

Abstract
The rhythmic contraction of the Caenorhabditis elegans pharynx is unique in that the network of 12 neurons, including two M3 neurons, that regulate the contraction is known. The neurotransmitters secreted by these cells, and the target cells responding to these chemical signals, are not known. Here, we describe an approach to obtain this missing information and use the M3 cells as an example. Electrical recordings (electropharyngeograms) were used in conjunction with temporally and spatially defined application of neurotransmitters via photolysis of inactive, photolabile precursors. To illustrate the technique we used pharyngeal preparations in which the two M3 neurons are intact and preparations in which they were removed by laser irradiation. Removal of M3 neurons results in the loss of the small negative peaks in the electropharyngeograms and an increase in time during which the pharynx remains contracted. We demonstrate that the application of glutamate by photolysis of caged glutamate to a pharynx from which the two M3 neurons were removed produces effects similar to those observed before removal of the M3 neurons. In control experiments, photolytic release from photolabile precursors of carbamoylcholine, a stable and well characterized analog of acetylcholine, or of γ-aminobutyric acid, from photolabile precursors did not have this effect. The response depended on the amount of glutamate released. By reducing the size of the photolytic beam, glutamate was released at several different locations of the pharynx. Two areas of the pharynx mainly respond to the application of glutamate; one corresponds to the pm4 muscle cells in the metacorpus, and the other to the junction between muscle cells pm5 in the isthmus and pm6 in the terminal bulb.
The nematode Caenorhabditis elegans is a model widely used for studying the development and function of the nervous system (1). The complete nervous system of the adult hermaphrodite consists of 302 neurons, which are known at the resolution of the electron microscope (2, 3). Twenty of the neurons are in the C. elegans pharynx (3). The rhythmical pumping of water by the pharynx in and out of the animal is important for the efficient concentration and grinding of the bacteria, which comprise the food source of the nematode. The pharynx consists of three functional sections, the corpus, isthmus (I), and terminal bulb (TB) (3) (Fig. 1A), which, respectively, concentrate, transfer, and grind up bacteria (4, 5). The contraction of the pharynx can be visually observed (4, 5). More conveniently, it is possible to record the electrical activity of the pharyngeal muscles and obtain electropharyngeograms (EPGs) (6, 7) (Fig. 2). These recordings of transient currents reflect changes in the membrane potential of the pharyngeal muscles and their contraction and relaxation. By using laser irradiation to ablate specific cells (8, 9), 12 pharyngeal neurons, including two M3 neurons, have been shown to affect pharyngeal function (10–13). How a group of neurons communicate by chemical signals to regulate the rhythmical contractions of a muscle like the pharynx is an unsolved problem of interest to many areas of science and medicine, including cardiology.
Figure 1.
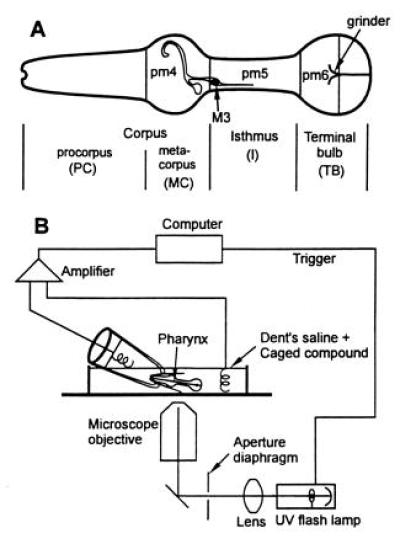
(A) Anatomy of the C. elegans pharynx (modified from ref. 3). Anterior is to the left. The pharynx is divided into three major functional parts, the corpus in the front of the nematode [subdivided into procorpus (PC) and metacorpus (MC)], I, and TB, and contains 20 neurons and 37 muscle cells (3). The location of the nucleus of the M3 motor neuron on the left side of the pharynx is shown; another M3 motor neuron is in the corresponding location on the right side of the pharynx. The location and extent of three of the large muscle types are indicated: pm4 in the MC, pm5 in the I area, and pm6 in the front region of the TB. The process of the M3 neuron to pm4 and pm5 muscle cells is drawn as an open line in this figure. (B) Diagram of the instrumentation.
Figure 2.
