

Abstract
High-temperature origin-of-life theories require that the components of the first genetic material are stable. We therefore have measured the half-lives for the decomposition of the nucleobases. They have been found to be short on the geologic time scale. At 100°C, the growth temperatures of the hyperthermophiles, the half-lives are too short to allow for the adequate accumulation of these compounds (t1/2 for A and G ≈ 1 yr; U = 12 yr; C = 19 days). Therefore, unless the origin of life took place extremely rapidly (<100 yr), we conclude that a high-temperature origin of life may be possible, but it cannot involve adenine, uracil, guanine, or cytosine. The rates of hydrolysis at 100°C also suggest that an ocean-boiling asteroid impact would reset the prebiotic clock, requiring prebiotic synthetic processes to begin again. At 0°C, A, U, G, and T appear to be sufficiently stable (t1/2 ≥ 106 yr) to be involved in a low-temperature origin of life. However, the lack of stability of cytosine at 0°C (t1/2 = 17,000 yr) raises the possibility that the GC base pair may not have been used in the first genetic material unless life arose quickly (<106 yr) after a sterilization event. A two-letter code or an alternative base pair may have been used instead.
Keywords: nucleobase hydrolysis/RNA world/chemical evolution
A high-temperature origin of life (80°–110°C) is widely favored (1–6) because hyperthermophiles, which grow at temperatures between 80° and 110°C, are claimed to be the oldest organisms on the Earth (7), although there are dissenting opinions (8–11). Added support for this theory comes from atmospheric models depicting an early warm (≈85°–110°C) Earth (12, 13). Models for an even higher temperature origin include proposals that life arose in the 350°C submarine vents (14–17) or between 150° and 250°C involving temperature and pH gradients (18).
For a compound to be used in the first living organism it needs to be sufficiently stable so that the balance between synthesis and degradation does not result in vanishingly small concentrations. Previous studies have shown that a major problem with an origin of life between 250°–350°C is the stability of the presumed components of the first organisms, where the half-lives for decomposition are at most a few minutes (19–22). These data, however, do not deal with an origin of life between 80°–110°C, where problems with the stability of RNA previously have been pointed out (23).
We show here that the rapid rates of hydrolysis of the nucleobases A, U, G, C, and T at temperatures much above 0°C would present a major problem in the accumulation of these presumed essential compounds on the early Earth. A high-temperature origin of life involving these compounds therefore is unlikely. These results are applicable to any origin-of-life theory in which life begins with the evolution of a self-replicating genetic system capable of undergoing Darwinian evolution. A high-temperature origin of life involving compounds other than those discussed here or involving the evolution of metabolic cycles before the evolution of the first genetic material may be possible, but also would be subject to stability criteria.
MATERIALS AND METHODS
The adenine, diaminopyrimidine, diaminopurine, and aminoimidazole carboxamide were from Aldrich. The hypoxanthine, 5-methylcytosine, and 5-hydroxymethylcytosine were from Calbiochem, and the guanine, cytosine, uracil, thymine, xanthine, isoguanine, and 4,5,6-triaminopyrimidine were from Sigma.
Analysis of all samples was performed on a Beckman model 110B HPLC system using a YMC (Kyoto) ODS-AQ analytical reversed-phase column and a Kratos absorbance detector set at 260 nm. The mobile phase for all experiments, except for those involving guanine, was a pH 4.8, 0.1 M phosphate buffer. For guanine, a 0.1 M phosphate, pH 2.5 buffer was used. This provided better separation between guanine and its hydrolysis product xanthine. Products were identified by retention time, coinjection with a known sample, and UV absorption.
Decomposition products were identified when possible. They are: for adenine, hypoxanthine, aminoimidazole carboxamide, and 4,5,6-triaminopyrimidine; for guanine, xanthine; for hypoxanthine, aminoimidazole carboxamide; for cytosine, uracil; for diaminopyrimidine, cytosine, isocytosine, and uracil; and for diaminopurine, guanine, isoguanine, and xanthine. No UV-absorbing products were observed from the decomposition of xanthine and uracil.
Rates of decomposition were calculated based on the disappearance of the base with at least 10% decomposition. Where possible, reactions were followed to at least two half-lives. Because of the long heating time involved for some (C and A) of the low-temperature reactions, some first-order rates were calculated from the appearance of decomposition products, rather than the disappearance of the starting material.
