

Abstract
Primase and helicase activities of bacteriophage T7 are present in a single polypeptide coded by gene 4. Because the amino terminal region of the gene 4 protein contributes to primase activity, we constructed a truncated gene 4 encoding the N-terminal 271-aa residues. The truncated protein, purified from cells overexpressing the protein, is a dimer in solution; the full-length protein is a hexamer. Although the fragment is devoid of dTTPase and helicase activities, it catalyzes template-directed synthesis of di-, tri-, and tetranucleotides. The rates for tetraribonucleotide synthesis and for dinucleotide extension on a 20-nucleotide template are similar for the full-length and truncated proteins. However, the activity of the primase fragment is unaffected by dTTP whereas the primase activity of the full-length protein is stimulated >14-fold. The primase fragment is defective in the interaction with T7 DNA polymerase in that primer synthesis cannot be coupled to DNA synthesis.
The replisome of bacteriophage T7 is composed of relatively few proteins. The multifunctional gene 4 protein encoded by the phage exemplifies this economical use of genetic information. Gene 4 encodes two collinear proteins from separate in-frame translational start sites, a 63-kDa and a 56-kDa protein. The smaller protein translocates 5′-to-3′ on single-stranded DNA (ssDNA) and unwinds duplex DNA (helicase activity) in a reaction coupled to the hydrolysis of a nucleoside triphosphate (1–3). The larger 63-kDa protein has all of the activities found in the 56-kDa protein and, in addition, catalyzes the template-directed synthesis of oligoribonucleotides (primase activity) (4–6) for use as primers by T7 DNA polymerase (7, 8). The N-terminal 63-aa residues not present in the 56-kDa protein contain a cys4 zinc finger required for interaction with primase recognition sites (9–11). The presence of helicase and primase activities in the 63-kDa protein simplifies reconstruction of the T7 replisome (12), but the competitive effects of multiple activities have complicated the characterization of the DNA primase.
The identification of domains within the 63-kDa gene 4 protein is one approach to obtaining a primase protein without helicase activity. The zinc finger essential for primase activity is located in the unique 63-residue N terminus of the protein, but it alone is not sufficient for primase activity. The 63-aa fragment is devoid of primase activity, and the 56-kDa protein catalyzes the synthesis of random diribonucleotides (9). The nucleotide binding site essential for helicase binding to ssDNA and for translocation is located near Lys-318 in the C-terminal half of the protein (13, 14). Other segments essential for functional helicase, one for hexamer formation and one for interaction with the polymerase, also are located near the C terminus. Limited proteolysis of the 63-kDa protein results in three major cleavages (15, 16). Using endoproteinase Glu-C, one cleavage, at Glu-52, releases a small N-terminal fragment containing the zinc finger, and a second cleavage releases a 3-kDa fragment from the C terminus (15). A third cleavage, at Glu-219, gives rise to two large fragments, of 20 and 33 kDa. On the basis of this cleavage site, the helicase domain has been assigned to the two C-terminal fragments (15). Likewise, based on sequence homology with other primases (17), the primase domain can be assigned to the large N-terminal fragment plus the small zinc finger fragment.
If the helicase and the primase reside in these separate domains, then the corresponding truncations of gene 4 should produce monofunctional helicase and primase proteins. Bird et al. (15) have confirmed this prediction by showing that the C-terminal domain is an active helicase. This helicase fragment can be expressed from a plasmid containing gene 4 but lacking the region predicted to encode the primase (15). We have constructed a plasmid encoding the N-terminal 271-aa fragment corresponding to the primase domain, after elimination the internal start codon for the 56-kDa gene 4 protein (18, 19), and we describe the properties of a gene 4 fragment lacking helicase activity but retaining primase activity.
MATERIALS AND METHODS
Reagents and Strains.
Oligonucleotides were obtained from Integrated DNA Technologies (Corralville, IA), and M13 ssDNA was from New England Biolabs. The 56-kDa and 63-kDa gene 4G64 (13) proteins and the T7 gene 2.5 protein (20) have been described. The 63-kDa gene 4G64 protein has a glycine substituted for methionine to eliminate the start site of the 56-kDa gene 4 protein but is indistinguishable from the wild-type protein (21). T7 DNA polymerase was provided by S. Tabor (Harvard Medical School). Escherichia coli DH5α was obtained from GIBCO/BRL, and strain BL21(DE3) was obtained from Novagen. Plasmid pGP4-G64S10 has been described (13).
Mutagenesis of T7 Gene 4.
Oligonucleotide primers g4R272Bam (5′ GCG CGC GGA TCC TCA TCA TAA CGA AAG AGC CGA TAC-3′) and gp4Nde 5′-AGA TAT ACA TAT GGA CAA TTC GC-3′ were used in the PCR to attach NdeI and BamHI restriction sites and to amplify T7 gene 4 from the plasmid pGP4-G64S10 (13). The 950-bp product was purified from an agarose gel, was digested with NdeI and BamHI, was purified, and was ligated into the NdeI and BamHI sites of pET24a plasmid (Novagen). In the resulting plasmid, pETg4P, the truncated gene 4 was under control of a T7(lac) promoter. The plasmid pETg4P was used to transform strain DH5α for sequencing and strain Bl21(DE3), for protein expression.
