

Abstract
Severely elevated levels of total homocysteine (approximately millimolar) in the blood typify the childhood disease homocystinuria, whereas modest levels (tens of micromolar) are commonly found in adults who are at increased risk for vascular disease and stroke. Activation of the coagulation system and adverse effects of homocysteine on the endothelium and vessel wall are believed to underlie disease pathogenesis. Here we show that homocysteine acts as an agonist at the glutamate binding site of the N-methyl-d-aspartate receptor, but also as a partial antagonist of the glycine coagonist site. With physiological levels of glycine, neurotoxic concentrations of homocysteine are on the order of millimolar. However, under pathological conditions in which glycine levels in the nervous system are elevated, such as stroke and head trauma, homocysteine’s neurotoxic (agonist) attributes at 10–100 μM levels outweigh its neuroprotective (antagonist) activity. Under these conditions neuronal damage derives from excessive Ca2+ influx and reactive oxygen generation. Accordingly, homocysteine neurotoxicity through overstimulation of N-methyl-d-aspartate receptors may contribute to the pathogenesis of both homocystinuria and modest hyperhomocysteinemia.
Keywords: [excitotoxicity, glycine binding site of N-methyl-d-aspartate receptor, glutamate binding site of N-methyl-d-aspartate receptor, homocysteine]
Elevated levels of homocysteine in the blood predispose to arteriosclerosis and stroke. In children with the relatively rare condition of homocystinuria, levels of total homocysteine approach millimolar concentrations. However, more modest levels (≈15–50 μM) are found very commonly in the general population (a condition known as hyperhomocysteinemia) (1, 2), and a concentration of up to 10 μM has been measured in brain (3). Indeed, it has been recently estimated that as many as 47% of patients with arterial occlusions manifest these modest elevations in plasma homocysteine (1, 2). Included among the many causes are genetic alterations in enzymes such as cystathionine β-synthase, a defect found in 1–2% of the general population, and deficiencies in vitamins B6, B12, and folate, whose intake is suboptimal in perhaps 40% of the population (4). The strength of the association between homocysteine and cerebrovascular disease appears to be greater than that between homocysteine and coronary heart disease or peripheral vascular disease (1, 5). Current theories on homocysteine arteriosclerosis do not explain this predilection, nor do they give insight into the cognitive deficits seen in some patients. In the present study, we show that homocysteine causes direct neurotoxicity by activating the N-methyl-d-aspartate (NMDA) subtype of glutamate receptor. Excessive stimulation of these receptors is known to mediate brain damage in focal ischemia (6, 7). Thus homocysteine may not only be associated with the vascular injury leading to stroke but may also participate in the ensuing neurotoxic response in the brain.
MATERIALS AND METHODS
Homocysteine and Derivatives.
d,l-Homocysteine was used here because the l-form was not commercially available. However, based upon previous data, it is likely that only the l-form is active at the glutamate binding site of the NMDA receptor; therefore, the effective concentration of d,l-homocysteine as an agonist may be half that reported here (8, 9). In the human condition hyperhomocysteinemia, the effective concentration is defined as the total level of homocysteine and its oxidation products (in particular the disulfide homocystine and related mixed disulfides) in the blood and is commonly referred to as total homocysteine or homocyst(e)ine (10).
Cerebrocortical Cultures.
Cortical cultures of mixed neurons and glia were derived from embryonic (fetal day 15 or 16) Sprague–Dawley rats as described (11). Briefly, following dissociation in 0.027% trypsin, cerebral cortical cells were plated at a density of 4.5 × 105 per 35-mm dish containing poly-l-lysine-coated glass coverslips in DMEM with Ham’s F-12, and heat-inactivated iron-supplemented calf serum (HyClone) at a ratio of 8:1:1. After 15 days in culture (when the astrocyte layer had become confluent), the cultures were treated with cytosine arabinoside for 72 hr. The culture medium was replenished three times weekly. Cultures were incubated at 36°C in a 5% CO2/95% air humidified atmosphere. The cultures were used for experiments at room temperature (21–24°C) 3–4 weeks after plating. Neurons could be reliably identified by morphological criteria under phase-contrast optics, as later confirmed by patch-clamp recording. To permit physiology experiments (calcium imaging or patch-clamping) under normal room air conditions, the culture medium was exchanged for a solution based on Hanks’ balanced salts before a recording session. The Hanks’ saline consisted of 137.6 mM NaCl, 1 mM NaHCO3, 0.34 mM Na2HPO4, 5.36 mM KCl, 0.44 mM KH2PO4, 1.25–2.5 mM CaCl2, 5 mM Hepes, and 22.2 mM dextrose, adjusted to pH 7.2 with 0.3 M NaOH.
