

Abstract
Substantial evidence now exists that intrinsic free-radical scavenging contributes to the receptor-independent neuroprotective effects of estrogens. This activity is inherently associated with the presence of a phenolic A-ring in the steroid. We report a previously unrecognized antioxidant cycle that maintains the “chemical shield” raised by estrogens against the most harmful reactive oxygen species, the hydroxyl radical (•OH) produced by the Fenton reaction. In this cycle, the capture of •OH was shown to produce a nonphenolic quinol with no affinity to the estrogen receptors. This quinol is then rapidly converted back to the parent estrogen via an enzyme-catalyzed reduction by using NAD(P)H as a coenzyme (reductant) and, unlike redox cycling of catechol estrogens, without the production of reactive oxygen species. Due to this process, protection of neuronal cells against oxidative stress is also possible by quinols that essentially act as prodrugs for the active hormone. We have shown that the quinol obtained from a 17β-estradiol derivative was, indeed, able to attenuate glutamate-induced oxidative stress in cultured hippocampus-derived HT-22 cells. Estrone quinol was also equipotent with its parent estrogen in reducing lesion volume in ovariectomized rats after transient middle carotid artery occlusion followed by a 24-h reperfusion. These findings may establish the foundation for a rational design of neuroprotective antioxidants focusing on steroidal quinols as unique molecular leads.
Keywords: hydroxyl radical, ischemia, prodrug
Epidemological (1) and basic science studies (2) underscore the powerful neuroprotective functions of estrogens. Estrogens play this protective role through several routes (3, 4), although their exact mode of action has remained elusive. The genomic pathway involving the binding to the cognate intracellular receptor proteins [estrogen receptor (ER)α or -β] and, thus, leading to gene transcription (5) has been implicated into the promotion of neuronal survival by estrogens (6). In addition to this classical effect promoted by estrogen response elements, ERs may also directly interact with several intracellular signaling pathways (cAMP response element-binding protein, mitogenactivated protein kinase/extracellular signal-regulated kinase, phosphatidylinositol-3-kinase, etc.) that affect the transcription of many other genes targeting neuroprotective actions in specific ways without interfering with the endocrine effects of the steroid (7). In addition, altered expression of the Bcl-2 family of proteins (8) that are important modulators of neuronal apoptosis, attenuation of glutamate-receptor activation and modulation of intracellular calcium concentrations through interaction with α-amino-3-hydroxy-5-methyl-4-izoxazolepropionate/kainiteand/or N-methyl-D-aspartate receptors (9, 10) have been associated with the neuroprotective effect of estrogens. Finally, estrogens could also serve as free-radical scavengers (i.e., by an ER-independent mechanism) in preventing nerve-cell death induced by various oxidative insults (11–14).
Oxidative stress is associated with the pathology of numerous neurodegenerative diseases and aging (15, 16). The brain is a specialized organ that concentrates metals necessary for normal neurological functions (17). Trauma, ischemia, and many other insults of neuropathological origin can release nonprotein-bound metal ions such as Fe2+ from damaged cells (18) and thereby increase oxidative stress in the CNS (19) through the generation of reactive oxygen species (ROS). The ROS hydrogen peroxide (H2O2) arising through spontaneous or enzyme catalyzed dismutation of superoxide (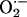 ) is a reactant in the reaction involving the excess metal ion, because hydroxyl radical (•OH, the most damaging ROS) is produced in the process H2O2 + M(n-1)+ → Mn+ + •OH+ OH–, where M is a redox-active metal such as iron [Fenton reaction (20)], copper, or manganese.
) is a reactant in the reaction involving the excess metal ion, because hydroxyl radical (•OH, the most damaging ROS) is produced in the process H2O2 + M(n-1)+ → Mn+ + •OH+ OH–, where M is a redox-active metal such as iron [Fenton reaction (20)], copper, or manganese.
Substantial evidence now exists that estrogen neuroprotection is related to or complemented by the intrinsic free-radical scavenging antioxidant capacity of the hormone due to its A-ring phenolic hydroxy group (21). However, this mechanism, including the chemical nature and fate of the products derived from the radical-scavenging reaction(s), has not been elucidated. Here we report our findings on a previously unrecognized in vitro and in vivo antioxidant cycle for estrogens that may contribute to the “chemical shield” (22) for neurons against the most harmful ROS, the hydroxyl radical (•OH) implicated in the etiology of several neurodegenerative diseases, aging, and stroke (15, 16).