In all cases the concentration of the nucleobase used was 1 × 10−3 M except for the case of xanthine in which a 1 × 10−4 M solution was used, and guanine, in which a saturated solution was filtered with a 0.22 μM acetate filter (approximately 2.5 × 10−5 M).
The following buffers were used for the pH rate profiles of adenine and guanine: 1 M HCl and 0.1 M HCl were used for pH 0 and 1. The other buffers used were 0.05 M formate for pH 3; 0.05 M acetate for pH 4 and 5; 0.05 M phosphate for pH 6, 7, and 8; and 0.05 M carbonate for pH 9 and 10. The ionic strength of all solution was adjusted to 0.2 with NaCl except for pH 0. The pH values were measured both before and after heating to make sure that they did not drift. All Arrhenius curves were conducted at pH 7 using a 0.05 M phosphate buffer, μ = 0.2. Because pH changes with temperature, pH values were adjusted accordingly by using the equation ΔpH = ΔpK, and the pK values of Robinson and Stokes (24).
Solutions were heated in sealed borosilicate ampoules in a series of Thermolyne 16500 dry baths, which held a constant temperature ±0.2°C as measured by a Hewlett Packard 2804A quartz thermometer. In some cases a white precipitate of silica formed inside of the ampoules. This generally was accompanied by a slight increase in pH and probably is caused by the hydration of silica.
Oxygen was found to affect the distribution of the products of decomposition, although the effect on the overall rate was small. All samples were degassed by three cycles of freezing, with a mixture of dry ice and ethanol, and thawing under vacuum. The tubes then were sealed under vacuum.
The decomposition of samples by bacterial contamination was also a constant problem in these experiments, especially with the insoluble guanine. Therefore all of the samples were sterilized at 100°C for 15 min immediately after preparation.
RESULTS
The stability of A to hydrolysis previously has been cited as a major factor that would limit its availability for use in the first genetic material (25). These data, however, are based on the rate of hydrolysis of A in acid (26) where the ΔH‡ is much smaller than at neutral pH. To establish the stability of adenine at neutral pH we have measured the pH rate profile at several temperatures. The pH rate profiles for the decomposition of adenine under anaerobic conditions at 100°, 120°, and 145°C are shown in Fig. 1, Upper. The profiles exhibit a flat region from pH 5–10, and there appear to be at least four reactions leading to decomposition. These include (AH+)(H+), (AH+)(H2O), (A)(H2O), (A)(OH−), or their kinetic equivalents.
Figure 1.

pH rate profile for the decomposition of adenine (Upper) and guanine (Lower).
The pH rate profiles for the decomposition of guanine at 100°, 120°, and 145°C are shown in Fig. 1, Lower. Like the pH rate profile for adenine, the pH rate profile for guanine exhibits a relatively flat region from pH 5–10. There also appear to be at least four reactions leading to the decomposition. These include (GH+)(H+), (GH+)(H2O), (G)(H2O), (G)(OH−), or their kinetic equivalents.
The stability of cytosine previously has been cited as a factor that would limit its availability on the early Earth (27). The pH rate profile for the hydrolysis of cytosine has been reported previously by Garret and Tsau (28) and also exhibits a flat region from pH 4–9.
Fig. 2 shows the Arrhenius curves and equations for the decomposition of A, U, G, C, and T at pH 7. The Arrhenius curves and equations for the alternative bases xanthine, hypoxanthine, and diaminopyrimidine are shown in Fig. 3. At pH 7 and 100°C, the rate of hydrolysis of diaminopurine is 1.01 × 10−8 s−1, isoguanine is 4.1 × 10−7 s−1, 5-methylcytosine is 8.9 × 10−7 s−1, and 5-hydroxymethylcytosine is 5.8 × 10−7 s−1.
Figure 2.

Arrhenius plot for the decomposition of A, U, G, C, and T, pH 7. ▵, data from Garret and Tsau (10). Equations are as follows: log k(A) = − 5902/T + 8.15; log k(U) = − 7649/T + 11.76; log k(G) = − 6330/T + 9.40; log k(C) = −5620/T + 8.69; log k(T) = − 7709/T + 11.24.
Figure 3.