Purification of the Primase Fragment.
Colonies of strain Bl21(DE3) containing pETg4P were inoculated into Luria–Bertani medium containing 60 μg/ml Kanamycin. At OD600 of 0.5, isopropyl β-d-thiogalactopyranoside was added to a final concentration of 1 mM, and the cells were grown at 37°C for 3 hr. The induced cells were harvested, washed, and collected by centrifugation. After the cell paste was suspended in Buffer A (50 mM Hepes, pH 7.5/1 mM EDTA/1 mM DTT), the cells were disrupted with sonic irradiation. The extract was clarified by centrifugation at 15,000 × g for 30 min, and the supernatant was designated Fraction I.
Fraction I was diluted to 10 mg/ml, and a 10% solution of streptomycin sulfate was added to a concentration of 1%. After centrifugation at 15,000 × g for 30 min, the supernatant contained the primase fragment (Fraction II).
Fraction II (19 ml) was loaded onto a column (4.9 cm2 × 10 cm) of DEAE Sepharose and washed with 400 ml of 50 mM Hepes (pH 7.5) and 0.1 mM DTT (Buffer B). The primase fragment was eluted with Buffer B containing a gradient of NaCl from 0 to 1 M. The fractions containing the primase fragment were combined (24 ml), and EDTA was added to a concentration of 1 mM. Ammonium sulfate (12 g) was added with stirring, and the precipitate was collected by centrifugation and dissolved in 5.5 ml of Buffer A (Fraction III).
Fraction IV (5 ml) was loaded onto a Sephacryl S-200HR column (4.9 cm2 × 50 cm) and was eluted with 50 mM Hepes, 1 mM EDTA, 0.1 mM DTT, and 50 mM NaCl. The fractions containing the primase fragment (38 ml) were combined (Fraction IV). Fraction IV was applied to a 5-ml HiTrap Blue Affinity column. The column was washed with 100 ml of Buffer A, 100 ml Buffer A containing 100 mM NaCl, and 100 ml of Buffer A containing 200 mM NaCl. The primase fragment was eluted with Buffer A containing 700 mM NaCl. The fractions containing the primase fragment (75 ml) were combined, and 32 g of ammonium sulfate were added with stirring. The precipitate was collected, was dissolved in 5 ml of Buffer A, and was dialyzed against Buffer A. Glycerol was added to a concentration of 50% (vol/vol), and the primase fragment (Fraction V) was stored at −40°C.
PAGE.
PAGE was performed with 10% polyacrylamide gels (22) by using a Mini-PROTEAN II Electrophoresis system (Bio-Rad). For denaturing electrophoresis, 0.1% SDS was included in the loading dye and electrophoresis buffer, and samples were boiled for 5 min before loading.
Nuclotide Hydrolysis Assay.
The hydrolysis of nucleoside triphosphates was monitored by using thin-layer chromatography as described (23). Products were measured by using a Fuji BAS1000 Bio-imaging Analyzer.
Helicase Assay.
Helicase activity of the T7 gene 4 proteins was measured by following the dissociation of [5′-32P]-labeled oligonucleotide annealed to M13 ssDNA. The 36-base oligonucleotide 5′-GGATCCGGGAATTCGTAATCGCCTAAGGCTAACGG-3′ contains an unpaired 3′-terminus because the 16 nucleotides on the 3′-end are not complementary to M13 DNA. Reactions were initiated by the addition of gene 4 protein or primase fragment, and the amounts of oligonucleotide displaced were determined (23).
Strand Displacement DNA Synthesis.
Helicase activity also can be measured by the ability of gene 4 proteins to stimulate the activity of T7 DNA polymerase on duplex templates (24). We have used a mini-circle containing a preformed replication fork (provided by J. Lee, Harvard Medical School) that consists of a duplex 70-nt circle containing a single 3′-hydroxyl terminus and a 5′-ssDNA tail. Reactions (20 μl) were preformed as described (25, 26) with either 100 nM 63-kDa gene 4 protein or primase fragment. Radioactive nucleotides incorporated into DNA were measured as described (8).
Oligoribonucleotide Synthesis Assay.
Oligoribonucleotide synthesis was measured as described (27, 28) in reactions containing either 100 nM 63-kDa gene 4 protein or primase fragment. Standard 10-μl reactions contained either M13 ssDNA or a 20-base oligonucleotide (5′-GGGTCACCGAGATCCTTCAG-3′) with 100 nM of either the 63-kDa gene 4 protein or primase fragment.
Oligoribonucleotide Extension Assay.
Oligonucleotide extension reactions using either 100 nM 63-kDa gene 4 protein or primase fragment were performed as described for the oligoribonucleotide synthesis assay except that AC dinucleotide was substituted for of ATP as indicated (29). When present, dTTP was included at 1 mM.
RNA-Primed DNA Synthesis Assay.