Materials and Methods
Materials. 10β-Hydroxyestra-1,4-dien-3,17-dione (E1-quinol) was prepared by the oxidation of estrone [3-hydroxyestra-1,3,5(10)-trien-17-one (E1)] with 3-chloroperbenzoic acid in the presence of benzoyl peroxide as a radical initiator and under light irradiation in refluxing dry dichloromethane (23). This method was also used to prepare, from the corresponding phenol [3-hydroxy-17β-butoxyestra-1,3,5(10)triene (17βOBu-E2)] (24), 10β-hydroxy-17β-butoxyestra-1,4-dien-3-one (17βOBu-E2-quinol). All other chemicals were purchased, unless specified otherwise, from Sigma–Aldrich.
Fenton Reaction. E1 (10–500 μM) was incubated for 2 min to 6 h in 1 ml of aqueous medium containing FeSO4 (300 μM to 3 mM) and H2O2 (1–100 mM) at a pH range of 3–7. The solutions were extracted with dichloromethane (3 × 1 ml). For reactions done under acidic pH, the combined organic extract was washed acid free with water, dried over sodium sulfate, and the solvent was evaporated under nitrogen stream. Samples were analyzed by liquid chromatography (LC)/atmospheric-pressure chemical ionization (APCI)-MS.
In Vitro Metabolism Studies. E1 or E1-quinol (100 μM) was incubated at 37°C in rat (Sprague–Dawley) brain homogenate (20%, wt/vol) in 0.1 M PBS, pH 7.4. The protein content of the brain homogenates was determined by spectrophotometry (25) using BSA as a reference. Tissue-free incubations were done by dissolving 1 mM of reducing agent (NADH, NADPH, glutathione or ascorbic acid, respectively) and 100 μM of E1-quinol in this medium. To the aliquots (0.5 ml), removed, glacial acetic acid (50 μl) and ethyl acetate (3 × 0.5 ml) were added, and the mixture was vortexed for 1 min and centrifuged at 7,200 × g for 5 min. The organic layer was removed, and the solvent was evaporated at room temperature under nitrogen stream. The sample residue was dissolved in the LC mobile phase, and analyzed by LC/APCI-MS.
In Vivo Metabolism. Experiments were performed according to a procedure involving in vivo cerebral microdialysis (26) in the rat hippocampus. Perfusion of the probes with the solution containing E1-quinol (10 pmols/microliter) in artificial cerebrospinal fluid was done at 1.0 μl/min for 12 h. Sample preparation for LC/MS analyses included extraction of the collected microdialysates with ethyl acetate. The evaporated residues were analyzed by LC/APCI-MS/MS.
LC/MS Analyses. LC separation was done by using a Supelco (Bellefonte, PA) 5 cm × 2.1-mm-i.d. Discovery HS C-18 reversed-phase column with 0.25 ml/min water/methanol/2-propanol/acetic acid/dichloromethane (53:35:5:5:2, vol/vol) as a mobile phase. The sample residues were dissolved in 40 μl of mobile phase, respectively, and 5 μl of the solution was injected for analysis. Mass spectra were recorded on a quadrupole ion-trap instrument (LCQ, ThermoFinnigan, San Jose, CA) by using positive-ion APCI as the method of ionization. MS/MS and MS3 product-ion scans were obtained after collision-induced dissociation with helium as the target gas. Compound identification was based on retention time (tR), APCI mass spectra, MS/MS, and MS3 with authentic compounds as references. E1, E1-quinol, and 2,3-dihydroxyestra-1,3,5(10)-trien-17-one (2-OH-E1) levels were determined by LC/APCI-MS/MS and calibration with solutions of known concentrations of the analytes extracted for analyses. As an internal standard, 1,3,5(10)-estratrien-17α-ethynyl-17β-ol (ethynylestradiol, Steraloids, Newport, RI) was added before each sample extraction.
Hydrogen Peroxide Production in Rat Brain Homogenate. An in vitro assay method (27) was adapted for the measurement of H2O2 production in tissue. The compounds (100 μM) were incubated for 30 min at 37°C in rat brain homogenate (male Sprague–Dawley, 2%, wt/vol, in 0.1 M PBS, pH 7.4) containing 200 μM NADPH and 20 μM sodium azide in a final volume of 100 μl. Two milliliters of a reagent solution (100 μM xylenol orange, 250 μM ferrous ammonium sulfate, and 100 μM sorbitol in 25 mM sulfuric acid) was added to the mixture, and the absorbance at 560 nm was recorded after keeping the sample at room temperature for 45 min. The amount of H2O2 produced was quantified by using a calibration curve based on assaying a serially diluted aqueous H2O2 standard solution.