Arrhenius plot for the decomposition of xanthine, hypoxanthine, and diaminopyrimidine. The Arrhenius equations are as follows: xanthine, log k = −6230/T + 9.42; hypoxanthine, log k = − 5270/T + 7.95; and diaminopyrimidine, log k = − 5341/T + 7.31.
DISCUSSION
Calculation of the half-lives for the rates of decomposition of the nucleobases A, U, G, C, and T clearly shows that these compounds are not stable on a geologic time scale at temperatures much above 0°C.
At 350°C, the half-lives for hydrolysis are between 2 and 15 sec, and at 250°C they are between 1 and 35 min. These rates are so fast that it would be impossible for these compounds to accumulate to significant levels, making a heterotrophic origin of life at these temperatures highly unlikely.
At 100°C the half-lives for the decomposition of the nucleobases are still very short. The half-life for A is 1 yr, G is 0.8 yr, U is 12 yr, and T is 56 yr. C is shortest of all with a half-life of only 19 days. Therefore unless these compounds were used immediately after their synthesis, an origin of life at ≈100°C is also unlikely.
Even at 25°C, the rate of hydrolysis of the compounds are fast on the geologic time scale. The half-lives for A and G are ≈10,000 yr, whereas that of C is only 340 yr.
At 0°C, the half-life of A is 6 × 105 yr, G is 1.3 × 106 yr, U is 3.8 × 108 yr, and T is 20 × 108 yr. These rates are comparable to the present rate of destruction of organic matter in sea water as it passes through the hydrothermal vents every 107 yr (29). This has been cited as a major limiting factor in the build-up of organic molecules on the early Earth (30, 31), and suggests that at 0°C, A, G, U, and T are sufficiently stable for a low-temperature origin of life. The case for C, however, is different, and is discussed below.
Rates of Synthesis vs. Rates of Decomposition.
It is necessary to consider the rates of synthesis as well as the rate of decomposition of the nucleobases to get the steady-state nucleobase concentrations. If the rates of synthesis are temperature dependent and have activation energies similar to those for decomposition, then the steady-state concentrations of the nucleobases would be independent of temperature. For example, the concentration of A is given by:
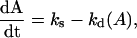 |
where A is the concentration of adenine, ks the rate of synthesis, and kd the rate of decomposition of the nucleobases. At steady state dA/dt = 0, and
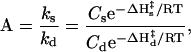 |
where Cs and Cd are the Arrhenius constants. If the ΔH‡ values for ks and kd are the same, then ks/kd and the steady-state concentration of A are independent of temperature. The value of kd is strongly dependent on temperature (i.e., ΔH‡ = 28 kcal for A). The rate of synthesis, ks, consists of two steps, the production of the precursor HCN and its reaction to produce A. The rate of production of A is temperature dependent with ΔH‡ = 20 kcal (32), but the reaction is very concentration dependent and only proceeds when [HCN] > 0.1 M. (33). At the necessary concentrations, the reaction is fast (hours at 100°C and a few months at −20°C). Therefore the rate-limiting step is the production of HCN by photochemical and electric discharge processes. Because these production rates are independent of temperature, the rate of synthesis of A will be independent as well.
Actually the rate of A synthesis at 100°C is less than that at T < 0°C. This is because HCN is hydrolyzed more rapidly to NH4HCO2 at higher temperatures, leading to low steady-state HCN concentrations. More importantly, the HCN cannot be concentrated at elevated temperatures (>0°C) as it can at lower temperatures (<0°C) by eutectic freezing, so adenine synthesis appears to be restricted to low temperatures. Similar considerations to these would apply to guanine, and to a lesser extent the pyrimidines.
Other Factors Affecting Rates of Decomposition.
The rates of decomposition measured here have not been corrected for general acid general base effects of the buffer on the reaction rate. These effects are generally small. For example, at the buffer concentrations used (0.05 M) the rate of decomposition for C is only ≈1.3 times faster for phosphate, and ≈1.5 times faster for acetate, than at zero buffer concentration (28, 34). Even relatively large changes in rate (10- to 100-fold), however, would not affect our conclusions on high-temperature theories.