Oligoribonucleotide synthesis catalyzed by the gene 4 protein was examined by measuring the ability of the oligonucleotides to serve as primers for T7 DNA polymerase (30). Radiolabeled nucleotides incorporated into DNA were measured as described (8).
RESULTS
A Plasmid Encoding the Primase Fragment.
Ilyina et al. (17) have shown that there is significant homology between the N-terminal residues of the 63-kDa gene 4 protein and other prokaryotic DNA primases. Based on amino acid alignments, the helicase activity should be contained within the region between residues 272 and 566. Consequently, we have constructed a gene 4 encoding two stop codons after the codon for leucine residue 271. The primase domain was amplified by using the PCR from plasmid pGP4-G64S10 (13), which lacks the initiation codon for the 56-kDa gene 4 protein. The upstream primer introduced an NdeI site at the 5′-end of gene 4. The downstream primer changed codon 272 to a stop (TGA) codon and introduced a BamHI site at the 3′-end of the truncated gene. After digestion with NdeI and BamHI, the PCR product was ligated into the vector pET24a and was placed under control of a T7(lac) promoter to create the plasmid pETG4-PF.
Purification and Physical Properties of the Primase Fragment.
Over-production of the primase fragment was achieved by induction of E. coli cells harboring plasmid pETG4-PF encoding the primase fragment under the control of the T7(lac) promoter. After growth and lysis of the induced cells, the primase fragment was purified to apparent homogeneity (Materials and Methods). Purification was followed by analysis of proteins on a denaturing polyacrylamide gel (Fig. 1A). The primase fragment represents 40% of the protein present in the lysate (Fraction I) as shown in Fig. 1A, lane 2. After removal of DNA by precipitation with streptomycin sulfate (Fig. 1A, lane 3), the protein was fractionated by chromatography on DEAE-Sepharose (Fig. 1A, lane 4), followed by gel filtration (Fig. 1A, lane 5) and finally by chromatography on Hitrap Blue affinity resin (Fig. 1A, lane 6). Homogeneous protein (≈20 mg/l of cell culture) was obtained.
Figure 1.

SDS and native PAGE of the primase fragment. (A) Proteins were separated on a 10% polyacrylamide gel in the presence of 1% SDS. Lanes: 1, 2 μg each of protein standards; 2, 20 μg of Fraction I, a cleared lysate of BL21(DE3) cells containing the plasmid pETg4-PF that were induced with isopropyl β-d-thiogalactopyranoside for 3 hr; 3, 20 μg of Fraction II, the supernatant after the precipitation of nucleic acids with streptomycin sulfate; 4, 5 μg of Fraction III, the DEAE-Sepharose chromatography pool; 5, 5 μg of Fraction IV, the pool from the Sephacryl S200HR column; 6, 5 μg of Fraction V, the pure primase fragment after chromatography on a Hi-trap Blue affinity column. (B) Proteins were analyzed on a native polyacrylamide gel run under nondenaturing conditions. Lanes: 1, 2 μg of primase fragment (Fraction V); 2, 2 μg of the 63-kDa gene 4 protein. The positions of the same protein standards shown in A are indicated.
There are several noteworthy differences between the expression and purification of the primase fragment and full-length gene 4 protein. First, whereas the primase fragment comprised 40% of the total protein, the full-length protein is expressed only to a maximum of 5–10% (21, 31). Second, the primase fragment is more soluble in extracts; more than half of the 63-kDa gene 4 protein is lost to inclusion bodies compared with <20% of the primase fragment. Third, the primase fragment does not purify together with DNA. This finding is not surprising because amino acid changes located in the helicase domain decrease the ability of the protein to bind DNA (13, 16). Finally, the primase fragment does not bind to an ATP agarose column, unlike the 63-kDa protein (6). This result is likely caused by the loss of the conserved nucleotide binding site in the helicase domain.
The purified primase fragment appears homogeneous on both a denaturing (Fig. 1A) and a nondenaturing (Fig. 1B) polyacrylamide gel. The primase fragment migrates as a single species with a mass of 60 kDa. Under the same conditions, the 63-kDa protein migrates as two species, a 63-kDa monomer and a 252-kDa hexamer. This result suggests that the primase fragment exists in solution only as a dimer. It was suggested that the gene 4 hexamer is formed from a combination of three dimers because gene 4 proteins containing amino acid substitutions for His475 or Asp-485 formed dimers but not hexamers (23).
Absence of Translocation and Helicase Activities.
Gene 4 protein catalyzes the ssDNA-dependent hydrolysis of dTTP (3, 4, 21, 32). The energy of hydrolysis is used to fuel the 5′-to-3′ translocation of the protein (5) and to unwind duplex DNA (2, 33, 34). The primase fragment has no detectable dTTPase activity whereas the full-length protein catalyzes the hydrolysis of dTTP at a rate of 1.4 μmol/min/mg (Fig. 2).
Figure 2.