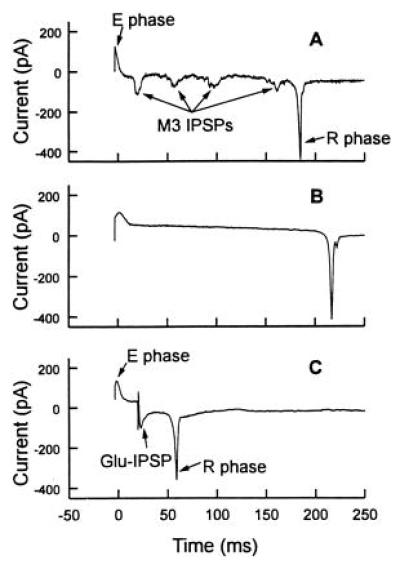
(A) Part of a typical EPG recorded from a wild-type (N2) C. elegans pharynx (6, 7). This portion of the EPG corresponds to a single pumping action of the pharynx, a contraction followed by a relaxation. It contains three phases, called E, P, and R. The E or excitation phase, which is the positive spike in the EPG, appears when the basal membranes of pharyngeal muscle cells change to a more positive transmembrane potential (depolarize). The E-phase may consist of one or two transients. These recordings show only the latter part of the E-phase, because the E-spike was used to trigger the recording by the pclamp software. The P or plateau phase is defined as the period between the E-phase and R-phase; during this period, the membrane potential remains depolarized and contraction proceeds. In this period, there are negative spikes (inhibitory postsynaptic potentials, IPSPs) caused by the M3 motor neurons. The R or repolarization phase initiates muscle relaxation and the membrane potential returns to negative values. The R-phase typically consists of two negative spikes. The first R-phase spike is caused by the transmembrane potential of corpus muscles returning to more negative values (repolarization), and the second by repolarization of TB muscles (6). The pump duration (time between the E-phase and the R-phase) is about 180 ms in this trace. (B) EPG from a nematode whose M3 neurons were killed by laser ablation (6, 8, 9). All IPSPs disappeared and the pump duration increased to about 220 ms in this trace. (C) EPG from the same nematode as in B with glutamate added to the pharynx by photolysis of 0.5-mM caged glutamate 20 ms after the E spike. The diameter of the illuminated area was about 200 μm, and the light energy was about 70 μJ. A glutamate-induced IPSP (Glu-IPSP) similar to the M3-induced IPSP was regenerated after photolysis of caged glutamate. The sharp spike prior to the Glu-IPSP is an artifact caused by the discharge of the UV flash lamp. This artifact existed in all the photolysis experiments and it is an indication of the time photolysis occurred.
Neither the neurotransmitters secreted by specific cells in the circuit nor the target cells which respond to these chemical signals are known. Here we describe an approach that can be used to (i) identify excitatory and inhibitory neurotransmitters that affect the circuit of pharyngeal neurons of C. elegans, (ii) locate the neurons that secrete the neurotransmitters and the cells that contain the responding receptors, and (iii) determine the effects of neurotransmitter concentration and time of application on contraction of the pharynx. To illustrate the approach we have used pharyngeal preparations from which M3 neurons were removed by laser irradiation (6). The M3 neurons are responsible for small negative spikes in the EPG (Fig. 2A) and control the time interval between contraction and relaxation of the pharynx (6, 12) The new approach involves the synthesis and development of photolabile or caged compounds that are biologically inert—i.e., act neither as activator nor as inhibitor—but that can be converted into an active neurotransmitter within microseconds by photolysis (14, 15). For example (16): ![]()
Pre-equilibration of receptors with caged neurotransmitters followed by photolytic uncaging in the microsecond time region avoids the problems encountered when rapid perfusion techniques are used to apply the neurotransmitter and diffusional access of neurotransmitters to their receptor binding sites is slow compared with the rate of receptor desensitization (17–19). When existing rapid perfusion techniques are used to equilibrate cell surface receptors with neurotransmitter and receptor desensitization is more rapid than ligand–receptor equilibration, only a small fraction of receptors (5% in the experiments cited) are converted to the open-channel form which may be too little to be detected (19). This problem is avoided with photolysis of caged neurotransmitters because, as during the release of neurotransmitter at neuronal junctions in intact animals, the concentration of neurotransmitter rises within microseconds to concentrations sufficient to open receptor channels before the receptors desensitize (15, 19). Additionally, using caged neurotransmitters (14, 16, 20, 21), one can obtain spatial resolution regarding the location of receptors in neurons and muscle cells (reviewed in refs. 22 and 23) because the laser light used for photolysis can be focused to a spot with a size on the order of micrometers, and the photolysis of the caged neurotransmitters occurs in the microsecond time region (14, 16, 21, 24, 25). Laser–pulse photolysis of caged neurotransmitters, therefore, provides both high temporal (reviewed in ref. 19) and spatial resolution (reviewed in refs. 22 and 23).
