

Abstract
In this study the authors compared the affect of vapor phase cigarette smoke (CS) versus cigarette smoke extract (CSE) on the lungs and upper airway of C57BL/6 mice. The authors found that CSE treatment significantly increased neutrophil influx (P < .001), baseline ciliary beat frequency (CBF) (P < .05), and protein kinase C activity compared to CS and controls. Isoproterenol increased CBF with CS exposure, but decreased CBF with CSE (P < .01). Isoproterenol increased protein kinase A (PKA) activity in all groups except CSE. CSE exposure induced inflammatory cell bronchiolitis. These data indicate that CSE exposure has differential effects on the lungs and tracheal epithelium compared to CS exposure.
Ciliary motility provides the driving force for mucociliary clearance. Normal function of the airway mucociliary clearance system prevents the attachment and colonization of pulmonary pathogens in the upper airways. Mucociliary dysfunction caused by impaired ciliary motility results in mucus accumulation, airway obstruction, and bacterial colonization, increasing the likelihood of infection in the lower airway [1-3]. Mucociliary clearance is affected by exposure to a number of environmental agents [4, 5], including cigarette smoke. Exposure to cigarette smoke (CS) has been linked to the development of lung cancer, emphysema, chronic bronchitis, and chronic obstructive pulmonary disease (COPD) [6]. Smokers are at a higher risk for pulmonary infection [7]. Chronic exposure to tobacco smoke is associated with increased ciliary abnormality [8] and reduced mucociliary clearance [9]. The increased risk for infection seen in smokers may be due to alterations in ciliary function in the upper airway.
Several studies have revealed that ciliary activity is controlled by phosphorylation of axonemal proteins [10-12]. Protein kinase C (PKC) and cyclic adenosine monophosphate (CAMP)-dependent protein kinase (PKA) are important regulators of airway ciliary beat frequency (CBF). Agents that increase PKC are associated with decreased CBF [13-15]. Alternatively, agents associated with increased CBF also increase PKA activity [16]. In past studies, we have shown that exposure of ciliated epithelial cells to cigarette smoke extract (CSE) in culture and whole cigarette smoke in vivo induces an increase in epithelial cell PKC activity [15, 17-19]. Many animal models of exposure to cigarette smoke components exist. Most recently, Miller and colleagues have developed a method of exposure consisting of intranasal administration of CSE [20]. This method was shown to induce an inflammatory response similar to that seen with whole cigarette smoke exposure with mild pathological alterations in the lung in BALB/c mice. Intranasal administration of CSE may be an alternative in vivo method of exposure to CS components without going through the lengthy process of exposing animals to whole cigarette smoke.
We conducted a comparative study to evaluate both methods side by side in C57BL/6 mice. The goal of this study was to evaluate the affects of using CSE as an alternate exposure method to cigarette smoke components in the C57BL/6 strain of mice. We were interested in characterizing the affects of CSE in this strain because of its past use in whole smoke exposure studies and its popularity as a background strain for an increasing number of knockout mice.
Knowing that data from the Miller study showed the administration of CSE invokes an inflammatory response in the lung similar to that seen in CS exposure in, we hypothesized that CSE would have the same affect on kinase activation and ciliary motility in vivo as seen with CS. That is to say, CSE would increase PKC activity and not affect ciliary responsiveness to isoproterenol in the tracheal epithelium. To test this hypothesis, we treated C57BL/6 mice with either whole-body CS, CSE intranasally, or appropriate sham and phosphate-buffered saline (PBS) controls for 6 weeks. We found that CSE is a more potent inducer of inflammation in the lungs than CS with treatments of limited duration. However, treatment with CSE resulted in an increase in baseline CBF and upon stimulation with isoproterenol, CSE blunted PKA activation and decreases ciliary beat, whereas CS had no effect.
MATERIALS AND METHODS
Animals
C57BL/6 mice were purchased from NCI (Frederick, MD) at 7 to 8 weeks of age and maintained at the Omaha VA Animal Research Facility under standard housing conditions. Mice were acclimated to the facility for 1 week prior to the start of exposure and received water and standard rodent chow ad libitum for the entire course of the study. Following acclimation, mice were randomly placed into 1 of 4 treatment groups (n = 11−12 per group): (1) exposure to air; (2) exposure to whole cigarette smoke, (3) intranasal exposure to PBS; and (4) intranasal exposure to cigarette smoke extract (Table 1). To determine handling versus treatment effects, a 5th group of mice received no treatment or handling of any kind aside from regular weighing and cage changing. Mice were monitored daily and weighed weekly. All experimental protocols were reviewed and approved by the Institutional Animal Care and Use Committee of the Omaha Veterans Affairs Medical Center. All protocols conformed to the Guide for the Care and Use of Laboratory Animals [21].
TABLE 1.
Basic Experimental Design
| Treatment
|
||
|---|---|---|
| Cigarette smoke (CS) | Cigarette smoke extract (CSE) | |
| Administration route | Inhalation
Mice place in middle chamber of 3-chambered smoking device. Cigarettes lit 2 at a time and burned for 6–7 min, followed by a cycle of filtered air of the same length. Treatment was repeated until all 20 cigarettes were burned (~1–1.5 h) |
Intranasal
Mice anesthetized by exposure to isoflurane in a bell jar until effect. Mice held in vertical position. CSE placed on the edge of the nostril and inhaled by the mouse. |
| Dose | 20 cigarettes/day | 50 μL/day |
| Duration of treatment | 6 days/week for 6 weeks | 6 days/week for 6 weeks |
Note. See Materials and Methods for treatment details and description of control groups.