Digital Ca2+ Imaging.
Homocysteine-evoked increases in intracellular Ca2+ concentration ([Ca2+]i) were measured using digital imaging techniques on cultured cerebrocortical neurons loaded with fura-2 and incubated in nominally Mg2+-free Hanks’ balanced salt solution containing 1.25 mM Ca2+ and 1 μM tetrodotoxin, as we have described (11).
Patch-Clamp Recordings.
NMDA- and homocysteine-induced whole-cell currents were recorded using standard patch-clamp techniques, as we have described (12). Cortical neurons were voltage-clamped at −60 mV. Cells were continuously superfused in nominally Mg2+-free Hanks’ balanced salt solution with 1.25 mM Ca2+, 1 μM tetrodotoxin, 1 μM glycine, and usually 10 μM strychnine (used to avoid possible contamination by Cl− currents activated by glycine receptors). The pipette-filling solution contained 120 mM cesium chloride, 20 mM tetraethylammonium chloride, 2 mM MgCl2, 1 mM CaCl2, 2.25 mM EGTA, and 10 mM Hepes (pH was adjusted to 7.2 with 0.3 M NaOH). Drugs were administered using a gravity-driven, rapid application “sewer pipe system” or by pneumatic pipette.
Assessment of Neurotoxicity.
For experiments with “acute” exposure to homocysteine, cerebrocortical cultures were incubated in Earle’s balanced salt solution containing d,l-homocysteine and glycine at various concentrations in a 95% air/5% CO2 humidified environment at 37°C for 20 hr. Neuronal injury in these cultures was assessed by the leakage of lactate dehydrogenase, as described (11). Similar results were obtained when viability was assayed in this preparation by trypan blue exclusion and neuronal cell counting (11). For the “chronic” neurotoxicity experiments, because of the prolonged time (6 days) of the incubation in low concentrations of homocysteine, the cultures were maintained in their original growth medium (containing ≥50 μM glycine by HPLC analysis). This medium interferes with the lactate dehydrogenase assay; therefore, viability was assessed by trypan blue exclusion and neuronal cell counting. After exposure to trypan blue, the neuronal cells were fixed in 2.5% glutaraldehyde and counted in a masked fashion under ×200 magnification, as we have described (11, 13). Typically, in the control dishes, we counted ≈1,000 neurons in 20 fields at this magnification.
HPLC.
Analysis of thiol amino acids and their oxidation products used o-phthaldialdehyde derivatization following a published procedure (14). HPLC was then performed according to standard methods (15). Briefly, 50 μl of culture media was treated with 2-mercaptoethanol in acetonitrile and iodoacetate at pH 9.5 to reduce disulfides, alkylate free thiols, and precipitate protein. A portion of this solution was then derivatized with o-phthaldialdehyde and analyzed on a Kromasil 100–5C18 column (150 mm × 4.6 cm i.d.) using a Hewlett-Packard Model 1050 HPLC with a Hewlett–Packard model 1046A fluorescence detector (excitation at 240 nm, emission at 455 nm).
RESULTS
Pharmacological Characterization of Homocysteine Response at the NMDA Receptor.
We first applied homocysteine plus 1 μM glycine to rat cortical neurons in culture while monitoring responses during whole-cell recording with a patch electrode or digital Ca2+ imaging with fura-2. d,l-Homocysteine (5 mM) increased [Ca2+]i by 97 ± 13 nM in rat cortical neurons (mean ± SEM, n = 7; P < 0.001 by Student’s t test). The homocysteine-mediated increases in [Ca2+]i were blocked by a variety of NMDA receptor antagonists, including d-2-amino-5-phosphonopentanoate, dizocilpine, and 7-chlorokynurenate but not by the non-NMDA receptor antagonist 6-cyano-7-nitroquinoxaline-2,3-dione (Fig. 1a). However, under these conditions, homocysteine was considerably less potent and less efficacious than NMDA (Fig. 1b): The maximal effect of homocysteine in the presence of 1 μM glycine was ≈30% that of NMDA. This lower efficacy could be secondary to a partial agonist effect of homocysteine at the NMDA binding site. Alternatively, we reasoned that homocysteine might interact with a second site on the NMDA receptor to inhibit its activity. The latter possibility was confirmed in the following experiments probing interactions of homocysteine at the glycine coagonist binding site (16, 17).