ER Binding. Competition binding assays were performed by using an enzyme fragment complementation (EFC) method described in the HitHunter (Fremont, CA) EFC Estrogen Chemiluminescence Assay kit. Competing ligands at final concentrations ranging from 10 pM to 10 μM were incubated with 5 nM recombinant ERα or -β (Panvera, Madison, WI) and 17β-estradiol-conjugated enzyme donor for 1.5 h. The enzyme acceptor was then added for another 1.5 h, after which chemiluminescence substrate was added for another hour. Relative luminescence was determined by using a Biotek FL600 plate reader (Biotek Instruments, Winooski, VT). Sigmoidal standard curves were created by graphpad prism (Ver. 3.02 for Windows, GraphPad, San Diego) by using a four-parameter logarithmic curve to determine the concentration to reach IC50.
Neuronal Cell Survival in Vitro. All studies were done on mouse clonal hippocampal HT-22 cells. The cells were cultured in DMEM supplemented with 10% FBS under the usual conditions. Experiments were done in a 96-well culture plate containing ≈5,000 cells per well as determined by a Neubauer hemacytometer. The cells were incubated for 24 h before the compounds were added. 17βOBu-E2 and 17βOBu-E2-quinol were dissolved in absolute ethanol and added, respectively, into the culture media. Sodium glutamate (20 mM) in a solution of phosphate buffer was added together with the test compounds or 5 h later, then the cells were incubated for 24 h. Cell viability was quantified by the Calcein AM assay (28) in a phosphate buffer solution and given as viability = (% surviving after treatment – % surviving without treatment)/% surviving without treatment.
In Vivo Neuroprotection Against Ischemic Stroke in Animal Model. All animal procedures were approved by the University of North Texas Health Science Center Animal Care and Use Committee. Female Sprague–Dawley rats (200–250 g, Charles River Breeding Laboratories) were acclimatized for 3 days before surgery. Bilateral ovariectomy was performed 2 wk before middle cerebral artery occlusion (MCAO). Animals were anesthetized by i.p. injection of ketamine (60 mg/kg) and xylazine (10 mg/kg). Rectal temperature was maintained at 37.5 ± 0.5°C during the procedure. The middle cerebral artery was occluded for 1 h, and then suture was withdrawn for reperfusion. E1 and E1-quinol were dissolved in corn oil and administered at a dose of 200 μg/kg s.c. 2 h before onset of the 1-h MCAO.
Animals were decapitated 24 h after reperfusion. Brains were harvested and placed in a brain matrix for slicing (Harvard Apparatus). Seven slices were made at 3, 5, 7, 9, 11, 13, and 15 mm posterior to the olfactory bulb. Slices were incubated for 30 min in a 2% solution of 2,3,5-triphenyltetrazolium chloride at 37°C and then fixed in 10% formalin. The stained slices were photographed and subsequently measured for the ischemic lesion volume (image-pro plus 4.1, Media Cybernetics, Silver Spring, MD). In a separate experiment involving drug dosing and MCAO as described above, the animals were killed after 24 h of reperfusion, and their uteruses were removed, blotted, and weighed for comparison among the treatment groups.
Data Analysis. Data are expressed as mean ± SEM. Statistical evaluations were done by one-way ANOVA followed by post hoc Dunnett's (comparison to a single control group) or Student–Newman–Keuls test (multiple comparisons).
Results
Hydroxyl-Radical Scavenging by Estrogens. The crucial role of •OH radicals in ROS-induced damage prompted us to investigate experimentally the actual reaction of an estrogen model, E1, on its exposure to this ROS in an aqueous medium. The classical Fenton reaction is an ideal paradigm for these studies, because this chemistry is responsible for producing the toxic •OH radicals involved in several neuropathological processes (15). Kinetic studies showed that conversion of E1 (100 μM) on exposure to •OH progressed rapidly [e.g., with half-life and initial velocity of 2.5 min and 10–6 M·s–1, respectively, at 300 μM Fe(II) and 1.3 mM H2O2 at pH 3.0] with a second-order rate constant (k) of ≈20 M–1·s–1. Independent of the Fe and H2O2 concentration, pH, and incubation time, LC/APCI-MS revealed a single principal product.
The m/z 287 of the protonated molecule (MH+) in the APCI mass spectrum of this product confirmed the incorporation of an oxygen atom, apparently due to the capture of an •OH by the conjugate phenoxyl-radical (ArO•) of E1. Our effort to identify the oxygenated E1 formed under Fenton conditions on the basis of matching the LC retention times and mass spectra, including MS/MS and MS3, with those of the commercially available reference compounds (specifically the catechol E1s) was unsuccessful. Extending the search, we then considered 10(β)hydroxyestra-1,4-dien-3,17-dione (E1-quinol) formed chemically by, among others, a peracid-induced photooxygenation of E1 (23). In contrast with the catechol structure of the well-known metabolic products 2,3-dihydroxyestra-1,3,5 (10)-trien-17-one and 3,4-dihydroxyestra-1,3,5 (10)-trien-17-one (29), 10β-hydroxyestra-1,4-dien-3,17-dione is a quinol that confers a nonaromatic nature to the steroidal A-ring; thus, its (bio)chemistry is expected to be substantially different from that of the catechol estrogens. The metabolic formation of E1-quinol in vitro in rat-liver microsomal incubations has been described only very recently (30). As shown in Fig. 1, E1-quinol was identified unequivocally as the principal product of E1 resulting from the capture of the •OH radicals. E1-quinol was resistant to further oxidation by •OH radicals under Fenton conditions. We also determined that E1-quinol had no affinity (IC50 > 10 μM) to ERs (Table 1, which is published as supporting information on the PNAS web site, www.pnas.org); thus, it was not intrinsically “feminizing.”