Cigarette Smoke Exposure
Mice were exposed to the smoke of 20 1R3F reference cigarettes (University of Kentucky, Lexington) per day. Mice receiving cigarette smoke (CS) were gradually brought up to the target exposure in a period of 2 weeks and treated 6 days/week for 6 weeks. Treatment was administered by placing mice in a Plexiglas smoking chamber [22] measuring 65 cm long, 50 cm wide, and 45 cm high. The smoking chamber was divided into 3 sections. The first section, 10 cm in width, contained 2 holes to the outside to hold the lit cigarettes and draw in mainstream smoke, an outer hood and vent to capture the sidestream smoke, and a fan to mix the smoke with air before it reached the middle chamber through a set of inner holes. The mice were placed in the middle chamber in separate cages, 3 to 4 to a cage, containing several 1.0-cm holes on all sides of the cages to ensure adequate and equal exposure. Cages were rotated within the chamber for each treatment. Smoke was pulled through the middle chamber into a 3rd chamber before being drawn out of the exposure apparatus using a suction vacuum attached to the outside of the 3rd chamber. This resulted in smoke being dispersed throughout the middle chamber. Cigarettes were lit 2 at a time and burned for 6 to 7 minutes. This was followed by a cycle of filtered air of the same length. Treatment was repeated until all 20 cigarettes were burned (~1 to 1.5 hours). Sham treatments (air-exposed group) were conducted in the same manner in a similar apparatus for the same periods of time but mice were exposed to filtered room air. CS exposure was measured by determining carboxyhemoglobin (COHb) levels using an IL-682 CO-Oximeter (Instrumentation Laboratories, Lexington, MA). CS-exposed mice had a mean COHb of 10.14 (±0.27 [SEM]) versus air-exposed mice with a mean COHb of 0.53 (±0.07 [SEM]).
Cigarette Smoke Extract Treatment
Cigarette smoke extract (CSE) was produced by bubbling the smoke from one University of Kentucky 1R3F reference cigarette through 12.5 mL of sterile, pyrogen-free PBS (pH = 7.4). Smoke was drawn through the medium using a standard peristaltic pump at a rate of 50 mL/min. Cigarettes burned for 6 minutes. CSE was made fresh prior to each treatment and administered within 30 minutes. Mice were anesthetized by exposure to isoflurane (Min Rad, Bethlehem, PA) in a bell jar until effect. Mice were then held in a vertical position and 50 μL of CSE was placed on the edge of the nostril and inhaled by the mouse.
Administration of CSE in this manner has been shown to produce an exposure resulting in ~75% of deposition in the lower and upper airways [20]. Sham-treated mice received 50 μL of sterile, pyrogen-free PBS administered in the same fashion. Mice were treated for 6 days/week for 6 weeks. Table 1 shows a summary of the experimental design.
Serum Cotinine Levels
Serum cotinine levels were measured using a Double Antibody Nicotine Metabolite Radioimmunoassay kit (Diagnostic Products Corporation, Los Angeles, CA) following manufacturer’s instructions with slight modifications as follows: the 100 and 500 μg/mL standards were diluted with the 0 μg/mL standard to create the following standards: 0, 50, 100, 250, and 500 μg/mL; and 50 μL of serum was used in place of urine as previously described [23].
Bronchoalveolar Lavage (BAL)
Mice were sacrificed by pentobarbital overdose (Nembutol, Abbott Labs, Chicago, IL). BAL was performed in a closed thorax manner by exposing the trachea, nicking the bottom of the larynx and inserting a 3/4-inch 24-gauge cannula into the proximal trachea. The proximal end of the trachea was tied off and 0.6 mL of sterile PBS (Gibco, Grand Island, NY) was gently introduced to the lungs and recovered. This was repeated 3 times for a total volume of 1.8 mL. Return volume varied by <10% between samples. BAL fluid was centrifuged to remove cells. Cells obtained were placed on slides for determination of cell populations using a cytospin (Cytopro Cytocentrifuge; Wescor, Logan UT) and stained with DiffQuik (Dade Behring, Newark, DE). Differential counts were made with ≥400 cells/sample from 2 slides/mouse.
Trachea Harvesting and Treatment
Following BAL, tracheas were removed and maintained in a closed sterile 15-mL conical tube in serum-free M199 containing penicillin/streptomycin (100 units/100 μg per mL) (Gibco) and fungizone (2 μg/mL) (Gibco) at room temperature until processing (30 to 60 minutes). Tracheal rings were cut (width ≈0.5 mm) from the distal end of the trachea just proximal to the first bifurcation of the trachea into right and left mainstream bronchi. The rings were placed in Petri dishes containing serum-free M199 (Gibco) for CBF determinations. The remaining tracheal tissue was opened with a longitudinal cut to expose the ciliated epithelium and placed in a Petri dish containing serum-free M199. Following baseline CBF determination, both the rings and the remaining tracheal tissue were stimulated with isoproterenol at a final concentration of 100 μM. The rings and tissue were incubated for 30 minutes at 37°C, 5% CO2 then allowed to equilibrate at 25°C for 10 minutes. The final CBF reading was taken from the tracheal rings. The remaining tracheal tissue was removed from the medium and the ciliated epithelial cells were extracted with a sterile cell lifter (Fisher, Springfield, NJ) into cell lysis buffer as previously described [16]. The epithelial lysate was then immediately flash frozen in liquid nitrogen for kinase assays.
Ciliary Beat Frequency
The motion of the actively beating cilia on the tracheal ring was quantified using phase contrast microscopy, and computerized frequency spectrum analysis. During CBF measurement, tracheal rings were maintained at a constant temperature (24°C ± 0.5°C) by a thermostatically controlled heated stage, as the temperature gradient is known to affect CBF [24]. All observations were recorded for analysis with a Kodak Megaplus 310 analog/digital video camera (Eastman Kodak Motion Analysis Systems Division, San Diego, CA). Whole-field analysis was performed and the CBF determined by collecting data sampled at 40 Hz from 512 samples (12.8 seconds) and performing frequency spectrum analysis using the Sisson-Ammons Video Analysis (SAVA) system [25].
PKA Activity
PKA activity was assessed in a similar manner as described previously [16]. Briefly, a portion of the epithelial cell lysates was sonicated and centrifuged at 10,000 × g for 30 minites at 4°C. The assay employed is a modification of procedures previously described [26], with 130 μM PKA substrate heptapeptide (LRRASLG), 10 μM cAMP, 0.2 mM isobutylmethyxanthine, 20 mM magnesium-acetate, and 0.2 mM [γ-32P] adenosine triphosphate (ATP) in a 40-mM Tris-HCl buffer (pH 7.5). Samples (20 μL) were added to 50 μL of the above reaction mixture and incubated for 15 minutes at 30°C. Incubations were halted by spotting 50 μL of each sample onto P-81 phosphocellulose papers (Whatman, Hillsboro, OR). Papers were then washed 5 times for 5 minutes each in phosphoric acid (75 mM), washed once in ethanol, dried, and counted in nonaqueous scintillant as previously described [27]. Negative controls consisted of similar assay samples with or without the substrate peptide. A positive control of 0.4 ng/mL purified catalytic subunit from type I bovine PKA (Promega, Madison, WI) was included as a sample. Kinase activity was expressed in relation to total cellular protein assayed and calculated in picomoles of phosphate incorporated per minutes per milligram of protein (pmol/min/mg).