Figure 1.
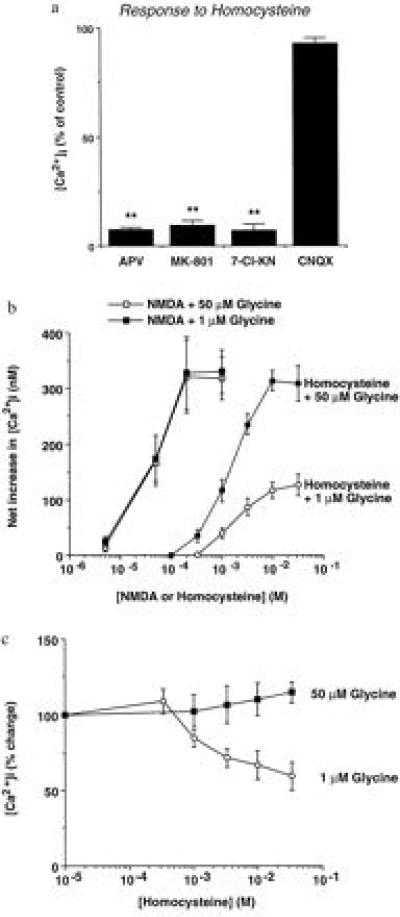
Homocysteine increases neuronal [Ca2+]i through NMDA receptor-channel activation. (a) Antagonism of homocysteine-mediated [Ca2+]i responses by NMDA receptor antagonists. Responses evoked by 5 mM d,l-homocysteine and 1 μM glycine were quantified in the absence and presence of NMDA- and non-NMDA-antagonists: d-2-amino-5-phosphonopentanoate (APV, 200 μM), dizocilpine (MK-801, 10 μM), 7-chlorokynurenate (7-Cl-KN, 10 μM), and 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX, 50 μM). Values in this and subsequent figures represent the mean ± SEM. Imaged fields contained five to seven neurons, each serving as its own control, and the results represent data from three separate experiments. Data are expressed as the percentage change compared with the control response (≈150 nM [Ca2+]i) elicited by 5 mM homocysteine plus 1 μM glycine. Responses to homocysteine/glycine in the presence of antagonist that were statistically smaller than those obtained to homocysteine/glycine alone are indicated by asterisks (∗∗, P < 0.01, analyzed with an ANOVA followed by a Scheffé multiple comparison of means). (b) Dependence on glycine of the concentration–response relationship of NMDA- and homocysteine-mediated increases in neuronal [Ca2+]i. The increase in homocysteine responses in the presence of elevated concentrations of glycine suggests that homocysteine is not only an agonist at the NMDA binding site but also a partial antagonist of the glycine site (data from six neurons in a representative experiment from a total of eight experiments). The open boxes have been displaced slightly for clarity. (c) Partial antagonism of NMDA responses by homocysteine. NMDA (100 μM)-mediated increases in [Ca2+]i in the presence of 1 or 50 μM glycine were measured at various concentrations of homocysteine (n = 3 experiments). To exclude an effect of rundown or desensitization, high or low glycine was applied randomly during each response. The results are consistent with the notion that homocysteine is a full agonist at the NMDA binding site and that its inhibition of NMDA responses is mediated functionally by partial antagonism of the glycine site (we cannot exclude the possibility that homocysteine acts as a partial agonist at this site in the absence of glycine).
When the glycine concentration was increased, the maximal effect of homocysteine increased (with no significant change in EC50) to match that of NMDA, both in digital calcium imaging experiments and in patch-clamp recordings (Figs. 1b and 2 a–d). With 10 μM glycine, the response to 10 mM d,l-homocysteine was increased 142 ± 32% (n = 4; data not shown), and with 50 μM glycine, the response was enhanced 181 ± 33% (Fig. 1b; n = 6). Moreover, in the presence of elevated glycine levels, 100–150 μM concentrations of l-homocysteine (20–300 μM d,l-homocysteine) evoked increases in neuronal [Ca2+]i or whole-cell currents (Figs. 1b and 2e). For example, approximately equivalent rises in [Ca2+]i were observed with 150 μM l-homocysteine and 10–20 μM NMDA, known neurotoxic concentrations in this preparation (11). Importantly, the dose–response curve for NMDA-induced [Ca2+]i changes was not altered by these increases in glycine concentration (Fig. 1b). Using the more sensitive technique of whole-cell recording in the presence of bicarbonate and elevated glycine, measurable NMDA receptor-mediated currents were evoked by 100 μM l-homocysteine (Fig. 2e). Taken together, these findings suggest that homocysteine competes with glycine at the glycine binding site of the NMDA receptor to decrease its activity, and in the presence of ≥50 μM glycine, homocysteine becomes a relatively high-affinity NMDA receptor agonist.