Fig. 1.

LC-MS analysis of the product formed from E1 on exposure to •OH generated by the Fenton reaction. (a) Extracted ion-current chromatogram (m/z 287); mass spectra are from the major LC peak at tR = 1.3 min; (b) full-scan APCI-MS; (c) MS/MS (MS2) product-ion scan from the molecular ion m/z 287 isolated as the precursor; and (d)MS3 product-ion scan of m/z 269 (the major product ion of the MS2, c). Fragmentation pattern and tR are identical to those of a synthetic 10β-hydroxyestra-1,4-dien-3,17-dione (E1-quinol).
Formation of Steroidal Quinol from Estrogen in Brain in Vitro. To probe whether the quinol structure also arises from E1 under biological Fenton conditions, we incubated E1 in rat-brain homogenate. The formation of E1-quinol in rat brain was indeed demonstrated via the incubation of E1 in this tissue homogenate, as shown in Fig. 2. E1-quinol was not only detected; it was by far the most abundant oxygenated product with a concentration about 15× higher after 30-min incubation than that of 2,3-dihydroxyestra-1,3,5(10)-trien-17-one (2-OH-E1), the major cytochrome P450 metabolite of this estrogen (29).
Fig. 2.

LC-MS analysis of product formed from E1 after 30-min incubation in rat-brain homogenate (20% wt/vol; protein content, 18.6 mg/ml). The chromatographic trace displayed is a selected MS/MS reaction monitoring (SRM) of m/z 287 → m/z 269 to indicate oxygenated E1. E1-quinol is present at a concentration ≈15× higher than the major cytochrome P450 metabolite 2,3-dihydroxyestra-1,3,5(10)-trien-17-one (2-OH-E1).
Effect of Steroidal Quinol on H2O2 Production. The redox cycling between the cytochrome P450-generated catechol metabolites of estrogens and their respective quinones produce free radicals, which is believed to be the main molecular event in the development of carcinogenic effect of the catechol estrogens (27, 31). Therefore, we wanted to confirm whether such prooxidant process took place for the E1-quinol in the brain. We addressed this issue by comparing the production of H2O2 in rat-brain homogenate between E1-quinol and the profound prooxidant, estra-1,5(10)-dien-3,4,17-trione (estrone-3,4-quinone) (3,4-E1Q). Although 3,4-E1Q significantly increased H2O2 production in brain tissue as expected, E1-quinol unquestionably lacked the prooxidant effect (Fig. 3); actually, it reduced oxidative stress compared with the control.
Fig. 3.

Effect of steroidal quinol vs. quinone on H2O2 production in rat brain homogenate. E1-quinol (shaded bar), unlike other E1 metabolites such as estra-1,5 (10)-dien-3,4,17-trione (estrone-3,4-quinone) (3,4-E1Q) (filled bar), does not induce oxidative stress. Addition of E1-quinol reduces H2O2 formation in rat-brain homogenate, whereas 3,4-E1Q is a profound prooxidant (n = 5–10). *, P < 0.05.
Reductive Aromatization of Steroidal Quinols. We incubated E1-quinol in rat-brain homogenate and found that even after a short incubation time (2.5 min), a significant level of E1 was present (Fig. 4 a and b). Our subsequent query was about the endogenous reducing agent involved in the process. When E1-quinol was incubated in the presence of, e.g., glutathione or ascorbate, we failed to detect E1. However, E1 was clearly present when the experiments were carried out in the presence of NADH and especially NADPH. Enzymes available in brain tissue homogenates further catalyzed the latter reaction (Fig. 4c) [an apparent rate of 177 pmol·min–1·(mg protein)–1 was measured in NADPH-supplemented rat brain homogenate]. By using the cerebral microdialysis technique (26), we also unambiguously confirmed that reduction of E1-quinol to the parent E1 also takes place in the brain in vivo.
Fig. 4.