PKC Activity
The remaining epithelial lysate was used to determine PKC activity. The assay employed was a modification of procedures previously described using 900 μM PKC substrate peptide (Peninsula, Belmont, CA), 12 mM calcium-acetate, 8 μM phosphatidyl-l-serine, 24 μg/mL phorbol 12-myristate 13-acetate, 30 mM dithiothreitol, 150 μM ATP, 45 mM magnesium acetate, and 10 μCi/mL [γ-32P] ATP (ICN Biomedicals, Costa Mesa, CA) in a Tris-HCl buffer (pH 7.5). Samples (20 μL) were added to 40 μL of the above reaction mixture and incubated for 15 minutes at 30°C. Spotting 50 μL of each sample onto P-81 phosphocellulose papers (Whatman, Clifton, NJ) halted incubations. Papers were then washed 5 times for 5 minutes each in phosphoric acid (75 mM), washed once in ethanol, dried, and counted in nonaqueous scintillant as previously described. Kinase activity was expressed in relation to total cellular protein assayed and calculated in picomoles of phosphate incorporated per minutes per milligram.
Histopathological Examination
Changes in mouse lung pathology were examined under conditions of air, PBS, CS, and CSE treatment. The lungs from mice in each treatment group were fixed with 10% neutral buffered formalin (pH 7.2) at the time of sacrifice using a 20-gauge catheter placed into the trachea and secured with silk suture. Lungs were dissected out and immediately inflated under 25 cm H2O pressure. Following fixation, lungs were paraffin embedded, serial sectioned (3 to 4 μm) and stained with hematoxylin and eosin (H&E) for histological analysis.
Statistical Analysis
Results are expressed as the mean ± the standard error of the mean (SEM) of the indicated number of animals in each group. One-way analysis of variance (ANOVA) was used to determine the significance of differences among the group means followed by Tukey’s multiple comparison test or student’s t test where appropriate. A probability of less than .05 was accepted as significant.
RESULTS
Mouse Weight, Condition, and Serum Cotinine Levels
Mice were weighed weekly for the duration of the treatment. Mice in all groups gained weight normally and at a similar rate. At no time was there a significant difference in the weight, or weight gain for either the CS group or the CSE group compared to their respective controls (Figure 1A). Serum cotinine levels in the CSE-exposed mice were significantly higher than those in mice exposed to CS (35.3 ± 3.1 ng/mL and 24.7 ± 2.6 ng/mL respectively; P < .05) (Figure 1B). Data from the group of control mice that were not handled to determine treatment effects was similar in all experimental parameters to the air-exposed group. Therefore, data for this group is not shown.
FIGURE 1.
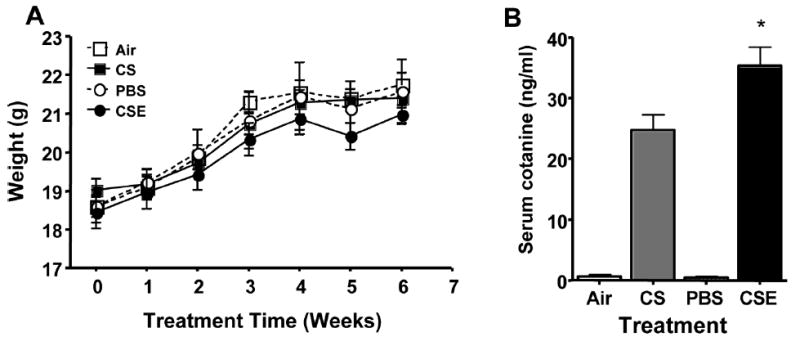
(A) Weight gain in mice exposed to air or whole cigarette smoke (CS) by inhalation, or intranasal administration of phosphate-buffered saline (PBS), or cigarette smoke extract (CSE). There were no significant differences in the weight or rate of weight gain for any of the treatment groups. (B) Serum cotinine levels in the CSE exposed mice were significantly higher than those in mice exposed to CS (P < .05).
BAL Cell Increases in CSE-, PBS-, and CS-Treated Mice
To compare the inflammatory response induced by CS or CSE treatment, total BAL cells were counted. Mice treated with CSE had the greatest increase in total BAL cell numbers (Figure 2), significantly higher than mice receiving CS (P < .05), PBS (P < .05), or air (P < .001). PBS treatment induced an increase in total BAL cells greater than that seen in the CS-exposed mice and significantly higher compared to the air-treated group (P < .05). Treatment with CS increased the total number of BAL cells significantly over control animals receiving air (P < .05). The percentage of macrophages in the BAL was significantly decreased in the PBS- and CSE-treated mice (Figure 3A) compared to the CS-treated group (P < .001). CS treatment did not induce a significant change in the percent macrophages compared to the air control group. The total number of macrophages increased significantly in the CSE-treated group despite the decrease in percentage (P < .05, CSE versus CS; P < .01, CSE versus Air) (Figure 3B). Mice treated with PBS also had an increase in the number of macrophages in the BAL although not significantly higher than the CS group. Although the percentage of macrophages for the CS group was not significantly different than that seen in the air-treated group, the number of macrophages in the BAL was increased by CS treatment compared with the air group (P < .05) (Figure 3B). Treatment with CSE induced a dramatic increase in the percentage of neutrophils in the BAL, significantly higher than all other groups (CSE versus PBS, P < .05; CSE versus CS and air, P < .001) (Figure 3C). Interestingly, PBS alone induced a significantly higher percentage of neutrophils in the BAL than was seen with the CS- and air-exposed groups (P < .001). The number of neutrophils in the BAL was greatest in the CSE-treated group (P < .001) and increased to a lesser degree in the PBS-treated mice (P < .05 PBS versus CS and air (Figure 3D).
FIGURE 2.