We used three photolabile receptor ligands (caged ligands) in our study: caged carbamoylcholine (14) (carbamoylcholine is a well-characterized and stable analog of acetylcholine), caged glutamate (16), and caged γ-aminobutyric acid (GABA) (21).
MATERIALS AND METHODS
Nematode Culturing.
Worms were cultured under normal conditions at 20°C (26) and grown on a lawn of Escherichia coli strain OP50 (27). Agar plates seeded with E. coli were kindly provided by K. J. Kemphues’ laboratory (Cornell University). The wild type used was the C. elegans variety Bristol, strain N2. A mutant strain MT6308, eat-4(ky5), from C. I. Bargmann (University of California, San Francisco) and J. M. Kaplan (Massachusetts General Hospital) (R. Y. N. Lee, University of Texas Southwestern Medical Center) was also used in the experiments.
EPG Recording.
The EPG recording apparatus used (Fig. 1B) was similar to the one described in ref. 7. The C. elegans (adult hermaphrodite) pharynx was exposed by dissecting the head away from the rest of the nematode. The head was then sucked into an electrode (20–40 μm diameter), which was pulled from a glass pipette (World Precision Instruments, Sarasota, FL) and filled with buffer solution that was in contact with a wire electrode leading to the EPC-7 amplifier headstage, and immersed in a bath solution. Another (ground) electrode was also placed in the bath solution. Dent’s saline (7) (140 mM NaCl/6.0 mM KCl/3.0 mM CaCl2/1.0 mM MgCl2/5.0 mM Hepes, pH 7.4) was used for the bath and the suction electrode solutions. The signal was recorded through a Patch Clamp L/M–EPC-7 amplifier combined with pclamp software, version 5.5 (Axon Instruments, Foster City, CA). The pharynx was viewed under a Zeiss Axiovert 35 microscope and manipulated by a Narishige micromanipulator. Data were analyzed by using a PC computer and origin software, version 3.5 (Microcal, Northampton, MA).
Laser Ablation.
Laser ablation of cells was performed as described in references 6 and 9. The laser beam was supplied by a small nitrogen laser (VSL-337i; Laser Science, Cambridge, MA) coupled with a dye laser module (DLM-110, coumarin 440 laser dye; Laser Science). The laser light was introduced into the microscope by a VSL-LMA microscope adapter (Laser Science) and reflected to a neuron in a young nematode by a dichroic beam-splitter (FT-510; Zeiss).
Synthesis and Photolysis of Caged Neurotransmitters.
The N-(α-carboxy-2-nitrobenzyl) group (14) was used in the synthesis of caged carbamoylcholine (14), caged glutamate (16), and caged GABA (21).
Experimental Setup.
A diagram of the apparatus used for EPG recording and photolysis of caged neurotransmitters is presented in Fig. 1B. A caged neurotransmitter (100 μl of a 1-mM solution) was added to the bath solution (100 μl) and allowed to equilibrate with the dissected pharynx for about 5 min. A pulse (0.1-ms width) of UV light from an Oriel (Stamford, CT) flash lamp was used to photolyze the caged neurotransmitter through the epifluorescence pathway of the Zeiss microscope. A chromatic beam-splitter (FT-390; Zeiss) was used to eliminate light with a wavelength greater than 390 nm. The size of the beam was adjusted by using microscope objective lenses with differing magnification (LD Achroplan ×40/0.6 for the experiments in Figs. 2C, 3, and 4A, and Plan-Neofluar ×100/1.3 oil for the experiment in Fig. 4B, both UV transparent) and by changing the size of the field aperture diaphragm of the microscope. The pulse energy was measured by using a Joule meter (model ED-200; Gentec, Palo Alto, CA).
Figure 3.