The effects of high pressure on reaction rates are of concern to prebiotic processes in the deep sea and near hydrothermal systems where the pressure may be as high as 300 atm (1 atm = 101.3 kPa). In general, the effects of pressure on reaction rates are small and are given by the equation:
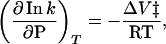 |
where ΔV‡ is the volume of activation. Typical values for ΔV‡ are around −20 to −40 cm3/mol (34–36). However, even an extremely large value of −100 cm3/mol only changes the reaction rate by a factor of ≈2.5 at 300 atm (3,000 m depth) and 100°C. At 1,000 atm and 100°C, the change in rate increases to a factor of only ≈25. For positive ΔV‡ values the rates decrease by the same factor.
The presence of other compounds in the prebiotic ocean also may have had an affect on reaction rates. The deamination of cytosine to uracil, for example, has been shown to be catalyzed by sulfite (37, 38). Other nucleophiles may act similarly. Nucleophilic addition to the C5-C6 double bond also may increase the rate of hydrolysis of uracil. Therefore the stability of these compounds may be considerably shorter than our values and needs to be corrected for these reactions.
Minerals and mineral surfaces also may affect the rates of decomposition of these compounds. However, it is difficult to predict whether a given mineral or minerals will stabilize on destabilize these compounds. The hydrolysis of adenine to hypoxanthine, for example, has been shown to dramatically increase in the presence of Cu2+-montmorillonite (39).
Purines and pyrimidines have been found in sediment cores from both ocean and lake basins (40–42), some dating back as far as 25 × 106 yr (43). However, these measurements are complicated by uncertainties in temperature, microbial activity, and sedimentation rates. The presence of cytosine in 25 × 106-yr-old sediments, however, is surprising and may be the result of contamination or decomposition under anhydrous conditions (43, 44).
The Instability of Cytosine.
The instability of C has long been recognized (27, 28, 45–48). For example, it is not found in the Murchison meteorite, whereas A, U, G, xanthine, and hypoxanthine are (49, 50). At 0°C, the half-life for the decomposition of C is 17,000 yr, which is still very short on the geologic time scale.
The instability of cytosine, even at 0°C, raises a serious question of whether it would have been a suitable base for the first genetic material. A steady-state calculation for the relative amounts of C and U in the prebiotic ocean can be done if we assume that all U is produced from the hydrolysis of C (k1 = 4.1 × 10−5 yr−1 at 0°C), and that the only other loss channels are the hydrolysis of U (k2= 1.9 × 10−9 yr−1 at 0°C) and the destruction of organic compounds by passage through the 350°C hydrothermal vents (k3 = 6.9 × 10−8 yr−1 at 0°C). At steady state:
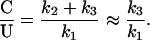 |
At 0°C, this gives a C/U ratio of ≈1/600. This ratio is so high that the enolic form of U(0.01% of U), which binds with G (51), would be comparable (1/15) to the concentration of C. 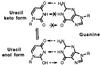
This would require proofreading corrections that would not be likely for the first replicative system. If the rate though the vents was 10 times faster in the past, because of higher heat flow, the ratio would be 1/60, which still would be highly error prone.
One solution to this problem with fidelity may be that the origin of life took place very rapidly after a sterilization event. Such an event would reset the C/U ratio, allowing for a favorable ratio in the first ≈105 yr, although at the expense of low initial concentrations of C. This suggests that if life arose using C, and its base pair G, it must have done so very quickly (<100,000 yr), shortening the previous proposal (31) that life may have evolved in as little as 107 yr.
Another possibility is that some concentration mechanisms were available that raised the concentration of C but not U, or that C may have been stabilized by the incorporation into a hydrogen-bonded polymer structure. This is the case for cytosine in double-stranded DNA, which hydrolyzes to uracil ≈140 times slower than C in single-stranded DNA (52). It is also possible that the early ocean was largely frozen with the aqueous phase at the NaCl-H2O eutectic (−21°C). This would increase the half-life of C to 8.0 × 105 yr. However, in the absence of any concentration or protective mechanisms, the instability of C suggests that it may not have been used in the first genetic molecule, and this raises questions as to whether G was also absent.
Alternatives To GC.
One possibility is that the first genetic material contained a two-letter code consisting of A and U, A and T (53), or A and I (inosine) (54). Although a genetic system based on a two-letter code, in principle, is possible, and attractive in the sense that it is simple, such a system would have more limited coding capabilities than a 2-bp system (53). In addition, a genetic material coded by only two bases would be very sticky, in that it would easily hydrogen-bond to itself. This could lead to problems in early replication.