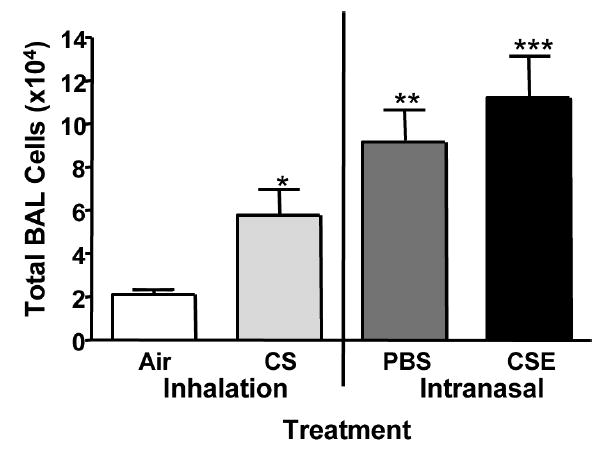
Total BAL cell numbers. Mice were treated with whole body exposure to air or whole cigarette smoke (CS) or received intranasal administration of phosphate-buffered saline (PBS) or cigarette smoke extract (CSE). Mice treated with CSE had the greatest total number of BAL cells, significantly greater than all other groups (*P < .05, CS versus air; **P < .05 PBS versus air; ***P < .05, CSE versus CS and PBS; ***P < .001 CSE versus air).
FIGURE 3.
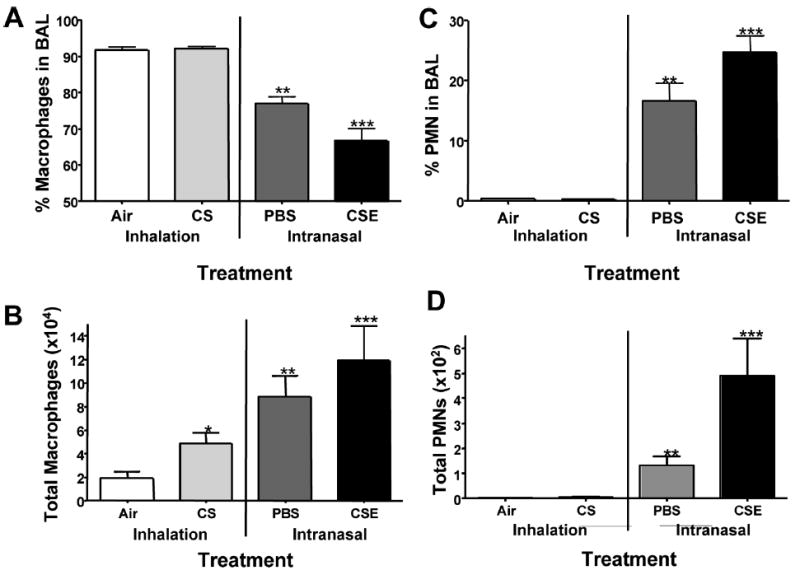
Macrophages and neutrophils (PMNs) in the BAL. (A) CSE treatment reduced the percentage of macrophages in the BAL compared to all other groups (***P < .001 CSE versus CS, air; ***P < .01, CSE versus PBS). PBS treatment also decreased the percentage of macrophages recovered in the BAL (**P < .01) over CS- and air-treated groups. (B) Despite the decrease in the percentage of macrophages, treatment with CSE, PBS, and CS significantly increased the total number of macrophages (***P < .05, CSE versus CS; P < .01, CSE versus Air; **P < .05 PBS versus air; P < .05 CS versus Air). (C) The percentage of PMNs was increased in CSE treated mice compared to PBS (***P < .05), CS and air (***P < .001). PBS treatment also induced a significant increase in the percentage of neutrophils (**P < .001 PBS versus CS and air). (D) The number of PMNs was significantly higher in the CSE treated group compared to all other treatment groups (***P < .001). PBS also had a significant increase in the number of PMNs compared to the CS and air treatment groups (**P < .05).
CSE Exposure Increases Baseline CBF and Alters Isoproterenol Responsiveness
Baseline and isoproterenol-induced changes in CBF were measured to determine if CSE exposure would affect resting CBF- and β-agonist–induced changes in CBF. Baseline CBF for the CS- and air-treated control groups were comparable at 10.8 Hz (± 0.31 [SEM]) and 10.71 Hz (±0.27 [SEM]), respectively (Figure 4). PBS-treated mice had a slightly higher mean CBF at 11.04 Hz (±0.43 [SEM]), but not significantly different from CS- and air-treated mice. Mice receiving CSE had an increase in baseline CBF of approximately 1.5 Hz over all other groups with a mean baseline CBF of 12.47 ± 0.31 Hz (P < .05). Isoproterenol induced an increase in CBF of +1.53 Hz (±0.59 [SEM]) in control mice receiving air (Figure 5). CS exposure had no effect on isoproterenol responsiveness as CBF in mice receiving CS increased +1.46 Hz (±0.49 [SEM]). Interestingly, treatment with PBS blunted the cilia response to isoproterenol producing a slight decrease in CBF of −0.40 Hz (±0.67 [SEM]). Cilia from mice treated with CSE paradoxically had a significant decrease in CBF in response to isoproterenol stimulation (−1.71 ± 0.60 Hz).
FIGURE 4.
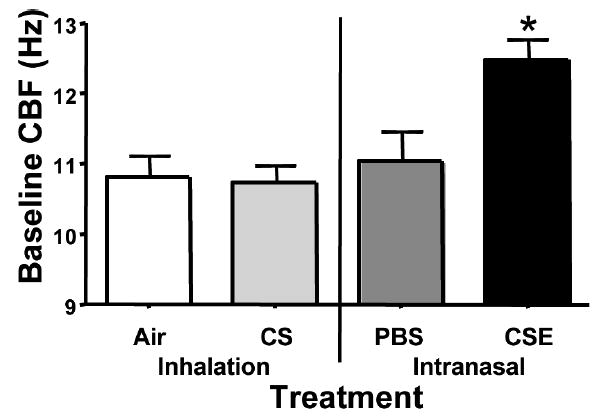
Baseline (unstimulated) ciliary beat frequency. CBF was measured in all samples prior to stimulation with isoproterenol. Baseline CBF was significantly higher in the mice treated with CSE compared to all other groups (*P < .05).
FIGURE 5.