Reductive enzyme-catalyzed regeneration of E1 from E1-quinol in rat brain tissue. (a) The chromatographic traces displayed are the SRM of m/z 287 → m/z 269 for E1-quinol and the SRM of m/z 271 → m/z 253 for E1. (b) The peak at tR = 4.5 min is unequivocally identified, on the basis of coelution with an authentic reference compound and identical APCI, MS/MS (shown in the chart together with the origin of the major fragments observed), and MS3 spectra, as E1. (c) Conversion of E1-quinol to E1 involves NAD(P)H as a reductant and is enzyme-catalyzed in brain tissue. Initial conversion rates to E1 were measured at 100 μM starting E1-quinol concentration in rat-brain homogenate, tissue-free NADPH solution (1 mM), and NADPH-supplemented (1 mM) rat-brain homogenate, respectively (n = 3). *, P < 0.05.
Protection of Cultured Neurons Against Oxidative Insults. We compared survival of cultured hippocampus-derived (HT-22) cells exposed to an oxidative stress (28) in the presence of a phenolic estrogen derivative and its quinol product. We again confirmed that 17βOBu-E2-quinol had no intrinsic affinity to ERs (IC50 > 10 μM), whereas the parent phenolic A-ring compound showed binding to the ERα and -β (Table 1). The feasibility of the conversion of 17βOBu-E2-quinol to 17βOBu-E2 by NADPH, which is essential for the proposed prodrug mechanism, has also been demonstrated (Fig. 8, which is published as supporting information on the PNAS web site). Although 17βOBu-E2 displayed neuroprotection at ≥10–7 M (24), the quinol (17βOBu-E2-quinol) was also neuroprotective against glutamate-induced oxidative stress on the neuronal cells at a concentration of ≥10–6 M (Fig. 5). A shift in the dose–response curve between the parent phenolic 17βOBu-E2 and its quinol in the cytotoxicity studies was observed when quinol was added simultaneously with the start of the oxidative insult. However, pretreatment of the cultured HT-22 cells with the quinol before their oxidative stressing by glutamate improved cell survival, and the shift in the dose–response curve compared with the parent phenolic compound diminished.
Fig. 5.

In vitro neuroprotection of hippocampus-derived HT-22 neuronal cells against glutamate exposure achieved by the nonphenolic 17βOBu-E2-quinol (▪, solid line), compared with that of the phenolic A-ring parent compound 17βOBu-E2 (♦, dotted line) (n = 6–20). *, P < 0.05.
In Vivo Neuroprotection Against Ischemic Stroke. When administered before ischemia, E1 significantly reduced infarct volume by 53% (P < 0.05) compared with control after transient MCAO followed by 24-h reperfusion in ovariectomized rats. In this experimental model for stroke, E1-quinol was equipotent with the parent estrogen in reducing lesion (Fig. 6a). On the other hand, whereas uterine weight of the animals doubled in the group treated with E1, no differences were obtained from the control group for the animals that received E1-quinol treatment (Fig. 6b).
Fig. 6.

Brain lesion volumes (a) and uterine weights (b) in ovariectomized rats after a transient MCAO on administration of E1, E1-quinol (200 μg/kg, s.c., respectively; 2 h before MCAO), and a vehicle control. Lesion volumes and uterine weights were measured after 24 h of reperfusion (n = 4–11). *, P < 0.05.
Discussion
There is abundant in vitro, in vivo, and clinical evidence supporting the role of estrogen in neuronal survival. The hormone apparently regulates many aspects of neuronal function. With the plethora of potentially neuroprotective pathways activated by estrogens (2–4), it is unlikely that a single mechanism is responsible for their neuroprotective action, and the consensus is that both rapid nongenomic and delayed genomic effects are needed for long-term neuroprotection. The importance of each pathway may also vary with neuronal type, developmental stage, type of receptor expressed, extracellular environment, and specific neurological condition. Our report focuses mainly on estrogens as potential free-radical scavengers that prevent oxidative neuronal cell death via an ER-independent mode of action.
Our studies indicated that the formation of nonphenolic quinols by direct •OH scavenging is a key element of the neuroprotective “chemical shield” erected by estrogens (22) against this harmful ROS (Figs. 1 and 2). In our studies, E1 was used as a model estrogen. [This choice was based on the evaluation of the analytical method we decided to use; E2 exhibited extensive water loss (–18 units, base peak at m/z 255) from the protonated molecule (MH+, m/z 273, relative abundance <5% of the base peak) in the APCI mass spectra, which complicated the structure identification of reaction products. On the other hand, E1 showed no decomposition with this method of ionization (32)]. We found that E1 converted to E1-quinol on exposure to •OH generated by the Fenton reaction both in a cell-free environment and in rat brain homogenate, where its apparent rate of formation was 1.5 pmol·min–1·(mg·protein)–1. Although metabolic formation of E1-quinol in rat-liver microsomes in vitro has also been reported recently (30), our results imply that metabolic enzymes are not necessary for steroidal quinol formation, but exposure to •OH continuously generated in the tissue homogenate is responsible for its formation through direct radical scavenging.