Figure 2.
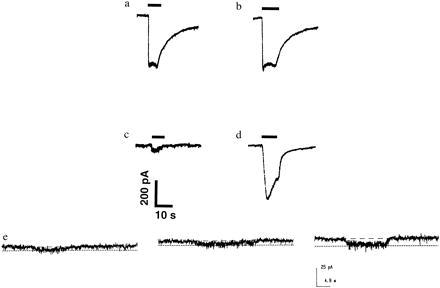
Whole-cell recording of homocysteine-evoked currents are increased by glycine. (a) Current evoked by 200 μM NMDA plus 1 μM glycine. (b) Current evoked by 200 μM NMDA plus 50 μM glycine. (c) The current evoked by 10 mM d,l-homocysteine (or 5 mM l-homocysteine, a maximal stimulus) with 1 μM glycine was relatively small. (d) Increasing the glycine concentration to 50 μM increased the magnitude of the current evoked by homocysteine on the same cortical neuron as in c. This action was not voltage-dependent as similar effects were seen at a holding potential of +40 mV (data not shown). Possibly due to rundown of the currents, 50 μM glycine did not always increase the maximal homocysteine-evoked current to the same level as the maximal NMDA-induced current in all neurons tested (n = 7). However, in each case the effect was qualitatively similar to that observed during the calcium imaging experiments. (e) Micromolar homocysteine evoked measurable currents in the presence of glycine and bicarbonate. When 200 μM d,l-homocysteine (equivalent to 100 μM l-homocysteine) was applied in Hanks’ balanced salt solution, a small macroscopic current was observed (left trace). To simulate physiological conditions, 24 mM bicarbonate was added, and then the same micromolar concentration of homocysteine elicited a somewhat larger whole-cell current (center trace; similar to the effect of bicarbonate on cysteine-induced currents noted previously; ref. 9). Application of 50 μM glycine in addition to the bicarbonate further increased the homocysteine-activated current to 200% (right trace). In e, the bath solution was made somewhat acidic (pH = 7.0 rather than 7.2) to more closely simulate ischemic conditions.
If this hypothesis were true, then homocysteine would be expected to partially antagonize NMDA-mediated increases in neuronal [Ca2+]i in the presence of 1 μM glycine. In contrast, homocysteine would not be expected to inhibit NMDA responses in the presence of 50 μM glycine. Indeed, these predictions were borne out empirically in the experiment illustrated in Fig. 1c. At maximal concentrations, homocysteine decreased the response to 30 μM NMDA by 32 ± 5.3% in the presence of 1 μM glycine (n = 3; P < 0.05 by Student’s t test).
Pathophysiological Relevance of Homocysteine to Brain Injury.
Our observations raise the important pathophysiological question of whether homocysteine contributes to neuronal damage after a stroke, perhaps precipitated by homocysteine-induced vascular injury. Therefore, we next performed a series of experiments to determine the neurotoxic potential of homocysteine. Reminiscent in duration and concentration of glutamate-induced neurotoxicity (18), acute exposure to 1 mM d,l-homocysteine (equivalent to 500 μM l-homocysteine) produced delayed (24 hr) neuronal damage by the criteria of lactate dehydrogenase leakage or the inability to exclude trypan blue [25 ± 10% death (mean ± SEM, n = 5 experiments, P < 0.05)]; this neurotoxicity was observed in the presence of normal (≈1 μM) glycine. However, after brain ischemia (19) or head trauma (20), glycine levels, which are normally ≈150 μM in plasma, approach 100 μM in cerebrospinal fluid and in cortical superfusates. Additionally, damage continues for several days or longer after an initial vascular insult (21, 22). In an attempt to model this component of neurotoxicity in the absence of potentially confounding endothelial cell injury (a paradigm that cannot be achieved in vivo), we incubated cerebrocortical cultures, containing neurons and glia but no endothelial cells, in homocysteine for 6 days in the presence of ≥50 μM glycine. Under these conditions, dose-dependent neurotoxicity was observed at concentrations of homocysteine as low as 10 μM (Fig. 3a). Dizocilpine or memantine (12, 23) ameliorated this injury, confirming that the neurotoxic damage was mediated by overstimulation of the NMDA receptor (Fig. 3b). d-2-Amino-5-phosphonopentanoate was similarly effective. As reactive oxygen species can be generated extracellularly by homocysteine (ref. 24 and references therein) or following excessive NMDA receptor stimulation (25), we tested the effects of superoxide dismutase and catalase on neurotoxicity. We found that preincubation in superoxide dismutase/catalase offered an intermediate degree of protection from neuronal damage (Fig. 3b). These studies emphasize the potential contribution by reactive oxygen species to homocysteine’s mechanism of cellular insult.