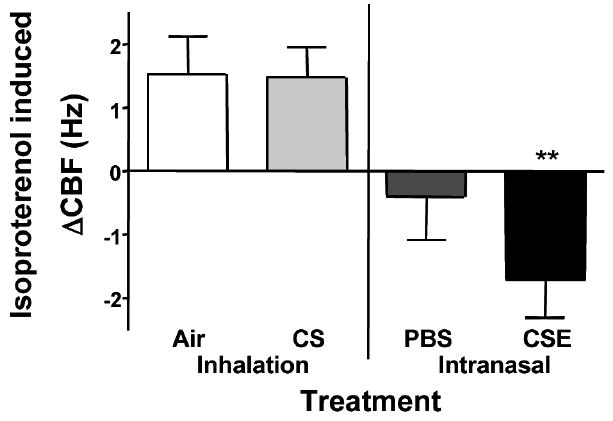
Isoproterenol induced change in CBF. Following baseline CBF measurement, tracheal rings from treated mice were stimulated with 100 μM isoproterenol for 40 minutes. CBF increased normally in tracheal rings from mice treated with air or smoke. CBF did not increase in tracheal rings from mice treated with PBS. CBF decreased significantly in response to isoproterenol stimulation in the tracheal rings from mice treated with CSE (**P < .01).
Isoproterenol-Stimulated PKA Activity is Blunted in Tissues from CSE-Treated Mice
Isoproterenol stimulation of ciliated tracheal epithelial cells has been shown to result in an increase of PKA activity [16]. Upon stimulation with isoproterenol, mice treated with air, CS, and PBS had a 2-fold or greater increase in PKA activity compared to unstimulated tracheal tissues (P < .001; Figure 6). CSE-treated mice had a nominal, but not significant, increase in PKA activity in response to isoproterenol when compared to unstimulated tissues from mice receiving the same treatment.
FIGURE 6.
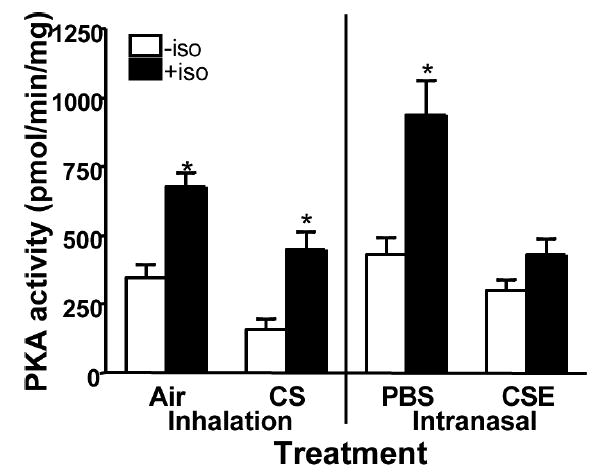
Isoproterenol stimulated PKA activity. PKA activity was measured in tracheal tissue from treated mice with (filled bars) or without (open bars) 100 μM isoproterenol. Isoproterenol stimulation significantly increased the amount of PKA activity in the ciliated tracheal epithelium of mice treated with air, CS, and PBS (*P < .05). No significant increase in PKA activity was measured in response to isoproterenol in the ciliated tracheal epithelium of mice treated with CSE.
CSE is a Potent Inducer of PKC Activity in the Ciliated Tracheal Epithelium
Exposure to CSE in cell culture in vitro has been shown to increase PKC activity in ciliated airway epithelial cells. The tracheal epithelium from mice treated with CS had significantly higher PKC activity (98.62 ± 9.65 pmol/min/mg) compared to the air-exposed group (41.22 ± 5.7 pmol/min/mg; Figure 7). The mice receiving PBS also had an increase in PKC activity similar to that seen in the whole smoke exposed group (76.4 ± 7.67 pmol/min/mg). CSE treatment had the greatest effect on PKC activity with tissues from mice in this treatment group having greater than 2 times the activity seen in the CS exposed group (257.0 ± 14.98 pmol/min/mg) (P < .001).
FIGURE 7.
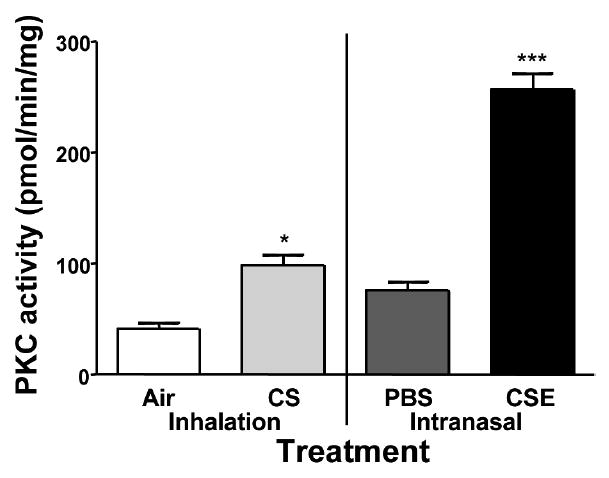
PKC activity in the ciliated tracheal epithelium. PKC activity in the ciliated tracheal epithelium of treated mice was measured. PKC activity was greatly increased and significantly higher in the tissue from CSE treated mice in comparison to all other groups (***P < .001). Tissues from CS treated mice had an increase in PKA activity significantly higher than that measured in Air treated mice (*P < .05), but not PBS treated mice.
CSE Exposure Increases Inflammatory Cell Bronchiolitis
To compare the inflammatory response involved by CS or CSE treatment, lung pathology was examined. Mice treated with CSE demonstrated a significant increase in inflammatory cell bronchiolitis (Figure 8). A milder bronchiolitis was observed in the lungs of mice exposed to CS or to instilled PBS compared to control and air-exposed mice. These data suggest that whereas instillation of liquid vehicle (PBS) into the lungs provides some inflammation, exposure to CSE provides a significantly greater inflammatory cell recruitment than exposure to CS.
FIGURE 8.