Absence of dTTP hydrolysis by the primase fragment. Nucleotide hydrolysis assays were performed by using 5 mM [α-32P]dTTP as the substrate. Rates of nucleotide hydrolysis were determined for reactions containing various amounts of the 63-kDa gene 4 protein (•) or the primase fragment (○). Protein concentrations are reported in terms of protein monomers.
Although the lack of dTTPase ensures an absence of helicase activity, we also have examined helicase activity in two assays. We measured the ability of the primase fragment to displace an oligonucleotide annealed to M13 ssDNA (24). In direct comparison with 63-kDa gene 4 protein, no displacement of the oligonucleotide was observed, even at concentrations 50-fold in excess of that required for the full-length protein (data not shown). In addition, we have measured the helicase activity of the primase fragment indirectly in an assay in which T7 DNA polymerase activity depends on helicase activity to expose ssDNA template on dsDNA. The DNA template was a 70-nt circular oligonucleotide annealed with a linear 110-nt oligonucleotide to form a replication fork (25, 26). In the presence of full-length gene 4 protein, DNA polymerase catalyzed the polymerization of nucleotides on this duplex template at a rate of 20 to 40 pmol/min whereas no detectable (<0.5 pmol/min) synthesis was found with the primase fragment, even at a 100-fold higher protein concentration.
Template-Directed Oligonucleotide Synthesis.
On a ssDNA template containing a T7 primase recognition site, 5′-GTC-3′, the 63-kDa gene 4 protein catalyzes the synthesis of the dinucleotide pppAC from precursors ATP and CTP (28). Although essential for recognition, the cytosine in the recognition sequence is not copied into the primer. T7 primase extends these dinucleotides to tri- and tetranucleotides at the recognition sequences 5′-(G/T)GGTC-3′ and 5′-GTGTC-3′ (5, 35).
Like the full-length protein, the primase fragment catalyzes template-directed synthesis of oligoribonucleotides. In the absence of ssDNA, no synthesis of oligoribonucleotides is detected in assays containing ATP, [α-32P]CTP, and either the 63-kDa protein (Fig. 3, lane 1) or the primase fragment (Fig. 3, lane 2). When M13 ssDNA is present, both the 63-kDa protein (Fig. 3, lane 3) and the primase fragment (Fig. 3, lane 4) catalyze the synthesis of oligoribonucleotides. In all of the reactions, the same concentration of primase fragment or 63-kDa protein was used based on the mass of a monomer. It is important to note that both proteins synthesized similar amounts and similar lengths of oligonucleotides. By using a synthetic ssDNA template of defined sequence, we can identify these oligonucleotides. On a template containing the recognition sequence 5′-GGGTC-3′, both the 63-kDa protein (Fig. 3, lane 5) and the primase fragment (Fig. 3, lane 6) synthesize similar amounts of pppAC, pppACC, and pppACCC.
Figure 3.

Template-directed oligoribonucleotide synthesis. Incorporation of [α-32P]rCTP into oligoribonucleotides was analyzed in reactions containing ATP, CTP, and 100 nM of primase (monomer concentration). Products of reactions were separated on a 25% polyacrylamide gel containing 2 M urea and visualized by using autoradiography. Reactions analyzed in lanes 1 and 2 contained no DNA template and 63-kDa gene 4 protein and primase fragment, respectively. Lanes 3 and 4 show the products of reactions containing 0.1 mg/ml M13 DNA and either the 63-kDa gene 4 protein (lane 3) or the primase fragment (lane 4). A 20-base oligonucleotide with a primase recognition site at the 3′-end (5′-GGGTCX15-3′) was present at 10 μM in the reactions analyzed in lanes 5 and 6. The reaction analyzed in lane 5 contained the 63-kDa gene 4 protein, and the reaction in lane 6 contained the primase fragment. The identity and sequence of the oligoribonucleotide products is indicated.
Although the primase fragment and the full-length protein catalyze the synthesis of oligonucleotides at similar rates, significant differences are apparent when dTTP is added to the reaction mixture. dTTP increases the affinity of the gene 4 protein for ssDNA, and its hydrolysis provides the energy for helicase translocation. Consequently, the primase activity of the 63-kDa gene 4 protein is stimulated greatly by dTTP. Stimulation is greatest when long templates such as M13 DNA are used as templates because translocation increases the frequency with which the protein encounters primase recognition sites (21). To minimize the effect of translocation, we have examined the effect of dTTP on oligonucleotide synthesis by using a 20-base template containing a primase recognition site (Fig. 4). In reactions containing [α-32P]CTP and ATP, the addition of dTTP increased the rate of oligoribonucleotide synthesis by the 63-kDa protein up to 11-fold but had no effect on synthesis catalyzed by the primase fragment. In addition, the average length of the products in the reaction catalyzed by the 63-kDa protein increases on addition of dTTP. The synthesis of tetranucleotides is stimulated >22-fold in the presence of dTTP whereas the synthesis of dimers is increased only 4-fold (Fig. 4, Inset). The ratio of dimers:tetramers synthesized by the primase fragment is not altered by the addition of dTTP.
Figure 4.