The molecular capture of the •OH inactivates the steroid as an estrogen. However, the parent phenolic A-ring estrogens are efficiently regenerated from the resultant quinols in brain tissue by enzyme-catalyzed reduction with NAD(P)H as a coenzyme (Fig. 4); thus, a cycle exists. Our work provided biochemical evidence for the existence of a previously undescribed antioxidant cycle for phenolic A-ring estrogens. The phenoxyl radical produced during the detoxification of the •OH via a radical exchange reaction in, among others, the chain-breaking reaction of lipid peroxidation (LOO· → LOOH) scavenges •OH to produce a bioreversible quinol that rapidly converts to the parent estrogen via a NAD(P)H-dependent enzyme-catalyzed reductive aromatization to perpetuate the antioxidant action, as shown in Fig. 7. Unlike redox cycling of catechol estrogens (27, 31), this reductive aromatization proceeds without the production of ROS (Fig. 3). This conceptualization of our findings establishes a mechanism whereby estrogens can provide neuroprotection against free-radical damage within a very short (seconds to minutes) time frame and can as well sustain neuroprotective cascades that require protein synthesis such as the estrogen-inducible bcl-2 expression (33).
Fig. 7.

Schematic illustration of how estrogens provide a chemical shield to neurons from •OH exposure by an antioxidant cycle. After the direct scavenging of this most harmful ROS, the phenolic A-ring estrogen is dearomatized to quinol that is rapidly recycled to the parent estrogen through an enzyme-catalyzed reductive aromatization process.
With in vitro and in vivo experiments, we have also shown that steroidal quinols are plausible prodrugs for neuroprotective phenolic A-ring compounds, which further supports the existence of the cyclic mechanism we discovered. Because most natural estrogens, including E2 and E1, manifest neuroprotection at relatively high concentration (≥10–5 M) in cultured HT-22 cells, we selected a synthetic estrogen analogue as the parent compound (24) for the chemical synthesis of the corresponding quinol. The observation that a nonphenolic quinol protected the hippocampus-derived HT-22 neurons against glutamate-induced oxidative stress supported our hypothesis for its reductive conversion to a neuroprotective phenolic A-ring steroid (Fig. 5). The shift in the dose–response curve between the parent 17βOBu-E2 and its quinol in our cytotoxicity studies was due to the treatment schedule. We added the quinol simultaneously with the start of the oxidative insult; therefore, the neuroprotective E2 derivative evolved in time through activation from the prodrug form. Until an adequate concentration of 17βOBu-E2 is reached during this activation period, oxidative stress eventually kills a fraction of the cells. On the other hand, pretreatment of the cultured HT-22 cells with the quinol before their oxidative stressing by glutamate improved cell survival, and the shift in the dose–response curve compared with the parent phenolic compound diminished (data not shown).
We chose experimental stroke for in vivo evaluation. Acute restoration of blood flow after ischemia leads to the production of ROS (34–36), which are directly toxic to neurons. Therapeutic nonenzymatic scavenging of free radicals may, therefore, be a viable strategy for the reduction of ischemic cerebral tissue damage. In animal models, natural estrogens used at supraphysiological concentrations (37, 38) and estrogen analogues with no affinity to ERs (39) are neuroprotective. Thus, the observed effect is thought to be partly due to an antioxidant mechanism. E1-quinol showed a decrease of reperfusion-associated ischemic damage equivalent to that of the parent estrogen in the in vivo paradigm chosen (Fig. 6a), which was another strong indication to the conversion of the compound to phenolic A-ring steroid in sufficient concentrations to mediate neuroprotection. On the other hand, E1-quinol is apparently sequestered rapidly and selectively by, e.g., the brain followed by an in situ reductive conversion to the pharmacologically active estrogen, whereas its distribution into and/or reduction in the uterus may not take place to a significant extent to affect this organ through ER-mediated (endocrine) pathways with the dosing regimen used in our study (Fig. 6). Therefore, quinols may be implicated as pharmaceutically more acceptable prodrugs (less lipophilic, more resistant to oxidative metabolism, safer/less toxic side effects, etc.) than the parent phenolic/estrogenic compounds (23, 40, 41) when used for neuroprotective therapy.