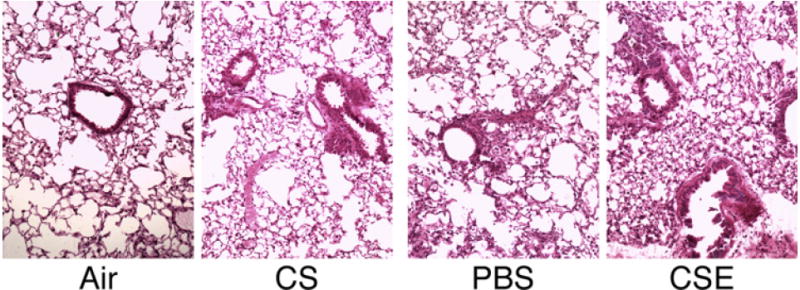
Lung pathology in smoke exposed mice. H&E staining of a section of lung from mice exposed to air or to whole cigarette smoke (CS) by inhalation, intranasal administration of phosphate buffered saline (PBS), or cigarette smoke extract (CSE). Increased mild bronchiolitis is observed in the lungs of mice exposed to CS or PBS compared to air-exposed control mice. CSE-exposed mice lungs demonstrate a significant increase in inflammatory cell bronchiolitis compared to PBS-treated mice.
DISCUSSION
Exposure of mice to cigarette smoke as a model of human smoking began in the 1940s [28] and continues today. Modern exposure methods using whole cigarette smoke are often labor intensive and of extended duration. As an alternative to whole smoke, Miller and colleagues published a method of exposure to cigarette smoke components involving intranasal administration of CSE [20]. In this model, inflammation occurs in the lungs with increased inflammatory cytokine levels and mild COPD-like pathology in the lower airway in a relatively short period of time. To determine if the effects of CSE exposure on the upper airway would be comparable to that seen with CS exposure, we conducted a comparative study to investigate the effects of CSE versus CS on ciliary function and kinase activity in the upper airway. Although the CSE treatment method used was basically the same in our study as in Miller’s, there are a number of differences between the two studies.
Miller ran a panel of different mouse strains to determine which strain to use based on a high sensitivity to CSE treatment and chose to use BALB/c mice. Despite the C57BL/6 mouse strain’s less robust response to allergens and pathogens with regards to pulmonary inflammation [29-32], we chose to use C57BL/6 mice for two reasons: (1) they have in the past been successfully used in a number of smoke exposure models; and (2) an increasing number of knockout mice are bred onto the C56BL/6 background. Having determined the utility of CSE treatment in this strain of mice allows us to further investigate the affects of loss of signaling components and inflammatory mediators in the development of inflammation and disease in the lungs using CSE in future studies with knockout mice.
Another difference between Miller’s work and our study is the vehicle for the CSE. From the data shown in the Miller study, it appears that the RPMI treatment alone has an inflammatory affect on the lungs. Although Miller shows no data from non–RPMI-treated mice, the percentage of neutrophils in the RPMI-treated groups is higher than that seen in a normal BALB/c or C57BL/6 mouse [30, 33, 34]. For this reason, we chose to use PBS with the assumption that the high neutrophil percentages seen in the Miller study for the control group could be avoided using a physiologic salt solution. In addition, we cannot rule out the possibility that CSE made with PBS differs in some way from CSE made in RPMI. However the similarity of the resultant inflammation with CSE made in PBS in our study, and that with CSE made in RPMI in the Miller study, suggests the affect of the two formulations is similar and no significant difference exists between them with regard to inflammatory components.
In Miller’s study, two different concentrations of CSE were used, CS-40 (40 cigarettes bubbled through 500 mL of RPMI) and CS-80 (80 cigarettes/500 mL RPMI). These two concentrations had the same affect on the lungs with respect to neutrophil influx, cytokine levels, and pathology in adult mice. The concentration we used was equivalent to the CS-40. We felt this concentration would be sufficient due to the fact that the only difference between treating with CS-40 and CS-80 specifically noted in the Miller study was a higher number of total cells in the BAL fluid, with that occurring mainly in young mice (4 weeks of age).
Although CSE treatment times in the Miller study varied from 1 to 40 days, we chose to treat the mice for 6 weeks based on a study published by Gairola, in which a time course of CS exposure in C57BL/6 mice shows that 6 weeks is the time point at which an increase in BAL cells occurs [33], and on two other rodent studies showing the development of pathological changes in the lungs occurring at this time point using a similar smoke exposure regimen [22].
Following a 6-week exposure, we found that CSE induced a greater influx of inflammatory cells into the lungs compared to the CS exposure. We think this is interesting and postulate that the degree of exposure occurring with each method is most likely not similar. Intuitively, the concentration of smoke components inhaled by the CS-treated mice may be considered dilute compared to that in the CSE exposure. We do not know how the retention time of the smoke components compare between the 2 groups, but it has been shown that low levels of cigarette smoke exposure correlate with increased clearance rates [35], whereas heavy exposures result in decreased clearance rates [19]. In our study, the fact that the inflammatory response was greater in the CSE group suggests that treatment with CSE results in a greater concentration of cigarette smoke components being administered. As a comparison of exposures, we measured serum levels of the nicotine metabolite cotinine. Mice exposed to CSE had significantly higher serum cotinine levels than CS-exposed mice. Cotinine levels in serum are dose dependent and increase when the number of cigarettes smoked increases. Higher cotinine levels in the sera correlates with a heavier exposure to cigarette smoke [36-38]. These data support the conclusion that using our methods, treatment with CSE results in a heavier exposure to cigarette smoke components over the same treatment length.
Data from our nonhandled control shows that the instillation of PBS devoid of CS components produces a similar effect as that seen with the RPMI controls in the Miller study with regard to inflammatory cell populations in the lung. Treatment with PBS alone resulted in a high percentage of neutrophils recovered in the BAL fluid. This effect of liquid instillation in the lung has been shown to occur in other studies in which lavage with sterile saline solution in vivo induces an inflammatory response in the lungs characterized by an influx of neutrophils in a number of animal species [39-41] and in humans [42]. However, the inflammatory response induced by PBS was less than that of CSE made in PBS, as seen with RPMI and CSE made with RPMI. The resultant inflammatory response caused by PBS instillation in the lung is significantly greater than that caused by CS treatment. These data suggest that some measure of the inflammatory response induced by CSE, whether made in PBS or RPMI, is likely due in part to the instillation of liquid in the lung.