Do The Purine And Pyrimidine Biosynthetic Pathways Suggest That GC Came After AU?
A metabolic argument can be given that guanine and cytosine were incorporated into the genetic material after adenine and uracil (Fig. 4). The biosynthesis of purine nucleoside monophosphates is from aminoimidazole carboxamide riboside monophosphate. The present pathways are indicated in Fig. 4 by solid arrows, and the dotted arrow shows a one-step pathway, instead of the present three-step pathway for the synthesis of GMP. A two-step pathway through XMP is also possible. The shorter route would be expected if GMP, or guanine, were used when the pathways first developed.
Figure 4.

The biosynthesis of purine nucleoside monophosphates from aminoimidazole carboxamide riboside monophosphate. The present pathways are indicated by solid arrows. The dotted arrow shows a one-step pathway for the synthesis of GMP, instead of the present three-step pathway. A two-step pathway through XMP is also possible. The shorter routes would be expected if GMP, or guanine, was used when the pathways developed.
Although this type of metabolic argument frequently is used to infer historical biochemical sequences, these arguments are weak in that the origin of the metabolic pathways may have been considerably later than the first use of these bases in the genetic material.
Alternative Bases.
If the first genetic material contained a four-base code, then the simplest solution to the instability of C is to use a modified C. 5-Methylcytosine (5-MeC) and 5-hydroxymethylcytosine (5-HMC) were found to be less stable than C at pH 7 and 100°C (t1/2 5-MeC = 9 days, t1/2 5-HMC = 13 days). N4-methylcytosine is estimated to have t1/2 = ≈38 days at 100°C based on the rate of hydrolysis of N4-methyldeoxycitidine (55).
Another possibility is that the first genetic material contained a base pair other than GC. A number of alternative base pairs have been proposed. These include isoguanine and isocytosine (53, 56), diaminopurine and U (56), pseudo-diaminopyrimidine (ribose bound to C5 rather than N1) and xanthine (56), A and urazole (1,2,4-triazole-3,5-dione) (57). An even more extreme alternative is suggested by an all-purine helix (54, 58, 59). Eschenmoser and Loewenthal (60) also have shown strong purine-purine binding interactions in their homo-DNA, a DNA analog.
On the basis of stability we can say that xanthine (t1/2 = 0.4 yr at 100°C, and 0.6 × 106 yr at 0°C), and diaminopurine (t1/2 = 2 yr at 100°C) are about as stable as A or G. Hypoxanthine (t1/2 = 12 days at 100°C, and 5,000 yr at 0°C) and isoguanine (t1/2 = 19 days at 100°C) are about as stable as C. The alternative pyrimidines isocytosine (t1/2 = 21 days at 100°C; ref. 61) and diaminopyrimidine (t1/2 = 42 days at 100°C, and 40,000 yr at 0°C) hydrolyze at about the same rate as cytosine. Therefore to get greater stability it may be necessary to use bases other than these pyrimidines (56, 57) or to use an all-purine helix (excluding hypoxanthine and isoguanine). In addition, the instability of all of the alternatives measured (t1/2 < 1 yr at 100°C) indicates that these compounds also would be excluded from use in a high-temperature origin of life.
Is There a Role for High Temperatures?
We are not suggesting that short-term, high-temperature processes (≈100°C) such as those that may have occurred in lagoons or on drying beaches did not play a role in the origin of life, but that the temperature of most of the Earth could not have been much above 0°C. However, even small portions of the Earth at high temperatures can lead to the rapid overall decomposition of organic compounds. For example, if 5% of the ocean is at 100°C and the remainder at 0°C, then assuming rapid mixing, the overall half-life for the decomposition of A will be ≈20 yr instead of the ≈106 yr at 0°C.
In one scenario, organic compounds would be stored at low temperatures (e.g., 0°C) and may be brought into higher (≈100°C) temperature regions (i.e., hot rocks, drying lagoons, low-temperature hydrothermal vents) for brief periods of time (<10 yr). Areas of extreme temperature, such as the hydrothermal vents (350°C), may be excluded from this view because of the very rapid rate of decomposition at these temperatures.
Conclusions.