Figure 3.
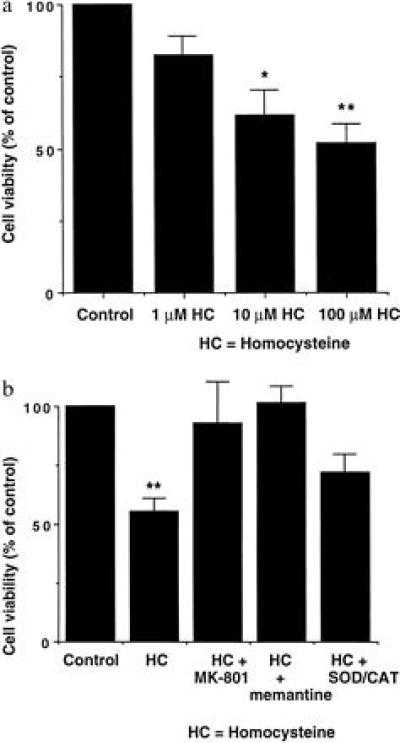
Homocysteine-mediated neuronal cell death. (a) Cultures incubated for 6 days in various concentrations of d,l-homocysteine (HC). Data are the means ± SEM of four replicate values from each of three pooled experiments. Asterisks indicate statistically different from control value (∗, P < 0.05; ∗∗, P < 0.01). (b) Neuroprotective effects of the NMDA antagonists dizocilpine (MK-801, 10 μM), memantine (12 μM), and superoxide dismutase/catalase (SOD/CAT, 50 units/ml each) from 100 μM d,l-homocysteine-mediated neurotoxicity. Cultures were incubated for 6 days in the presence of homocysteine before the assessment of neurotoxicity. Data are the means ± SEM from a representative experiment (performed with quadruplicate samples and repeated seven times). Asterisks indicate statistically different from control value and from the homocysteine groups treated with the NMDA antagonists dizocilpine or memantine (∗∗, P < 0.01). Note that although the superoxide dismutase/catalase value was not significantly different from the homocysteine group, it was also not significantly different from the control group (at either the P < 0.05 or 0.01 levels), indicating an intermediate save from homocysteine-induced neurotoxicity.
Identity of Homocysteine as the Neurotoxin.
Several substances related metabolically to homocysteine, such as homocysteic acid (homocysteine sulfonic acid) and cysteine (especially in the presence of bicarbonate), may also be neurotoxic, at least in part via their stimulation of NMDA receptors (8, 9, 26–30). Other structurally related sulfur amino acids, including cysteic acid, cysteine sulfinic acid, S-sulfo-cysteine, and homocysteine sulfinic acid, also merit consideration as neurotoxins (30). To rule out a contribution by these compounds, we performed HPLC analysis on o-phthaldialdehyde-derivatized fluids and capillary zone electrophoresis from cultures exposed acutely or chronically to homocysteine (14, 15). During acute exposures to homocysteine, none of the other sulfur amino acid derivatives (within the sensitivity of the assays, ≈1 μM) was different from controls (Fig. 4 a and b). Over the 6-day incubation period, we observed variable changes in cysteine and a peak most consistent with cysteine sulfinic acid, but neither showed a consistent rise over controls nor correlated with homocysteine-induced neurotoxicity (Fig. 4 c and d). Moreover, within the sensitivity of these assays, we observed no additional peaks compared with control, making it unlikely that some other sulfur derivative might be involved. Although our findings do not exclude low level oxidation of thiol or the intracellular metabolism of homocysteine to derivative higher oxides of sulfur, the concentrations of these substances would appear to be lower than their respective Km for the NMDA receptor; specifically, our results cannot be explained by an effect of homocysteic acid, although this compound is known to activate NMDA receptors (8, 26–28, 31).