In conclusion, we have demonstrated that the “chemical shield” erected by estrogens to protect neurons against •OH-radical inflicted damage involves a perpetuated antioxidant cycle through quinols as key intermediates devoid of estrogenic activity (i.e., they are nonfeminizing). Estrogens have been merely the starting point for the discovery of steroidal quinols as therapeutically useful compounds. We have implied that these key intermediates of the antioxidant cycle actually operate as prodrugs in a concentration range considered useful for, among others, nonenzymatic scavenging of free radicals to reduce ischemic cerebral tissue damage. These findings lay the foundation for a rational design of novel neuroprotective agents that may improve therapeutic safety compared with that of estrogens.
Supplementary Material
Acknowledgments
We thank Drs. Bruce S. McEwen (The Rockefeller University, New York) and William G. Luttge (McKnight Brain Institute, University of Florida) for thoughtful review of the project. This research was supported in part by National Institutes of Health Grants NS44765, AG10485, and RR12023.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: E2, estra-1,3,5(10)-trien-3,17β-diol; 17βOBu-E2, 3-hydroxy-17β-butoxyestra-1,3,5 (10)-triene; 17βOBu-E2-quinol, 10β-hydroxy-17β-butoxyestra-1,4-dien-3-one; LC, liquid chromatography; APCI, atmospheric-pressure chemical ionization; E1, 3-hydroxyestra-1,3,5(10)-trien-17-one; E1-quinol, 10β-hydroxyestra-1,4-dien-3,17-dione; ER, estrogen receptor; ROS, reactive oxygen species; SRM, selected-reaction monitoring; tR, retention time; MCAO, middle cerebral artery occlusion.
References
- 1.Blanchet, P. J., Fang, J., Hyland, K., Arnold, L. A., Mouradian, M. M. & Chase, T. N. (1999) Neurology 53, 91–95. [DOI] [PubMed] [Google Scholar]
- 2.Lee, S. B. & McEwen, B. S. (2001) Annu. Rev. Pharmacol. Toxicol. 41, 569–591. [DOI] [PubMed] [Google Scholar]
- 3.Green, P. S. & Simpkins, J. W. (2000) Int. J. Dev. Neurosci. 18, 357–358. [DOI] [PubMed] [Google Scholar]
- 4.Behl, C. (2002) Nat. Rev. Neurosci. 3, 433–442. [DOI] [PubMed] [Google Scholar]
- 5.Hall, J. M., Couse, J. F. & Korach, K. F. (2001) J. Biol. Chem. 276, 36869–36872. [DOI] [PubMed] [Google Scholar]
- 6.Wise, P. M., Dubal, D. B., Wilson, M. E., Rau, S. W., Bottner, M. & Rosewell, K. L. (2001) Brain Res. Rev. 37, 313–319. [DOI] [PubMed] [Google Scholar]
- 7.Lonard, D. M. & Smith, C. L. (2002) Steroids 67, 15–24. [DOI] [PubMed] [Google Scholar]
- 8.Garcia-Segura, L. M., Cardona-Gomez, P., Naftolin, F. & Chowen, J. A. (1998) Neuroreport 9, 595–597. [DOI] [PubMed] [Google Scholar]
- 9.Wong, M. & Moss, R. L. (1992) J. Neurosci. 12, 3217–3225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weaver, C. E., Park-Chung, M., Gibbs, T. T. & Farb, D. (1997) Brain Res. 761, 338–341. [DOI] [PubMed] [Google Scholar]
- 11.Behl, C., Skutella, T., Lezoulac'h, F., Post, A., Widmann, M., Newton, C. & Hoelsboer, F. (1997) Mol. Pharmacol. 51, 535–541. [PubMed] [Google Scholar]
- 12.Green, P. S., Bishop, J. & Simpkins, J. W. (1997) J. Neurosci. 17, 511–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moosmann, B. & Behl, C. (1999) Proc. Natl. Acad. Sci. USA 96, 8867–8872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vedder, H., Anthes, N., Stumm, G., Wurz, C., Behl, C. & Krieg, J. C. (1999) J. Neurochem. 72, 2531–2538. [DOI] [PubMed] [Google Scholar]
- 15.Halliwell, B. (2001) Drugs Aging 18, 685–716. [DOI] [PubMed] [Google Scholar]