It is logical to assume that one, or a combination, of the components of cigarette smoke in the CSE, are responsible for the increased baseline CBF through an as yet unknown mechanism. The lack of PBS stimulation of baseline beating would support this conclusion. One possible explanation may be that the exposure to nicotine is responsible for the increase in baseline CBF [43]. Alternatively, increased inflammatory cytokine release by local epithelial cells and infiltrating alveolar macrophages may be the cause of the baseline change due to CSE. Upon stimulation, macrophages recruited during pulmonary inflammation are known to secrete proinflammatory cytokines [44]. Exposure to proinflammatory cytokines has been shown to produce a sustained increase in CBF [45-47]. Continual exposure of the ciliated epithelium to increased levels of proinflammatory cytokines during CSE treatment via periciliary fluid brought up by mucociliary action might result in an increased baseline CBF.
It is well established that β-agonist stimulation of ciliated cells induces an increase in CBF [24, 48, 49]. We did not anticipate that exposure of the upper airway in vivo to CS or CSE would have an effect on isoproterenol-stimulated cilia beat. However, we found that intranasal instillation of CSE induced a significant slowing of ciliary motility upon isoproterenol treatment. Interestingly, the β-agonist response was blunted in PBS-treated mice. The mechanism responsible for the decreased cilia beat in the CSE-treated mice occurring with isoproterenol stimulation is unknown at this time. The slowing of cilia beat only occurred with the CSE-treated mice where a combination of PKA inhibition and PKC activation existed concurrent with stimulation of the β-adrenergic receptor. This unique combination of events may allow alteration of intracellular Ca2+ levels in the cell, which is known to affect CBF [50-52]. Alternatively, alteration of phosphorylation on the axoneme may be occurring. Cross-talk and regulation of signaling by PKA and PKC occur in many cell types in either a synergistic or antagonistic fashion [53-57]. It is possible that the phosphorylation of a target protein on the axoneme by PKA produces a conformation in which a phosphorylation target of PKC is masked. Recently it has been reported that phosphorylation of the alpha-1 subunit of kidney Na,K-ATPase by PKC is modulated by a cAMP-stimulated pathway and possibly by the state of phosphorylation of the PKA phosphorylation site, Ser-938 [53]. There is evidence that both PKA and PKC have phosphorylation targets on the axoneme with phosphorylation by PKC, resulting in decreased CBF [14, 58]. One possible explanation is that with PKA activity inhibited, phosphorylation of the PKA target in the axoneme does not occur. This may allow the phosphorylation of a target protein by PKC on the axoneme resulting in decreased beating.
PKA is an important regulator of airway CBF. Those agents associated with increased CBF also increase PKA activity [16]. Neither exposure to air nor to CS altered basal or isoproterenol-induced PKA activity in the tracheal ciliated epithelium of treated mice. PBS treatment had no effect on isoproterenol’s ability to induce PKA activity in the tissues from mice in this group. It is interesting to note that the CBF response to isoproterenol was blunted despite the increased PKA activity. We have not observed this disconnect between increased PKA and CBF in airway cells before. PKA signaling is compartmentalized in a number of cell types [59]. The assay we employ measures total epithelial cell PKA activity. It is possible that PBS affects the PKA activity localized in close physical proximity to the axoneme and therefore the effect of PBS on the PKA activity related to cilia beat is not detected. It is also possible that PBS instillation in the upper airway has a physical effect resulting in a mechanical alteration of β-agonist responsiveness that is only observed in the intact cell. Although we did not expect to see a blunting of isoproterenol-induced PKA activity in tissues from the CSE-treated mice, the lack of increased PKA activity correlates with the lack of increased CBF with isoproterenol stimulation.
We hypothesized that CSE would increase PKC activity in the ciliated tracheal epithelium in vivo. Although CS exposure increased PKC activity in the tracheal epithelium to a level significantly higher than that seen in air-exposed mice, exposure to CSE increased PKC activity to a level significantly higher than treatment with CS. How exposure to CS or CSE induces PKC activity is unknown at this time. Determining which PKC isoform(s) are upregulated may help to determine additional cellular components needed for CS and CSE to induce an increase in PKC activity.
Often times the relevance of animal models is called into question as to whether or not the exposure is similar or, if it is not, whether the results obtained are applicable to pathological development of disease in humans. The strain of mice used in murine models of smoke exposure may also affect the results achieved. Differing from the Miller study, our work uses C57BL/6 mice instead of BALB/c. A number of studies have shown strain differences in the pulmonary inflammatory response between BALB/c and C57BL/6 with the BALB/c strain seen as more sensitive; i.e., having a more robust response to allergens and pathogens [29-32]. Although we suspect it would, at this time, we have not evaluated epithelial kinase activity and CBF alterations in CSE-treated BALB/c or any additional mouse strain. This study was not undertaken to address of affects of CSE on C57BL/6 versus BALB/c, but to evaluate the utility of this method for mimicking short-term exposure to vapor phase smoke in the C57BL/6 strain.
Although CSE may be a way to bring about the development of pathology in a shorter time period, its resemblance to the smoke exposure experienced by smokers (vapor phase) is not equivalent. Not only the CSE exposure, but also the introduction of liquid into the lungs alter cellular function in the upper airway in an unexpected fashion in C57BL/6 mice. It is possible that cellular function and/or signaling in the other cells in the lungs may also be altered in a manner inconsistent with exposure to whole CS. Before this method is used to replace exposure to whole smoke, these differences must be taken into consideration.
Acknowledgments
This work was supported by 5T32AA007582-05 (MKE), 5R01AA008769-15 (JHS), and Veterans Administration Merit Review Award (TAW). Dr. Wyatt is an American Lung Association Career Investigator.