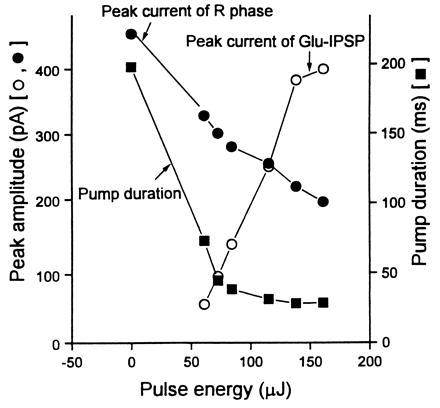
Data obtained with EPGs from MT6308 (eat-4) mutants that are defective in M3 function and that do not exhibit IPSPs in their EPGs (R. Y. N. Lee, personal communication; ref. 6). By varying the pulse energy (from 61 μJ to 161 μJ), the concentration of glutamate released by photolysis of 0.5-mM caged glutamate in an illuminated area of about 200 μm diameter was increased. The UV flash lamp was turned on 20 ms after the appearance of the E spike in the EPG. The Glu-IPSP increased and the pump duration and the R-phase spike current in the EPG decreased with increasing pulse energy used. On average five experiments were done for each data point shown. The SEM is similar to the size of the symbol in the figure. A similar relationship was obtained by changing the concentration of caged glutamate in the bath (from 0.25 mM to 1.0 mM) but keeping the pulse energy and illuminated area constant (data not shown).
Figure 4.
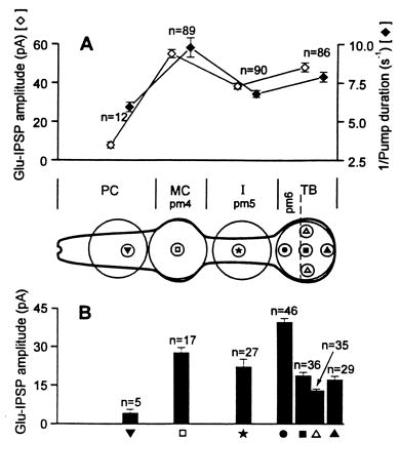
The diagram of the pharynx inserted between A and B shows the areas that were illuminated in the photolysis experiments. The large circles correspond to an illuminated area of 35 μm diameter (similar to the size of the TB of the adult hermaphrodite nematode which was used for EPG recording). The small circles correspond to an area of 7 μm. (A) The illuminated area is indicated by the large circles. The open (⋄) and solid (♦) diamonds represent the Glu-IPSP current amplitude and the reciprocal of pump duration, respectively. Caged glutamate was photolyzed in different locations (large circles) (PC, MC, I, and TB) of the C. elegans pharynx. All data were collected from eight nematodes. (B) The photolytic area is indicated by the small circles in the diagram of the pharynx. The current amplitudes of the Glu-IPSPs are shown. All data were collected from five nematodes in the experiments. The error bar is the SEM, and n is the total number of measurements. Statistical analysis of the current amplitude of Glu-IPSP [Wilcoxon’s two-sample test, two tailed, (29, 30)]: in A, MC vs. PC, MC vs. I, I vs. PC, and TB vs. PC, P < 0.002; MC vs. TB and TB vs. I, 0.02 > P > 0.002; in B, • is significantly different from other data points, P < 0.002.
RESULTS AND DISCUSSION
The most prominent features in EPGs (Fig. 2A) are the capacitive current transients associated with the contraction (E-phase) and relaxation (R-phase) of the pharyngeal muscles (6). Between the E-phase and R-phase small negative spikes, inhibitory post synaptic potentials (IPSPs) can be seen (Fig. 2A). These depend on the functional presence of the M3 motor neurons (6). The cell bodies of the two M3 neurons are located on either side of the pharynx just posterior to the MC (3) (Fig. 1A). Laser ablation of the M3 neurons, or mutation-induced lack of M3 function (6), led to the absence of the IPSPs and to an increase in the time interval between the contraction and relaxation of the muscle, the pump duration (Fig. 2B). The M3 neurons are necessary and sufficient for the IPSPs (6, 28). When all neurons controlling pharyngeal pumping except M3 and M4 were laser-ablated (6), the IPSPs could still be seen in the EPG. More recently, the EPGs recorded from M4-ablated nematodes displayed IPSPs (28). Electron microscope investigations indicate that the M3 motor neurons form synapses with muscle cells pm4 and possibly pm5 (Fig. 1A) (3). This suggests that M3 neurons communicate directly with muscle cells to generate IPSPs. We observed that if only the M3 on the left side of the pharynx or only the M3 on the right side was laser-ablated, the IPSPs still existed in the EPG but the peaks were smaller, indicating that both M3 neurons are responsible for the appearance of IPSPs.