Effect of dTTP on oligoribonucleotide synthesis. The products of oligonucleotide synthesis assays containing ATP, [α-32P]CTP, and 10 μM 20-base oligonucleotide (5′-GGGTCX15-3′) were separated on 25% polyacrylamide gel containing 2 M urea, and their amounts were measured by using a Fuji BAS1000 Bio-imaging Analyzer. The amounts of di-, tri-, and tetranucleotide synthesized were normalized assuming on labeled nucleotide per dinucleotide (pppAC), two labeled nucleotides per trinucleotide (pppACC), and three labeled nucleotides per tetranucleotide (pppACCC). The rates of oligonucleotide synthesis are shown for reactions catalyzed by the 63-kDa gene 4 protein (•) or the primase fragment (○). (Inset) The rates of dinucleotide (white), trinucleotide (gray), and tetranucleotide (black) synthesis by the 63-kDa gene 4 protein or the primase fragment in the presence and absence of 0.6 mM dTTP.
Because helicase translocation most likely plays a small role in the stimulation of oligoribonucleotide synthesis with short templates, the stimulation by dTTP likely results from an increased affinity for the template. We used a kinetic analysis to examine the affinity of the primase fragment for a 20-nt oligonucleotide template (5′-GGGTCX15-3′) in the presence and absence of dTTP. The effect of DNA template concentration on oligonucleotide synthesis is essentially identical for both proteins in the absence of dTTP (Fig. 5). In the absence of dTTP, a double-reciprocal analysis yields a Kapparent for the DNA of 5 ± 4 μM and 10 ± 8 μM for the 63-kDa gene 4 protein and the primase fragment, respectively. We conclude that the deletion of the helicase domain has no effect on the affinity of the primase for the template in the absence of dTTP. The 63-kDa protein is stimulated greatly by the presence of dTTP with a Kapparent for the DNA of 0.4 ± 0.3 μM, indicating a 12-fold increase in affinity.
Figure 5.

Effect template concentration on oligoribonucleotide synthesis. The products of oligonucleotide synthesis assays containing ATP and [α-32P]CTP were as described in Fig. 4. The rates of oligonucleotide synthesis were determined for reactions containing the indicated concentrations of 20-base oligonucleotide (3′-GGGTCX15) and either the primase fragment (○), the 63-kDa gene 4 protein (•), or the 63-kDa gene 4 protein in the presence of 1 mM dTTP (▴).
Dinucleotide Extension.
Although the primase fragment and the full-length enzyme have nearly identical kinetic properties with regard to oligoribonucleotide synthesis in the absence of dTTP, caution must be used in this comparison. The full-length protein functions as a hexamer whereas the primase fragment is a dimer. Likewise, the NTP hydrolysis site in the intact gene 4 protein, although preferring dTTP, can use ATP hydrolysis for translocation (4). Thus, the omission of dTTP may not eliminate binding of the 63-kDa protein to ssDNA.
In addition to catalyzing the de novo synthesis of oligoribonucleotides from NTPs, T7 DNA primase mediates the annealing of dinucleotides at recognition sites and catalyzes their extension (29). For example, the 63-kDa protein extends 5′-AC-3′ bound to the primase recognition site 5′-GGGTC-3′ on ssDNA to 5′-ACCC-3′ in the presence of CTP. By replacing ATP with AC, only CTP is required for extension. Because CTP is not a substrate to fuel helicase translocation (4), oligonucleotide synthesis catalyzed by the primase fragment can be compared more directly to that by the full-length protein.
We have measured the amounts of tri- plus tetra-nucleotides synthesized by extension at increasing amounts of AC in the presence of [α-32P]CTP but in the absence of ATP (Fig. 6). At each concentration of dinucleotide, the amount of dinucleotide extended by the primase fragment was only 2 to 3× the amount extended by the 63-kDa protein. The addition of dTTP to the full-length protein stimulated 20-fold the extension reaction, and it altered the ratio of the two products. In the reactions using the primase fragment described in Fig. 6, 0.73 tetranucleotides were produced for each trinucleotide. This value is slightly less processive than that obtained with the full-length protein, where the ratio of trinucleotide to tetranucleotide was 1.0. When dTTP was present with the full-length protein, 4.1 tetranucleotides were produced for each trinucleotide.
Figure 6.

Extension of dinucleotides. The products of dinucleotide extension assays containing [α-32P]CTP and 10 μM 20-base oligonucleotide (5′-GGGTCX15) were analyzed as described in Fig. 4. The relative amounts of tri- and tetranucleotide synthesized were normalized assuming one labeled nucleotide per two labeled nucleotides per trinucleotide (ACC) and three labeled nucleotides per tetranucleotide (ACCC). Rates of dinucleotide extension are reported at various dinucleotide (AC) concentrations for reactions containing the primase fragment (○), the 63-kDa gene 4 protein (•), or the 63-kDa gene 4 protein in the presence of 1 mM dTTP (▴).
Interaction with T7 DNA Polymerase.