Most atmospheric models generally predict a warm early Earth with high levels of CO2 or other greenhouse gases. In the absence of greenhouse warming, however, the Earth’s oceans would have been frozen because of a 30% less luminous sun (62). Our kinetic data on the stability of the nucleobases indicate that a cold or frozen early Earth would be more favorable for the accumulation of the nucleobases and therefore for the origin of life. An early frozen Earth may have been melted numerous times as a result of a large meteor or comet impacts (63). However, very large impactors could boil the Earth’s oceans. The rates of hydrolysis at 100°C, for all of the nucleobases measured, suggest that an ocean-boiling impact event would completely decompose the nucleobases in addition to a number of other biologically important compounds. This would require the whole prebiotic process to begin again. Ocean-boiling impacts therefore are more damaging to prebiotic chemistry than to an early biosphere (64–66), where the survival of a single organism (e.g., in a crustal environment) would be sufficient to reestablish the entire ecosystem.
Other stability problems also point to a low-temperature origin of life and early evolution in the pre-RNA and RNA world. These include the stability of ribose (67), the decomposition of nucleosides (28, 68), and the hydrolysis of the phosphodiester bonds of RNA (23). Similar stability considerations would apply to any alternative pre-RNA backbone, e.g., peptide nucleic acids. All of these factors point to a low-temperature accumulation of organic compounds on the primitive Earth and a low-temperature origin of life. Therefore, atmospheric models suggesting a cool early Earth (≈ 0°C) rather than a warm one (12, 13) need to be considered.
Acknowledgments
We thank Jeff Bada, Antonio Lazcano, and Jim Lyons for their helpful comments. This work was supported by the National Aeronautics and Space Administration Specialized Center of Research and Training in Exobiology (NSCORT).
References
- 1.Woese C R. Microbiol Rev. 1987;51:221–271. doi: 10.1128/mr.51.2.221-271.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Russell M J, Hall A J, Cairns-Smith A G, Braterman P S. Nature (London) 1988;336:117. [Google Scholar]
- 3.Woese C R, Wächtershäuser G. In: Palaeobiology: A Synthesis. Briggs D E G, Crowther P R, editors. Oxford: Blackwell; 1990. pp. 3–9. [Google Scholar]
- 4.Pace N R. Cell. 1991;65:531–533. doi: 10.1016/0092-8674(91)90082-a. [DOI] [PubMed] [Google Scholar]
- 5.Schwartzman D, McMenamin M, Volk T. Bioscience. 1993;43:390–393. [PubMed] [Google Scholar]
- 6.Russell M J, Daniel R M, Hall A J, Sherringham J A. J Mol Evol. 1994;39:231–243. [Google Scholar]
- 7.Stetter K O. In: Evolution of Hydrothermal Ecosystems on Earth (and Mars?) Brock G R, Goode J A, editors. Chichester, U.K.: Wiley; 1996. pp. 1–10. [Google Scholar]
- 8.Miller S L, Lazcano A. J Mol Evol. 1995;41:689–692. doi: 10.1007/BF00173146. [DOI] [PubMed] [Google Scholar]
- 9.Doolittle R F. Proc Natl Acad Sci USA. 1995;92:2421–2423. doi: 10.1073/pnas.92.7.2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Forterre P, Confalonieri F, Charbonnier F, Duguet M. Origins Life Evol Biosphere. 1995;25:235–249. doi: 10.1007/BF01581587. [DOI] [PubMed] [Google Scholar]
- 11.Gogarten-Boekels M, Hilario E, Gogarten J P. Origins Life Evol Biosphere. 1995;25:251–264. doi: 10.1007/BF01581588. [DOI] [PubMed] [Google Scholar]
- 12.Kasting J F, Ackerman T P. Science. 1986;234:1383–1385. doi: 10.1126/science.11539665. [DOI] [PubMed] [Google Scholar]
- 13.Kasting J F. Science. 1993;259:920–926. doi: 10.1126/science.11536547. [DOI] [PubMed] [Google Scholar]
- 14.Corliss, J. B., Baross, J. A. & Hoffman, S. E. (1981) Oceanologica Acta 4, Suppl., 59–69.