In our experiments we used the dissected pharynx, which still contracts and has a normal EPG (7). Removal of the cuticle that covers the nematode facilitates access to pharmacological agents (7). The dissected pharynx of a nematode in which the M3 neurons were ablated was equilibrated with the caged receptor ligands for 5 min. Only free glutamate released by illuminating caged glutamate with a pulse of UV light from a flash lamp mimicked the action of M3 (Fig. 2). The average pump duration decreased and an IPSP-like current deflection (Glu-IPSP) was observed, provided the light pulse occurred just after the E-phase indicative of pharyngeal contraction (Fig. 2C). Fig. 3 shows that the amplitude of the IPSPs and the effect on pump duration increased with increasing amount of light to which caged glutamate was exposed—i.e., an increase in the amount of glutamate released. The illuminated area in this experiment had a diameter of 200 μm and covered the whole pharynx. In several cases (pulse energy above 160 μJ and caged glutamate concentration at 0.5 mM), the contraction terminated immediately after the UV pulse. No response was seen after an UV pulse if caged glutamate was absent or if the pharynx was irradiated but caged carbamoylcholine (14), or caged GABA (21), was used instead of caged glutamate (data not shown). This indicates that the responses observed were due to glutamate and not due to illumination alone or to the action of other receptor ligands such as carbamoylcholine or GABA, or to the caging group itself. In animals that lacked functional M3s, either due to a genetic abnormality (eat-4 mutant) or because both M3 cells were removed by laser ablation, no spontaneous IPSPs were seen (Fig. 2B) (6) but equivalent responses could still be elicited by the photolytic release of glutamate (Fig. 2C). These results, which show that glutamate induces a pharyngeal muscle response very similar to that caused by M3 activity, and the experiments mentioned above that indicate that M3 neurons communicate directly with the pharyngeal muscle cells (3, 6), suggest that the neurotransmitter secreted by M3 activates glutamate receptors.
Next we investigated the spatial distribution of glutamate sensitivity by reducing the size of the illuminated spot and positioning the spot over different areas of the pharynx. Fig. 4 illustrates the results. The illuminated area was first adjusted to have a diameter of about 35 μm (Fig. 4A). The effects of glutamate on the amplitudes of Glu-IPSP in different areas of the pharynx were compared (Fig. 4A). Photolysis of caged glutamate at the PC induced only a negligible response. Photolysis of caged glutamate at the MC, which is made up of the pm4 muscle cells adjacent to the M3 motor neurons, induced the strongest response. A second pharyngeal region which responds to photolysis of caged glutamate is the TB containing the pm6 muscle cells; the response is somewhat stronger than that of the I area containing the pm5 muscle cells. Next, repetition of the experiment with a smaller illuminated area (7 μm) (Fig. 4B) allowed us to localize the response in the TB to an area adjacent to the junction of muscle cells pm6 in the TB and pm5 in the I area. The size of the Glu-IPSP current is proportional to the average reduction of the pump duration (Fig. 4A). Experiments showed that in both the MC and the TB region the response to glutamate decreased with a decrease in the illuminated area, presumably because there were fewer glutamate receptors in the smaller area. The response was the same when glutamate was released to the left or to the right side of the pharynx when the size of the illuminated area was kept constant. This is consistent with the observation that M3 processes are on the left and right side of pm4 muscle cells (Fig. 1A), and either motor neuron can initiate IPSPs.
We also investigated the glutamate response in eat-4 mutant animals that possess morphologically normal M3 neurons but do not show spontaneously occurring IPSPs (Fig. 3). We found that illumination of the caged glutamate in areas of 35 μm (covering the MC, pm4 muscle cells) and 200 μm (covering the whole pharynx) diameter triggered IPSP events of a magnitude comparable to those seen in the wild type with the same illuminated areas. This finding suggests that the pharyngeal muscles express functional glutamate receptors at a normal density even in the absence of input from M3 neurons.