The biological role of the oligonucleotides synthesized by gene 4 primase is to serve as primers for T7 DNA polymerase. In the presence of the 63-kDa protein, ATP, and CTP there is a marked stimulation of DNA synthesis catalyzed by T7 DNA polymerase on M13 ssDNA. The ability of the polymerase to use the oligonucleotides so effectively is mediated by an interaction of the gene 4 protein with the polymerase (25). In the absence of gene 4 protein, DNA polymerase does not use tetranucleotides efficiently (7, 36). The primase fragment, although clearly capable of synthesizing tetraribonucleotides, is unable to provide these as functional primers for the T7 DNA polymerase. At high concentrations of primase fragment (>500 nM), a small but detectable stimulation of T7 DNA polymerase was observed, albeit at a rate 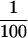 that of the observed with the full-length protein (Fig. 7). The preformed tetranucleotides synthesized by the primase fragment when purified are used efficiently as primers for DNA synthesis by the full-length primase and T7 DNA polymerase (data not shown).
that of the observed with the full-length protein (Fig. 7). The preformed tetranucleotides synthesized by the primase fragment when purified are used efficiently as primers for DNA synthesis by the full-length primase and T7 DNA polymerase (data not shown).
Figure 7.

Interaction between the primase fragment and T7 DNA polymerase. The reaction mixtures contained 12 μg/ml M13, 100 μM CTP, 100 μM ATP, and 300 μM each of [α-32P]dGTP, dATP, dCTP, and dTTP. After a preincubation at 30°C for 10 min, reactions were initiated by the addition of T7 DNA polymerase premixed with either the primase fragment or the 63-kDa gene 4 protein. Rates of incorporation of nucleotides into DNA are reported for reactions containing various amounts of the primase fragment (○) or the 63-kDa gene 4 protein (•).
DISCUSSION
The finding that gene 4 of bacteriophage T7 encodes both a helicase and a primase whereas these activities are usually encoded by separate genes in other organisms (17, 37) suggests that these activities reside in separate domains. Considerable indirect evidence has accumulated to support this hypothesis. First, gene 4 actually encodes two polypeptides, a 56- and a 63-kDa protein, the former arising from an internal translational initiation site (37). The 56-kDa protein has helicase activity but no primase activity (9). The 63-kDa protein is essential for T7 growth whereas the 56-kDa protein is not (18, 19).
Primase activity depends on the cys4 zinc finger located within the 63-aa residues that are unique to the full-length protein. However, the zinc finger itself is not sufficient for primase activity because the 56-kDa protein contains the active site for phosphodiester bond formation (9, 11). The fact that the 56-kDa gene 4 helicase contains the site for phosphodiester bond formation (9) suggests that the intact primase domain consists of the zinc finger plus an N-terminal portion of the 56-kDa protein. In support of this interpretation, electron micrographs of the full-length protein suggest a bilobal shape (38), and a C-terminal fragment has been isolated that has helicase activity (15). The primase fragment described here catalyzes template-directed oligonucleotide synthesis comparable to that of the 63-kDa protein.
The only significant difference between the primase activity of the full-length protein and the primase fragment occurs in the presence of dTTP. The full-length gene 4 protein binds ssDNA and translocates 5′ to 3′ along the DNA concomitant with the hydrolysis of dTTP. The binding of dTTP produces a conformational change in gene 4 protein (39) that affects both DNA binding and translocation. Both the binding and translocation of gene 4 protein can affect oligoribonucleotide synthesis. Binding to ssDNA, mediated through dTTP binding to the nucleoside binding site of the helicase domain, stimulates oligonucleotide synthesis by stabilizing the protein on ssDNA (22). The translocation activity provides an efficient mechanism by which the primase domain can be brought to primase recognition sites (5, 27). The fact that dTTP stimulates oligoribonucleotide synthesis on long DNA templates may be attributed primarily to increased helicase translocation. On oligonucleotides containing a recognition site, the stimulation of the 63-kDa protein by dTTP likely is caused by an increased affinity for ssDNA. Hence, in the presence of dTTP, the primase fragment and full-length protein have comparable activities at high DNA template concentrations whereas the activities differ greatly at low template concentrations.
Another noteworthy difference between the primase fragment and the full-length protein is their subunit composition. Wild-type gene 4 protein functions as a hexamer (23, 40) that surrounds the DNA (33, 38). In vitro the maximum helicase and primase activities are achieved only when a mixture of the two forms is present (6, 28, 31). We find that under conditions where the 63-kDa protein forms hexamers, the primase fragment forms dimers exclusively. This finding is not surprising because residue substitutions in the C-terminal helicase domain also disrupt the hexamer, resulting in a dimeric protein (23). These results suggest that the C-terminal helicase domain is required for one of the subunit interfaces in the hexamer and that a distinct subunit–subunit interaction is mediated by the N-terminal primase domain (23). In a similar manner, the dimeric T4 gp41 helicase assembles into hexamers on binding a nucleoside triphosphate (41).