- 15.Shock E L. Origins Life Evol Biosphere. 1990;20:331–367. [Google Scholar]
- 16.Holm N G. Origins Life Evol Biosphere. 1992;22:5–14. [Google Scholar]
- 17.Shock E L. In: Evolution of Hydrothermal Ecosystems on Earth (and Mars?) Brock G R, Goode J A, editors. Chichester, U.K.: Wiley; 1996. pp. 40–60. [Google Scholar]
- 18.Russell M J, Hall A J. J Geol Soc (London) 1997;154:377–402. doi: 10.1144/gsjgs.154.3.0377. [DOI] [PubMed] [Google Scholar]
- 19.Bernhardt G, Lüdemann H D, Jaenicke R, König H, Stetter K O. Naturwissenschaften. 1984;71:583–586. [Google Scholar]
- 20.White R H. Nature (London) 1984;310:430–432. doi: 10.1038/310430a0. [DOI] [PubMed] [Google Scholar]
- 21.Miller S L, Bada J L. Nature (London) 1988;334:609–611. doi: 10.1038/334609a0. [DOI] [PubMed] [Google Scholar]
- 22.Bada J L, Miller S L, Zhao M. Origins Life Evol Biosphere. 1995;25:111–118. doi: 10.1007/BF01581577. [DOI] [PubMed] [Google Scholar]
- 23.Lindahl T. Nature (London) 1993;362:709–715. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- 24.Robinson R A, Stokes R H. Electrolyte Solutions. London: Butterworth; 1959. pp. 517–540. [Google Scholar]
- 25.Shapiro R. Origins Life Evol Biosphere. 1995;25:83–98. doi: 10.1007/BF01581575. [DOI] [PubMed] [Google Scholar]
- 26.Frick L, Mac Neela J P, Wolfenden R. Bioorganic Chem. 1987;15:100–108. [Google Scholar]
- 27.Shapiro R. Origins Life Evol Biosphere. 1996;26:238–239. [Google Scholar]
- 28.Garret E R, Tsau J. J Pharmacol Sci. 1972;61:1052–1061. doi: 10.1002/jps.2600610703. [DOI] [PubMed] [Google Scholar]
- 29.Edmond J M, Von Damm K L, McDuff R E, Measures C I. Nature (London) 1982;297:187–191. [Google Scholar]
- 30.Stribling R, Miller S L. Origins Life Evol Biosphere. 1987;17:261–273. doi: 10.1007/BF02386466. [DOI] [PubMed] [Google Scholar]
- 31.Lazcano A, Miller S L. J Mol Evol. 1994;39:546–554. doi: 10.1007/BF00160399. [DOI] [PubMed] [Google Scholar]
- 32.Sanchez R, Ferris J, Orgel L E. J Mol Biol. 1967;30:223–253. [PubMed] [Google Scholar]
- 33.Sanchez R, Ferris J, Orgel L E. Science. 1966;153:72–73. doi: 10.1126/science.153.3731.72. [DOI] [PubMed] [Google Scholar]
- 34.Notari R E, Chin M L, Cardoni A. J Pharmacol Sci. 1970;59:28–32. doi: 10.1002/jps.2600590103. [DOI] [PubMed] [Google Scholar]
- 35.Taniguchi Y. In: High Pressure Chemical Synthesis. Jurczak J, Baranowski B, editors. Amsterdam: Elsevier; 1989. pp. 349–373. [Google Scholar]
- 36.Asano T. In: Organic Synthesis at High Pressure. Matsumoto K, Acheson R M, editors. New York: Wiley Interscience; 1990. pp. 7–75. [Google Scholar]
- 37.Shapiro R, Servis R E, Welcher M. J Am Chem Soc. 1970;92:422–424. [Google Scholar]
- 38.Hayatsu H. Prog Nucleic Acid Res Mol Biol. 1976;16:75–124. doi: 10.1016/s0079-6603(08)60756-4. [DOI] [PubMed] [Google Scholar]
- 39.Stras̆ak M, S̆ers̆en̆ F. Naturwissenschaften. 1991;78:121–122. [Google Scholar]
- 40.Shimoyama A, Hagishita S, Harada K. Geochem J. 1988;22:143–148. [Google Scholar]
- 41.Dungworth G, Thijssen M, Zuurveld J, Van Der Velden W, Schwartz A W. Chem Geol. 1977;19:295–308. [Google Scholar]
- 42.Van Der Velden W, Schwartz A W. Chem Geol. 1976;18:273–284. [Google Scholar]