CONCLUSIONS
Here we demonstrate that in combination with (i) knowledge of the position of specific cells in a circuit of cells controlling observable responses, (ii) the laser-ablation technique, and (iii) EPGs, the cells responding to a specific neurotransmitter and candidate cells releasing it can be identified with help of a laser-pulse photolysis technique employing caged neurotransmitters (14–16, 19–21, 24, 25, 31). Consistent with electron microscope investigations which indicated that the M3 motor neurons form synapses with muscle cells pm4 and possibly pm5 (3), we found that the MC region, which is made up of pm4 muscles, displayed the strongest response to the application of glutamate by photolysis of caged glutamate. The I-containing muscle cells pm5 also gave a definite response (Fig. 4A). We propose that action of the M3 motor neurons controls the length of the pump cycle by delivering a neurotransmitter that activates glutamate receptors on pm4 pharyngeal muscle cells of the MC and to less an extent to pm5 muscle cells of the I area. Two observations are pertinent. When the magnitude of the Glu-IPSP is increased by increasing the amount of released glutamate, (i) the negative spike in the EPG associated with relaxation of the muscle (R-phase) is decreased and becomes smaller than the Glu-IPSP, and (ii) the time interval between the Glu-IPSP and the R-phase is decreased (Fig. 2A) This suggests that repolarization of the pharynx leading to its relaxation is controlled by the two M3 neurons and presumably by glutamate receptors in the terminal bulb, which is innervated by as yet unidentified cell or cells.
Interestingly, the application of glutamate by photolysis of caged glutamate at the junction between muscle cells pm5 in the I area and pm6 in the TB also induced a strong response, even though there is no physical evidence for direct interaction of the M3 neurons with this area of the pharynx. Nevertheless, increasing the concentration of released glutamate in this area also increases the magnitude of the Glu-IPSP and reduces the time interval between the Glu-IPSP and R-phase. The cell or cells secreting the neurotransmitter which activates these receptors are still unknown.
The results presented indicate that the approach described can be used in the future to identify other cells that regulate the rhythmical pumping of the pharynx, to determine which specific neurotransmitter they secrete, and to identify which cells contain the responding receptors. This information is expected to be basic to an understanding of how a circuit of cells communicating by chemical signals controls the rhythmical contractions of a muscle like the pharynx. The caged neurotransmitters required for these studies can be synthesized using published procedures (14, 16, 21, 24, 25) or can be obtained from Molecular Probes.
Acknowledgments
We thank Profs. Kenneth J. Kemphues (Cornell University) and Monica A. Driscoll (Rutgers University) for their helpful comments and Dr. Kyle Gee (Molecular Probes) for gifts of the caged compounds used in the studies. H.L. thanks M. Wayne Davis, Dr. David M. Raizen, Dr. Joe Dent, and Dr. Raymond Y. N. Lee (members of L.A.’s laboratory) for teaching him the EPG recording and the laser ablation technique. These experiments were initiated while G.P.H. was on sabbatical leave in Prof. Robert Horvitz laboratory at the Massachusetts Institute of Technology. He is grateful to Profs. Horvitz and Cornelia Bargmann and members of the Horvitz’s laboratory for training in experiments with C. elegans. This work was supported by Grant NS 08527 awarded to G.P.H. by the National Institutes of Health. H.L. is supported by a National Institutes of Health postdoctoral fellowship. L.A. is supported by a fellowship from the Klingenstein Foundation.
ABBREVIATIONS
- EPG
electropharyngeogram
- IPSP
inhibitory postsynaptic potential
- Glu-IPSP
glutamate-induced IPSP
- PC
procorpus
- MC
metacorpus
- I
isthmus
- TB
terminal bulb
- GABA
γ-aminobutyric acid
- SEM
standard error of the mean
References
