuropsychological Assessment of Children (Psykologien Kustannus, Helsinki) (Finnish), revised version.
- 11.Denckla, M. B. & Rudel, G. R. (1976) Neuropsychologia 14, 471–479. [DOI] [PubMed] [Google Scholar]
- 12.Christensen, A.-L. (1982) Luria's Neuropsychological Test (Psykologien Kustannus, Helsinki) (Finnish).
- 13.Wolf, M. (1986) Brain Lang. 27, 360–379. [DOI] [PubMed] [Google Scholar]
- 14.Lindsberg, P. J., Carpén, O., Paetau, A., Karjalainen-Lindsberg, M.-L. & Kaste, M. (1996) Circulation 94, 939–945. [DOI] [PubMed] [Google Scholar]
- 15.Edelmann, L., Spiteri, E., Koren, K., Pulijaal, V., Bialer, M. G., Shanske, A., Goldberg, R. & Morrow, B. E. (2001) Am. J. Hum. Genet. 68, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sgambato, V., Vanhoutte, P., Pagès, C., Rogard, M., Hipskind, R., Besson, M. & Caboche, J. (1998) J. Neurosci. 18, 214–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cammarota, M., Bevilaqua, L. R. M., Ardenghi, P., Paratcha, G., Levi de Stein, M., Izquierdo, I. & Medina, J. H. (2000) Mol. Brain Res. 76, 36–46. [DOI] [PubMed] [Google Scholar]
- 18.Berman, D. E. (2003) Neurobiol. Learn. Mem. 79, 122–126. [DOI] [PubMed] [Google Scholar]
- 19.Smith, S. D., Kimberling, W. J., Pennington, B. F. & Lubs, H. A. (1983) Science 219, 1345–1347. [DOI] [PubMed] [Google Scholar]
- 20.Cardon, L. R., Smith, S. D., Fulker, D. W., Kimberling, W. J., Pennington, B. F. & DeFries, J. C. (1994) Science 266, 276–279. [DOI] [PubMed] [Google Scholar]
- 21.Grigorenko, E. L., Wood, F. B., Meyer, M. S. & Pauls, D. L. (2000) Am. J. Hum. Genet. 66, 715–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gayán, J., Smith, S. D., Cherny, S. S., Cardon, L. R., Fulker, D. W., Brower, A. M., Olson, R. K., Pennington, B. F. & DeFries, J. C. (1999) Am. J. Hum. Genet. 64, 157–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fisher, S. E., Marlow, A. J., Lamb, J., Maestrini, E., Williams, D. F., Richardson, A. J., Weeks, D. E., Stein, J. F. & Monaco, A. P. (1999) Am. J. Hum. Genet. 64, 146–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Field, L. L. & Kaplan, B. J. (1998) Am. J. Hum. Genet. 63, 1448–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Petryshen, T. L., Kaplan, B. J., Fu Liu, M. & Field, L. L. (2000) Am. J. Hum. Genet. 66, 708–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rabin, M., Wen, X. L., Hepburn, M., Lubs, H. A., Feldman, E. & Duara, R. (1993) Lancet 342, 178 (lett.). [DOI] [PubMed] [Google Scholar]
- 27.Froster, U., Schulte-Körne, G., Hebebrand, J. & Remschmidt, H. (1993) Lancet 342, 178–179 (lett.). [DOI] [PubMed] [Google Scholar]
- 28.Fagerheim, T., Raeymaekers, P., Tonnessen, F. E., Pedersen, M., Tranebjaerg, L. & Lubs, H. A. (1999) J. Med. Genet. 36, 664–669. [PMC free article] [PubMed] [Google Scholar]
- 29.Nopola-Hemmi, J., Myllyluoma, B., Haltia, T., Taipale, M., Ollikainen, V., Ahonen, T., Voutilainen, A., Kere, J. & Widén, E. (2001) J. Med. Genet. 38, 658–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fisher, S. E., Francks, C., Marlow, A. J., MacPhie, I. L., Newbury, D. F., Cardon, L. R., Ishikawa-Brush, Y., Richardson, A. J., Talcott, J. B., Gayan, J., et al. (2002) Nat. Genet. 30, 86–91. [DOI] [PubMed] [Google Scholar]
- 31.Rioux, J. D., Daly, M. J., Silverberg, M. S., Lindblad, K., Steinhart, H., Cohen, Z., Delmonte, T., Kocher, K., Miller, K., Guschwan, S., et al. (2001) Nat. Genet. 29, 223–228. [DOI] [PubMed] [Google Scholar]
- 32.Menasche, G., Pastural, E., Feldmann, J., Certain, S., Ersoy, F., Dupuis, S., Wulffraat, N., Bianchi, D., Fischer, A., Le Deist, F., et al. (2000) Nat. Genet. 25, 173–176. [DOI] [PubMed] [Google Scholar]
- 33.Pastural, E., Barrat, F. J., Dufourcq-Lagelouse, R., Certain, S., Sanal, O., Jabado, N., Seger, R., Griscelli, C., Fischer, A. & de Saint Basile, G. (1997) Nat. Genet. 16, 289–292. [DOI] [PubMed] [Google Scholar]
- 34.Enard, W., Przeworski, M., Fisher, S. E., Lai, C. S. L., Wiebe, V., Kitano, T., Monaco, A. P. & Pääbo, S. (2002) Nature 418, 869–872. [DOI] [PubMed] [Google Scholar]
- 35.Venter, J. C., Adams, M. D., Myers, E. W., Li, P. W., Mural, R. J., Sutton, G. G., Smith, H. O., Yandell, M., Evans, C. A., Holt, R. A., et al. (2001) Science 291, 1304–1351. [DOI] [PubMed] [Google Scholar]
- 36.Blatch, G. L. & Lässle, M. (1999) BioEssays 21, 932–939. [DOI] [PubMed] [Google Scholar]
- 37.Goldstein, L. S. (2001) Proc. Natl. Acad. Sci. USA 98, 6999–7003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ramarao, M. K., Bianchetta, M. J., Lanken, J. & Cohen, J. B. (2001) J. Biol. Chem. 276, 7475–7483. [DOI] [PubMed] [Google Scholar]
- 39.Ferrer, I., Friguls, B., Dalfó, E. & Planas, A. M. (2003) Acta Neuropathol. 105, 425–437. [DOI] [PubMed] [Google Scholar]