The physical association of the helicase and primase activities results in more efficient primer synthesis. In systems in which the primase and helicase functions reside in separate proteins, the proteins form a complex. In E. coli, the dnaG-encoded DNA primase and the dnaB-encoded helicase form a complex (42), and, in phage T4, the gp41 protein (helicase) combines in a 6:1 complex with the T4 gp61 protein (primase) when bound to DNA and ATP (43–45). It was proposed that the T7 gene 4 evolved after a gene fusion of two genes that were the common ancestors of the current bacterial and phage primase and helicase genes (17). Our results support this scenario by showing that the primase function can be isolated independently. The physical linkage of the two activities permits the primase to access recognition sites more frequently because of increased affinity for ssDNA and translocation along ssDNA. We show that, although beneficial, neither dTTP hydrolysis nor translocation is necessary for primase activity. Furthermore, the primase fragment contains nucleotide and DNA binding sites distinct from sites necessary for helicase activity. This dissection of the helicase/primase complex provides support for the model proposed by Kusakabe et al. (22) that primer synthesis can occur at template sites distal to where the helicase domain is bound.
The linkage of the primase and helicase may be even more important to facilitate interactions between other replication proteins. The T7 DNA replication complex is composed of, in addition to the gene 4 helicase/primase, the product of T7 gene 2.5, a ssDNA binding protein; the product of gene 5, a DNA polymerase; and E. coli thioredoxin, a processivity factor. Together, the proteins function as the minimal complex necessary for the replication of both strands of a linear duplex DNA molecule. The primase fragment alone inefficiently primes DNA synthesis by T7 DNA polymerase. Hence, the crucial interprotein interaction between the gene 4 and 5 proteins likely is disrupted by the deletion of the helicase motif.
The prerequisite unwinding and separation of the two strands of DNA to produce ssDNA templates for the priming of DNA synthesis and subsequent copying by DNA polymerase makes the physical association of primase and helicase activities critical. T7 has devised an elegant solution to this coordination by combining the helicase and primase functions into a single protein molecule. The isolation, purification, and characterization of the primase fragment of the T7 gene 4 protein makes possible studies of the T7 primase in the absence of competitive nucleotide or DNA binding effects.
Acknowledgments
We are grateful to Benjamin B. Beauchamp, Daochun Kong, Stephen M. Notarnicola, and Stanley S. Tabor for providing purified proteins and plasmids. This work was supported by United States Public Health Service Grant AI-06045 and by American Cancer Society Grant NP-1Z to C.C.R. and American Cancer Society Post-doctoral Fellowship PF-4452 to D.N.F.
ABBREVIATION
- ssDNA
single-stranded DNA
References
- 1.Kolodner R, Richardson C C. Proc Natl Acad Sci USA. 1977;74:1525–1529. doi: 10.1073/pnas.74.4.1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Matson S W, Tabor S, Richardson C C. J Biol Chem. 1983;258:14017–14024. [PubMed] [Google Scholar]
- 3.Bernstein J A, Richardson C C. J Biol Chem. 1988;263:14891–148999. [PubMed] [Google Scholar]
- 4.Matson S W, Richardson C C. J Biol Chem. 1983;258:14009–14016. [PubMed] [Google Scholar]
- 5.Tabor S, Richardson C C. Proc Natl Acad Sci USA. 1981;78:205–209. doi: 10.1073/pnas.78.1.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bernstein J A, Richardson C C. J Biol Chem. 1989;264:13066–13073. [PubMed] [Google Scholar]
- 7.Scherzinger E, Lanka E, Hillenbrand G. Nucleic Acids Res. 1977;4:4151–4163. doi: 10.1093/nar/4.12.4151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nakai H, Richardson C C. J Biol Chem. 1986;261:15208–15216. [PubMed] [Google Scholar]
- 9.Bernstein J A, Richardson C C. Proc Natl Acad Sci USA. 1988;85:396–400. doi: 10.1073/pnas.85.2.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hine A V, Richardson C C. Proc Natl Acad Sci USA. 1994;91:12327–12331. doi: 10.1073/pnas.91.25.12327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kusakabe T, Richardson C. J Biol Chem. 1996;271:19563–19570. doi: 10.1074/jbc.271.32.19563. [DOI] [PubMed] [Google Scholar]