Figure 4.
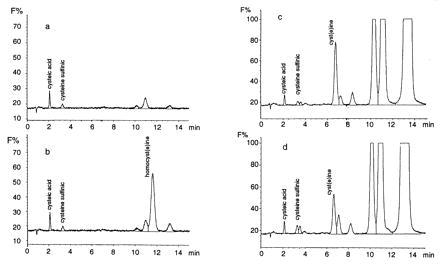
HPLC fluorescence chromatograms of o-phthaldialdehyde-derivatized culture media obtained from acute (5-min; a and b) or chronic (6-day; c and d) exposure to 100 μM homocysteine. The ordinate axis represents relative fluorescence (F%). (a) Chromatogram of control culture medium of mixed neuronal/glial cultures assayed 3 weeks after plating. The culture medium was changed on a Monday-Wednesday-Friday schedule and was assayed here just before a scheduled medium change. (b) Chromatogram of culture medium exposed to homocysteine for 5 min was similar to that of the control except for the presence of homocyst(e)ine. No other peak was consistently changed in three identical experiments; specifically, homocysteic acid was not found. (c) Chromatogram of control culture medium of mixed neuronal/glial cultures assayed 3 weeks after plating. In this case the medium was not changed for the previous 6 days. (d) Chromatogram of culture medium obtained identically to c, except exposed to homocysteine 6 days earlier. Under these conditions, oxidation of homocysteine to homocysteic acid was not encountered. The peaks appearing between 10 and 14 min in chromatograms c and d are not sulfated amino acids and did not comigrate as homocyst(e)ine on additional runs (not shown). By the end of the 6-day incubation, homocysteine had been metabolized and was no longer detectable. The experiment was repeated three times. Cysteic acid was added as an internal standard to all chromatograms and was not present if not added exogenously.
DISCUSSION
Homocysteine as an Excitotoxin.
Our findings show that homocysteine acts as a partial antagonist of the glycine site of the NMDA receptor and therefore inhibits NMDA receptor-mediated activity in the presence of normal concentrations of glycine. However, homocysteine is also an agonist at the glutamate site of the NMDA receptor and is therefore a potential excitotoxin. From our data one might infer that, under normal conditions in the central nervous system, neurotoxicity would not occur because glycine levels are in the low micromolar range, whereas homocysteine is ≈0.5 μM in the cerebrospinal fluid (32) and up to 10 μM in certain brain regions (3). Under conditions of normal glycine concentrations, the agonist (excitotoxic) action of homocysteine at the glutamate site of the NMDA receptor would only prevail if levels approached millimolar concentrations, a scenario pertinent to the rare disorder homocystinuria. Indeed, this mechanism may contribute to the cognitive changes and markedly increased risk of cerebrovascular disease in children and young adults with this disorder. However, during stroke (19) or head trauma (20), disruption of the blood–brain barrier results in exposure of the brain to near plasma levels of amino acids, including homocysteine and glycine. Therefore, in the case of stroke induced by the common disorder hyperhomocysteinemia, the brain is exposed to ≈15–50 μM total homocysteine for prolonged periods (1, 2, 33), in contrast to relatively short-lived pathological elevations in extracellular glutamate. This level of homocysteine was found to be neurotoxic in the present study in the presence of elevated glycine, which is also increased in the brain under these conditions (19, 20). Elevated glycine levels synergize with homocysteine to overstimulate NMDA receptors and contribute to neuronal damage. Given the epidemiology of hyperhomocysteinemia, this mechanism of neurotoxicity is likely to be operative in a substantial proportion of stroke patients. Additionally, glycine is increased in the central nervous system for other reasons, such as the enzyme-deficiency disease nonketotic hyperglycinemia. Thus, in conjunction with elevated glycine, micromolar concentrations of homocysteine that are shown to be neurotoxic in the present report and are within the physiological range in brain (3) could contribute to neuronal injury if allowed access to the extracellular space during tissue injury.