- 43.Rosenberg E. Science. 1964;146:1680–1681. doi: 10.1126/science.146.3652.1680. [DOI] [PubMed] [Google Scholar]
- 44.Milton A, Rosenberg E. Geochim Cosmochim Acta. 1964;28:1953–1959. [Google Scholar]
- 45.Shapiro R, Klein R S. Biochemistry. 1966;5:2358–2362. doi: 10.1021/bi00871a026. [DOI] [PubMed] [Google Scholar]
- 46.Ferris J P, Sanchez R A, Orgel L E. J Mol Biol. 1968;33:693–704. doi: 10.1016/0022-2836(68)90314-8. [DOI] [PubMed] [Google Scholar]
- 47.Lindahl T, Nyberg B. Biochemistry. 1974;13:3405–3410. doi: 10.1021/bi00713a035. [DOI] [PubMed] [Google Scholar]
- 48.Miller S L, Orgel L E. The Origins of Life on Earth. Englewood Cliffs, NJ: Prentice Hall; 1974. pp. 123–124. [Google Scholar]
- 49.Stoks P G, Schwartz A W. Nature (London) 1979;202:709–710. [Google Scholar]
- 50.Stoks P G, Schwartz A W. Geochim Cosmochim Acta. 1981;45:563–569. [Google Scholar]
- 51.Saenger W. Principles of Nucleic Acid Structure. New York: Springer; 1984. pp. 110–115. [Google Scholar]
- 52.Frederico L A, Kunkel T A, Shaw B R. Biochemistry. 1990;29:2532–2537. doi: 10.1021/bi00462a015. [DOI] [PubMed] [Google Scholar]
- 53.Rich A. In: Horizons in Biochemistry. Kasha M, Pullman B, editors. New York: Academic; 1962. pp. 103–126. [Google Scholar]
- 54.Crick F H C. J Mol Biol. 1968;38:367–379. doi: 10.1016/0022-2836(68)90392-6. [DOI] [PubMed] [Google Scholar]
- 55.Ehrlich M, Norris K F, Wang Y-H, Kuo K C, Gehrke C W. Biosci Rep. 1986;6:387–393. doi: 10.1007/BF01116426. [DOI] [PubMed] [Google Scholar]
- 56.Piccirilli J A, Krauch T, Moroney S E, Benner S A. Nature (London) 1990;343:33–37. doi: 10.1038/343033a0. [DOI] [PubMed] [Google Scholar]
- 57.Kolb V M, Dworkin J P, Miller S L. J Mol Evol. 1994;38:549–557. doi: 10.1007/BF00175873. [DOI] [PubMed] [Google Scholar]
- 58.Wächtershäuser G. Proc Natl Acad Sci USA. 1988;85:1134–1135. doi: 10.1073/pnas.85.4.1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zubay G. Chemtracts. 1991;2:439–449. [Google Scholar]
- 60.Eschenmoser A, Loewenthal E. Chem Soc Rev. 1992;21:1–16. [Google Scholar]
- 61.Robertson M P, Levy M, Miller S L. J Mol Evol. 1996;43:543–550. doi: 10.1007/BF02202102. [DOI] [PubMed] [Google Scholar]
- 62.Sagan C, Mullen G. Science. 1972;177:56–56. doi: 10.1126/science.177.4043.52. [DOI] [PubMed] [Google Scholar]
- 63.Bada J, Bigham C, Miller S L. Proc Natl Acad Sci USA. 1994;91:1248–1250. doi: 10.1073/pnas.91.4.1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Maher K A, Stevenson D J. Nature (London) 1988;331:612–614. doi: 10.1038/331612a0. [DOI] [PubMed] [Google Scholar]
- 65.Sleep N H, Zahnle K J, Kasting J F, Morowitz H J. Nature (London) 1989;342:139–142. doi: 10.1038/342139a0. [DOI] [PubMed] [Google Scholar]
- 66.Zahnle K J, Sleep N H. In: Comets and the Origin and Evolution of Life. Thomas P J, Chyba C F, McKay C P, editors. New York: Springer; 1997. pp. 175–208. [Google Scholar]
- 67.Larralde R, Robertson M P, Miller S L. Proc Natl Acad Sci USA. 1995;92:8158–8160. doi: 10.1073/pnas.92.18.8158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Garrett E R, Mehta P J. J Am Chem Soc. 1972;94:8542–8547. doi: 10.1021/ja00779a041. [DOI] [PubMed] [Google Scholar]