- 12.Debyser Z, Tabor S, Richardson C C. Cell. 1994;77:157–166. doi: 10.1016/0092-8674(94)90243-7. [DOI] [PubMed] [Google Scholar]
- 13.Notarnicola S M, Richardson C C. J Biol Chem. 1993;268:27198–27207. [PubMed] [Google Scholar]
- 14.Patel S S, Hingorani M M, Ng W M. Biochemistry. 1994;33:7857–7868. doi: 10.1021/bi00191a013. [DOI] [PubMed] [Google Scholar]
- 15.Bird L E, Hakansson K, Pan H, Wigley D B. Nucleic Acids Res. 1997;25:2620–2626. doi: 10.1093/nar/25.13.2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Washington M T, Rosenberg A H, Griffin K, Studier F W, Patel S S. J Biol Chem. 1996;271:26825–26834. doi: 10.1074/jbc.271.43.26825. [DOI] [PubMed] [Google Scholar]
- 17.Ilyina T V, Gorabalenya A E, Koonin E V. J Mol Evol. 1992;34:351–357. doi: 10.1007/BF00160243. [DOI] [PubMed] [Google Scholar]
- 18.Mendelman L V, Notarnicola S M, Richardson C C. Proc Natl Acad Sci USA. 1992;89:10638–10642. doi: 10.1073/pnas.89.22.10638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rosenberg A H, Patel S S, Johnson K A, Studier F W. J Biol Chem. 1992;267:15005–15012. [PubMed] [Google Scholar]
- 20.Kim Y T, Tabor S, Bortner C, Griffith J D, Richardson C C. J Biol Chem. 1992;267:15022–15031. [PubMed] [Google Scholar]
- 21.Mendelman L V, Notarnicola S M, Richardson C C. J Biol Chem. 1993;268:27208–27213. [PubMed] [Google Scholar]
- 22.Kusakabe T, Baradaran K, Lee J, Richardson C C. EMBO J. 1998;17:1542–1552. doi: 10.1093/emboj/17.5.1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Notarnicola S M, Park K, Griffith J D, Richardson C C. J Biol Chem. 1995;270:20215–20224. doi: 10.1074/jbc.270.34.20215. [DOI] [PubMed] [Google Scholar]
- 24.Lechner R L, Richardson C C. J Biol Chem. 1983;258:11185–11196. [PubMed] [Google Scholar]
- 25.Notarnicola S M, Mulcahy H L, Lee J, Richardson C C. J Biol Chem. 1997;272:18425–18433. doi: 10.1074/jbc.272.29.18425. [DOI] [PubMed] [Google Scholar]
- 26.Lee, J., Chastain, P. D., II, Kusakabe, T., Griffith, J. D. & Richardson, C. C. (1998) Mol. Cell, in press. [DOI] [PubMed]
- 27.Kusakabe T, Richardson C C. J Biol Chem. 1997;272:5943–5951. doi: 10.1074/jbc.272.9.5943. [DOI] [PubMed] [Google Scholar]
- 28.Mendelman L V, Richardson C C. J Biol Chem. 1991;266:23240–23250. [PubMed] [Google Scholar]
- 29.Kusakabe T, Richardson C C. J Biol Chem. 1997;272:12446–12453. doi: 10.1074/jbc.272.19.12446. [DOI] [PubMed] [Google Scholar]
- 30.Nakai H, Richardson C C. J Biol Chem. 1988;263:9831–9839. [PubMed] [Google Scholar]
- 31.Patel S S, Rosenberg A H, Studier F W, Johnson K A. J Biol Chem. 1992;267:15013–15021. [PubMed] [Google Scholar]
- 32.Hingorani M M, Washington M T, Moore K C, Patel S S. Proc Natl Acad Sci USA. 1997;94:5012–5017. doi: 10.1073/pnas.94.10.5012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hacker K J, Johnson K A. Biochemistry. 1997;36:14080–14087. doi: 10.1021/bi971644v. [DOI] [PubMed] [Google Scholar]
- 34.Ahnert P, Patel S S. J Biol Chem. 1997;272:32267–32273. doi: 10.1074/jbc.272.51.32267. [DOI] [PubMed] [Google Scholar]
- 35.Romano L J, Richardson C C. J Biol Chem. 1979;254:10476–10482. [PubMed] [Google Scholar]
- 36.Romano L J, Richardson C C. J Biol Chem. 1979;254:10483–10489. [PubMed] [Google Scholar]
- 37.Dunn J J, Studier F W. J Mol Biol. 1983;166:477–535. doi: 10.1016/s0022-2836(83)80282-4. [DOI] [PubMed] [Google Scholar]
- 38.Egelman H H, Yu X, Wild R, Hingorani M M, Patel S S. Proc Natl Acad Sci USA. 1995;92:3869–3873. doi: 10.1073/pnas.92.9.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yong Y, Romano L J. J Biol Chem. 1995;270:24509–24517. doi: 10.1074/jbc.270.41.24509. [DOI] [PubMed] [Google Scholar]
- 40.Patel S S, Hingorani M M. J Biol Chem. 1993;268:10668–10675. [PubMed] [Google Scholar]
- 41.Dong F, Gogol E P, von Hippel P H. J Biol Chem. 1995;270:7462–7473. doi: 10.1074/jbc.270.13.7462. [DOI] [PubMed] [Google Scholar]
- 42.Arai K, Kornberg A. Proc Natl Acad Sci USA. 1979;76:4308–4312. doi: 10.1073/pnas.76.9.4308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu C C, Alberts B M. J Biol Chem. 1981;256:2821–2829. [PubMed] [Google Scholar]
- 44.Hinton D M, Nossal N G. J Biol Chem. 1987;262:10873–10878. [PubMed] [Google Scholar]
- 45.Dong F, von Hippel P H. J Biol Chem. 1996;271:19625–19631. doi: 10.1074/jbc.271.32.19625. [DOI] [PubMed] [Google Scholar]