- 1.Wood W B. In: The Nematode Caenorhabditis elegans. Wood W B, editor. Plainview, NY: Cold Spring Harbor Lab. Press; 1988. pp. 1–16. [Google Scholar]
- 2.White J G, Southgate E, Thomson J N, Brenner S. Philos Trans R Soc London B. 1986;314:1–340. doi: 10.1098/rstb.1986.0056. [DOI] [PubMed] [Google Scholar]
- 3.Albertson D G, Thomson J N. Philos Trans R Soc London B. 1976;275:299–325. doi: 10.1098/rstb.1976.0085. [DOI] [PubMed] [Google Scholar]
- 4.Doncaster C C. Nematologica. 1962;8:313–320. [Google Scholar]
- 5.Seymour M K, Wright K A, Doncaster C C. J Zool Soc (London) 1962;201:527–539. [Google Scholar]
- 6.Raizen D M, Avery L. Neuron. 1994;12:483–495. doi: 10.1016/0896-6273(94)90207-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Avery L, Raizen D M, Lockery S. Methods Cell Biol. 1995;48:251–269. [PMC free article] [PubMed] [Google Scholar]
- 8.Sulston J E, White J G. Dev Biol. 1980;78:577–597. doi: 10.1016/0012-1606(80)90353-x. [DOI] [PubMed] [Google Scholar]
- 9.Bargmann C I, Avery L. Methods Cell Biol. 1995;48:225–250. doi: 10.1016/s0091-679x(08)61390-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Avery L, Horvitz H R. Cell. 1987;51:1071–1078. doi: 10.1016/0092-8674(87)90593-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Avery L, Horvitz H R. Neuron. 1989;3:473–485. doi: 10.1016/0896-6273(89)90206-7. [DOI] [PubMed] [Google Scholar]
- 12.Avery L. J Exp Biol. 1993;175:283–297. doi: 10.1242/jeb.175.1.283. [DOI] [PubMed] [Google Scholar]
- 13.Raizen D M. Ph.D. thesis. Dallas: Univ. of Texas Southwestern Medical Center; 1996. [Google Scholar]
- 14.Milburn T, Matsubara N, Billington A P, Udgaonkar J B, Walker J W, Carpenter B K, Watt W W, Marque J, Denk W, McCray J A, Hess G P. Biochemistry. 1889;28:49–55. doi: 10.1021/bi00427a008. [DOI] [PubMed] [Google Scholar]
- 15.Niu L, Grewer C T, Hess G P. In: Techniques in Protein Chemistry. Marshak D R, editor. VII. San Diego: The Protein Society/Academic Press; 1996. pp. 139–149. [Google Scholar]
- 16.Wieboldt R, Gee K R, Niu L, Ramesh D, Carpenter B K, Hess G P. Proc Natl Acad Sci USA. 1994;91:8752–8756. doi: 10.1073/pnas.91.19.8752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Udgaonkar J B, Hess G P. Proc Natl Acad Sci USA. 1987;84:8758–8762. doi: 10.1073/pnas.84.24.8758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Geetha N, Hess G P. Biochemistry. 1992;31:5488–5499. doi: 10.1021/bi00139a010. [DOI] [PubMed] [Google Scholar]
- 19.Hess G P, Niu L, Wieboldt R. Ann NY Acad Sci. 1995;757:23–39. doi: 10.1111/j.1749-6632.1995.tb17462.x. [DOI] [PubMed] [Google Scholar]
- 20.Wilcox M, Viola R W, Johnson K W, Billington A P, Carpenter B K, McCray J A, Guzikowski A P, Hess G P. J Org Chem. 1990;55:1585–1589. [Google Scholar]
- 21.Gee K R, Wieboldt R, Hess G P. J Am Chem Soc. 1994;116:8366–8367. [Google Scholar]
- 22.Katz L C, Dalva M B. J Neurosci Methods. 1994;54:205–218. doi: 10.1016/0165-0270(94)90194-5. [DOI] [PubMed] [Google Scholar]
- 23.Denk W, Delaney K R, Gelperin A, Kleinfeld D, Strowbridge B W, Tank D W, Yuste R. J Neurosci Methods. 1994;54:151–162. doi: 10.1016/0165-0270(94)90189-9. [DOI] [PubMed] [Google Scholar]
- 24.Niu L, Gee K R, Schaper K, Hess G P. Biochemistry. 1996;35:2030–2036. doi: 10.1021/bi9516485. [DOI] [PubMed] [Google Scholar]
- 25.Niu L, Wieboldt R, Ramesh D, Carpenter B K, Hess G P. Biochemistry. 1996;35:8136–8142. doi: 10.1021/bi952364n. [DOI] [PubMed] [Google Scholar]
- 26.Sulston J E, Hodgkin J A. In: The Nematode Caenorhabditis elegans. Wood W B, editor. Plainview, NY: Cold Spring Harbor Lab. Press; 1988. pp. 587–606. [Google Scholar]
- 27.Brenner S. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Raizen D M, Lee R Y N, Avery L. Genetics. 1995;141:1365–1382. doi: 10.1093/genetics/141.4.1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sokal R R, Rohlf F J. Biometry: The Principles and Practice of Statistics in Biological Research. New York: Freeman; 1981. pp. 429–437. [Google Scholar]
- 30.Rohlf F J, Sokal R R. Statistical Tables. 2nd Ed. New York: Freeman; 1981. pp. 80–81. [Google Scholar]
- 31.Matsubara N, Billington A, P, Hess G P. Biochemistry. 1992;31:5507–5514. doi: 10.1021/bi00139a012. [DOI] [PubMed] [Google Scholar]