It is also intriguing to speculate on additional mechanisms of homocysteine neurotoxicity. Indeed, the toxicity of cysteine may derive in part from reaction with bicarbonate (9) and in part from the disulfide cystine (34), which is transported into neurons in exchange for the extracellular transport of glutamate via the anionic cystine-glutamate transporter. In the latter case, the local rise in extracellular excitatory amino acids could then contribute to neurotoxicity. As homocysteine-induced currents increase in the presence of bicarbonate (Fig. 2e) and homocysteine is in equilibrium with its disulfide, similar mechanisms may well be operative. In particular, these effects could promote homocysteine agonist activity at substantially lower micromolar concentrations than those shown in Fig. 1b.
Therapeutic Implications.
Current approaches to treatment of hyperhomocysteinemia are aimed at lowering plasma levels of homocysteine and amelioration of vascular injury. Our findings suggest that down-regulation of the NMDA receptor might also be desirable, perhaps with clinically tolerated NMDA receptor antagonists or drugs that act downstream from the receptor to offer neuroprotection (7). Organic nitrates or related nitric oxide-donors would seem to offer unusual therapeutic potential as they can both attenuate homocysteine-induced vascular injury (24) and protect from NMDA receptor-mediated neurotoxicity (11).
Acknowledgments
We thank N. J. Sucher, D. Zhang, and Z.-H. Pan for helpful discussions. This work was supported in part by grants from the National Institutes of Health (to S.A.L. and J.S.S.) and a Pew Scholar’s Award (to J.S.S.).
ABBREVIATIONS
- NMDA
N-methyl-d-aspartate
- [Ca2+]i
intracellular Ca2+ concentration
References
- 1.Lindgren A, Brattström L, Norrving B, Hultberg B, Anderson A, Johannson B B. Stroke. 1995;26:795–800. doi: 10.1161/01.str.26.5.795. [DOI] [PubMed] [Google Scholar]
- 2.Perry I J, Refsum H, Morris R W, Ebrahim S B, Ueland P M, Shaper A G. Lancet. 1995;346:1395–1398. doi: 10.1016/s0140-6736(95)92407-8. [DOI] [PubMed] [Google Scholar]
- 3.Broch O J, Ueland P M. J Neurochem. 1984;43:1755–1757. doi: 10.1111/j.1471-4159.1984.tb06105.x. [DOI] [PubMed] [Google Scholar]
- 4.Stampfer M J, Malinow M R. N Engl J Med. 1995;332:328–329. doi: 10.1056/NEJM199502023320511. [DOI] [PubMed] [Google Scholar]
- 5.Perry T L, Bergeron C, Steele J C, McLachlan D R, Hansen S. J Neurol Sci. 1990;99:3–8. doi: 10.1016/0022-510x(90)90194-r. [DOI] [PubMed] [Google Scholar]
- 6.Simon R P, Swan J H, Griffiths T, Meldrum B S. Science. 1984;226:850–852. doi: 10.1126/science.6093256. [DOI] [PubMed] [Google Scholar]
- 7.Lipton S A, Rosenberg P A. N Engl J Med. 1994;330:613–622. doi: 10.1056/NEJM199403033300907. [DOI] [PubMed] [Google Scholar]
- 8.Olney J W, Price M T, Salles K S, Labruyere J, Ryerson R, Mahan K, Frierdich G, Samson L. Brain Res Bull. 1987;19:597–602. doi: 10.1016/0361-9230(87)90077-3. [DOI] [PubMed] [Google Scholar]
- 9.Olney J W, Zorumski C, Price M T, Labruyere J. Science. 1990;248:596–599. doi: 10.1126/science.2185543. [DOI] [PubMed] [Google Scholar]
- 10.Mansoor M A, Svardal A M, Schneede J, Ueland P M. Clin Chem. 1992;38:1316–1321. [PubMed] [Google Scholar]
- 11.Lipton S A, Choi Y-B, Pan Z-H, Lei S Z, Chen H-S V, Sucher N J, Loscalzo J, Singel D J, Stamler J S. Nature (London) 1993;364:626–632. doi: 10.1038/364626a0. [DOI] [PubMed] [Google Scholar]