- 16.Cuzzocrea, S., Riley, D. P., Caputi, A. P. & Salvemini, D. (2001) Pharmacol. Rev. 53, 135–159. [PubMed] [Google Scholar]
- 17.Bush, A. J. (2000) Curr. Opin. Chem. Biol. 4, 184–191. [DOI] [PubMed] [Google Scholar]
- 18.Bishop, G. M. & Robinson, S. R. (2001) Brain Res. 90, 175–187. [DOI] [PubMed] [Google Scholar]
- 19.Halliwell, B. (1992) J. Neurochem. 56, 1609–1623. [DOI] [PubMed] [Google Scholar]
- 20.Walling, C. (1975) Acc. Chem. Res. 28, 125–131. [Google Scholar]
- 21.Culmsee, C., Vedder, H., Ravati, A., Junker, V., Otto, D., Ahlemeyer, B., Krieg, J. C. & Krieglstein, J. (1999) J. Cereb. Blood Flow Metab. 19, 1263–1269. [DOI] [PubMed] [Google Scholar]
- 22.Wickelgren, I. (1997) Science 276, 675–678. [DOI] [PubMed] [Google Scholar]
- 23.Solaja, B. A., Milic, D. R. & Gasic, M. J. (1996) Tetrahedron Lett. 37, 3765–3768. [Google Scholar]
- 24.Prokai, L., Oon, S. M., Prokai-Tatrai, K., Abboud, K. A. & Simpkins, J. W. (2001) J. Med. Chem. 44, 110–114. [DOI] [PubMed] [Google Scholar]
- 25.Lowry, O. H., Rosenbrough, N. J., Farr, A. L. & Randall, R. J. (1951) J. Biol. Chem. 193, 265–275. [PubMed] [Google Scholar]
- 26.Prokai, L., Kim, H. S., Zharikova, A. D., Roboz, J., Ma, L., Deng, L. & Simonsick, W. J., Jr. (1998) J. Chromatogr. A. 800, 59–68. [DOI] [PubMed] [Google Scholar]
- 27.Nutter, L. N. Wu, Y.-Y., Ngo, E. O., Sierra, E. E., Gutierrez, P. L. & Abdul-Hajj, Y. J. (1994) Chem. Res. Toxicol. 7, 23–28. [DOI] [PubMed] [Google Scholar]
- 28.Green, P. S., Perez, E. J., Calloway, T. & Simpkins, J. W. (2000) J. Neurocytol. 29, 419–423. [DOI] [PubMed] [Google Scholar]
- 29.Lee, A. J., Kosh, J. W., Conney, A. H. & Zhu, B. T. (2001) J. Pharmacol. Exp. Ther. 298, 420–432. [PubMed] [Google Scholar]
- 30.Ohe, T., Hirobe, M. & Mashino, T. (2002) Drug Metab. Dispos. 28, 110–112. [PubMed] [Google Scholar]
- 31.Cavalieri, E. L., Stack, D. E., Devanesan, P. D., Todorovic, R., Dwivedy, I., Higginbotham, S., Johanson, S. L., Patil, K. D., Gross, M. L., Gooden, J. K., et al. (1997) Proc. Natl. Acad. Sci. USA 94, 10937–10942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ma, Y. C. & Kim, H. Y. (1997) J. Am. Soc. Mass Spectrom. 8, 1010–1020. [Google Scholar]
- 33.Nielsen, J. & Brinton, R. D. (2002) Endocrinology 143, 205–212. [DOI] [PubMed] [Google Scholar]
- 34.Forman, L. G., Liu, P., Nagele, R. G., Yin, K. & Wong, P. Y. (1998) Neurochem. Res. 23, 141–148. [DOI] [PubMed] [Google Scholar]
- 35.Peters, O., Back, T., Lindauer, U., Busch, C., Megow, D., Dreier, J. & Dirnagl, U. (1998) Cereb. Blood Flow Metab. 18, 196–205. [DOI] [PubMed] [Google Scholar]
- 36.Mason, R. B., Pluta, R. M., Walbridge, S., Wink, D. A., Oldfield, E. H. & Boock, R. J. (2000) J. Neurosurg. 93, 99–107. [DOI] [PubMed] [Google Scholar]
- 37.Yang, S. H., Shi, J., Day, A. L. & Simpkins, J. W. (2000) Stroke 31, 745–749. [DOI] [PubMed] [Google Scholar]
- 38.Shi, J., Yang. S. H., He, Z., Lucas, T. H., Buckley, D. L., Blackband, S. J., King, M. A., Day, A. L. & Simpkins, J. W. (2001) Stroke 32, 987–992. [DOI] [PubMed] [Google Scholar]
- 39.Liu, R., Yang, S. H., Perez, E., Yi, K. D., Wu, S. S., Eberst, K., Prokai, L., Prokai-Tatrai, K., Cai, Y. C., Covey, D. F., et al. (2002) Stroke 33, 2485–2491. [DOI] [PubMed] [Google Scholar]
- 40.Stella, V. J., Charman, W. N. A. & Naringrekar, V. H. (1985) Drugs 29, 455–473. [DOI] [PubMed] [Google Scholar]
- 41.Takata, J., Matsunaga, K. & Karube, Y. (2002) Toxicology 180, 183–193. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.