- 12.Chen H-S V, Pellegrini J W, Aggarwal S K, Lei S Z, Warach S, Jensen F E, Lipton S A. J Neurosci. 1992;12:4427–4436. doi: 10.1523/JNEUROSCI.12-11-04427.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lei S Z, Pan Z-H, Aggarwal S K, Chen H-S V, Hartman J, Sucher N J, Lipton S A. Neuron. 1992;8:1087–99. doi: 10.1016/0896-6273(92)90130-6. [DOI] [PubMed] [Google Scholar]
- 14.Cooper J D H, Turnell D C. In: Sulfur and Sulfur Amino Acids. Jacoby W B, Griffith O W, editors. Orlando, FL: Academic; 1987. pp. 141–143. [Google Scholar]
- 15.Jones B N, Paabo S, Stein S. J Liq Chromatogr. 1981;4:565–586. [Google Scholar]
- 16.Johnson J W, Ascher P. Nature (London) 1987;325:529–531. doi: 10.1038/325529a0. [DOI] [PubMed] [Google Scholar]
- 17.Kleckner N W, Dingledine R. Science. 1988;241:835–837. doi: 10.1126/science.2841759. [DOI] [PubMed] [Google Scholar]
- 18.Choi D W, Maulucci G M, Kriegstein A R. J Neurosci. 1987;7:357–368. doi: 10.1523/JNEUROSCI.07-02-00357.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Palmer A M, Marion D W, Botscheller M L, Bowen D M, DeKosky S T. NeuroReport. 1994;6:153–156. doi: 10.1097/00001756-199412300-00039. [DOI] [PubMed] [Google Scholar]
- 20.Phillis J W, Smith-Barbour M, O’Regan M H, Perkins L M. Neurochem Res. 1994;19:1125–1130. doi: 10.1007/BF00965145. [DOI] [PubMed] [Google Scholar]
- 21.Kirino T. Brain Res. 1982;239:57–69. doi: 10.1016/0006-8993(82)90833-2. [DOI] [PubMed] [Google Scholar]
- 22.Pulsinelli W A, Brierley L B, Plum F C. Ann Neurol. 1982;11:491–498. doi: 10.1002/ana.410110509. [DOI] [PubMed] [Google Scholar]
- 23.Bormann J. Eur J Pharmacol. 1989;166:591–592. doi: 10.1016/0014-2999(89)90385-3. [DOI] [PubMed] [Google Scholar]
- 24.Stamler J S, Osborne J A, Jaraki O, Rabbani L E, Mullins M, Singel D, Loscalzo J. J Clin Invest. 1993;91:308–318. doi: 10.1172/JCI116187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lafon-Cazal M, Pietri S, Culcasi M, Bockaert J. Nature (London) 1993;364:535–537. doi: 10.1038/364535a0. [DOI] [PubMed] [Google Scholar]
- 26.Sawada S, Takada S, Yamamoto C. Brain Res. 1982;238:282–285. doi: 10.1016/0006-8993(82)90798-3. [DOI] [PubMed] [Google Scholar]
- 27.Hablitz J J. Brain Res. 1982;247:149–153. doi: 10.1016/0006-8993(82)91040-x. [DOI] [PubMed] [Google Scholar]
- 28.Do K Q, Herrling P L, Streit P, Turski W A, Cuénod M. J Neurosci. 1986;6:2226–2234. doi: 10.1523/JNEUROSCI.06-08-02226.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Misra C H. Neurochem Res. 1989;14:253–257. doi: 10.1007/BF00971320. [DOI] [PubMed] [Google Scholar]
- 30.Lehmann A, Hagberg H, Sandberg M. Eur J Neurosci. 1993;5:1398–1412. doi: 10.1111/j.1460-9568.1993.tb00926.x. [DOI] [PubMed] [Google Scholar]
- 31.Patneau D K, Mayer M L. J Neurosci. 1990;10:2385–2399. doi: 10.1523/JNEUROSCI.10-07-02385.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hyland K, Bottiglieri T. J Chromatogr. 1992;579:55–62. doi: 10.1016/0378-4347(92)80362-t. [DOI] [PubMed] [Google Scholar]
- 33.Selhub J, Jacques P F, Bostom A G, D’Agostino R B, Wilson P W F, Belanger A J, O’Leary D H, Wolf P A, Schaefer E J, Rosenberg I H. N Engl J Med. 1995;332:286–291. doi: 10.1056/NEJM199502023320502. [DOI] [PubMed] [Google Scholar]
- 34.Murphy T H, Miyamoto M, Sastre A, Schnaar R L, Coyle J T. Neuron. 1989;2:1547–1558. doi: 10.1016/0896-6273(89)90043-3. [DOI] [PubMed] [Google Scholar]