Contributor Information
Margaret K. Elliott, Pulmonary, Critical Care and Sleep Medicine Section, University of Nebraska Medical Center, Omaha, Nebraska, USA
Joseph H. Sisson, Pulmonary, Critical Care and Sleep Medicine Section, University of Nebraska Medical Center, Omaha, Nebraska, USA
William W. West, Department of Pathology and Microbiology, University of Nebraska Medical Center, Omaha, Nebraska, USA
Todd A. Wyatt, Pulmonary, Critical Care and Sleep Medicine Section, University of Nebraska Medical Center, Omaha, Nebraska, USA; and Veterans Affairs Medical Center Research Service, Omaha, Nebraska, USA
References
- 1.LiPuma JJ. Expanding microbiology of pulmonary infection in cystic fibrosis. Pediatr Infect Dis J. 2000;19:473–474. doi: 10.1097/00006454-200005000-00016. [DOI] [PubMed] [Google Scholar]
- 2.Sohy C, Pilette C, Niederman MS, Sibille Y. Acute exacerbation of chronic obstructive pulmonary disease and antibiotics: what studies are still needed? Eur Respir J. 2002;19:966–975. doi: 10.1183/09031936.02.00291302. [DOI] [PubMed] [Google Scholar]
- 3.Daviskas E, Robinson M, Anderson SD, Bye PT. Osmotic stimuli increase clearance of mucus in patients with mucociliary dysfunction. J Aerosol Med. 2002;15:331–341. doi: 10.1089/089426802760292681. [DOI] [PubMed] [Google Scholar]
- 4.Gong H., Jr Health effects of air pollution. A review of clinical studies. Clin Chest Med. 1992;13:201–214. [PubMed] [Google Scholar]
- 5.Wolff RK. Effects of airborne pollutants on mucociliary clearance. Environ Health Perspect. 1986;66:223–237. doi: 10.1289/ehp.8666223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Proctor RN. The global smoking epidemic: a history and status report. Clin Lung Cancer. 2004;5:371–376. doi: 10.3816/CLC.2004.n.016. [DOI] [PubMed] [Google Scholar]
- 7.Brown R, Pinkerton R, Tuttle M. Respiratory infections in smokers. Am Fam Physician. 1987;36:133–140. [PubMed] [Google Scholar]
- 8.Verra F, Escudier E, Lebargy F, Bernaudin JF, De Cremoux H, Bignon J. Ciliary abnormalities in bronchial epithelium of smokers, ex-smokers, and nonsmokers. Am J Respir Crit Care Med. 1995;151:630–634. doi: 10.1164/ajrccm/151.3_Pt_1.630. [DOI] [PubMed] [Google Scholar]
- 9.Cohen D, Arai SF, Brain JD. Smoking impairs long-term dust clearance from the lung. Science. 1979;204:514–517. doi: 10.1126/science.432655. [DOI] [PubMed] [Google Scholar]
- 10.Brokaw CJ. Regulation of sperm flagellar motility by calcium and cAMP dependent phosphorylation. J Cell Biochem. 1987;35:175–184. doi: 10.1002/jcb.240350302. [DOI] [PubMed] [Google Scholar]
- 11.Satir P, Barkalow K, Hamasaki T. The control of ciliary beat frequency. Trends Cell Biol. 1993;3:409–412. doi: 10.1016/0962-8924(93)90092-f. [DOI] [PubMed] [Google Scholar]
- 12.Walczak CE, Nelson DL. Regulation of dynein-driven motility in cilia and flagella. Cell Motil Cytoskeleton. 1994;27:101–107. doi: 10.1002/cm.970270202. [DOI] [PubMed] [Google Scholar]
- 13.Wong LB, Park CL, Yeates DB. Neuropeptide Y inhibits ciliary beat frequency in human ciliated cells via nPKC, independently of PKA. Am J Physiol. 1998;275:C440–C448. doi: 10.1152/ajpcell.1998.275.2.C440. [DOI] [PubMed] [Google Scholar]
- 14.Salathe M, Pratt MM, Wanner A. Protein kinase C-dependent phosphorylation of a ciliary membrane protein and inhibition of ciliary beating. J Cell Sci. 1993;106:1211–1220. doi: 10.1242/jcs.106.4.1211. [DOI] [PubMed] [Google Scholar]
- 15.Wyatt TA, Schmidt SC, Rennard SI, Sisson JH. Acetaldehyde-stimulated PKC activity in airway epithelial cells treated with smoke extract from normal and smokeless cigarettes. Proc Soc Exp Biol Med. 2000;225:91–97. doi: 10.1046/j.1525-1373.2000.22511.x. [DOI] [PubMed] [Google Scholar]
- 16.Wyatt TA, Spurzem JR, May K, Sisson JH. Regulation of ciliary beat frequency by both PKA and PKG in bovine airway epithelial cells. Am J Physiol. 1998;275:L827–L235. doi: 10.1152/ajplung.1998.275.4.L827. [DOI] [PubMed] [Google Scholar]
- 17.Wyatt TA, Heires AJ, Sanderson SD, Floreani AA. Protein kinase C activation is required for cigarette smoke-enhanced C5a-mediated release of interleukin-8 in human bronchial epithelial cells. Am J Respir Cell Mol Biol. 1999;21:283–288. doi: 10.1165/ajrcmb.21.2.3636. [DOI] [PubMed] [Google Scholar]
- 18.Wyatt TA, Gentry-Nielsen MJ, Pavlik JA, Sisson JH. Desensitization of PKA-stimulated ciliary beat frequency in an ethanol-fed rat model of cigarette smoke exposure. Alcohol Clin Exp Res. 2004;28:998–1004. doi: 10.1097/01.ALC.0000130805.75641.F4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vander Top EA, Wyatt TA, Gentry-Nielsen MJ. Smoke exposure exacerbates an ethanol-induced defect in mucociliary clearance of Streptococcus pneumoniae. Alcohol Clin Exp Res. 2005;29:882–887. doi: 10.1097/01.alc.0000164364.35682.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miller LM, Foster WM, Dambach DM, Doebler D, McKinnon M, Killar L, Longphre M. A murine model of cigarette smoke-induced pulmonary inflammation using intranasally administered smoke-conditioned medium. Exp Lung Res. 2002;28:435–455. doi: 10.1080/01902140290096728. [DOI] [PubMed] [Google Scholar]
- 21.Institute of Laboratory Animal Resources, Commission on Life Sciences, National Research Council. Washington, DC: National Academy Press; 1996. Guide for the Care and Use of Laboratory Animals. [Google Scholar]
- 22.Li T, Molteni A, Latkovich P, Castellani W, Baybutt RC. Vitamin A depletion induced by cigarette smoke is associated with the development of emphysema in rats. J Nutr. 2003;133:2629–2634. doi: 10.1093/jn/133.8.2629. [DOI] [PubMed] [Google Scholar]
- 23.Perkins SL, Livesey JF, Escares EA, Belcher JM, Dudley DK. High-performance liquid-chromatographic method compared with a modified radioimmunoassay of cotinine in plasma. Clin Chem. 1991;37:1989–1993. [PubMed] [Google Scholar]
- 24.Sanderson MJ, Dirksen ER. Mechanosensitive and beta-adrenergic control of the ciliary beat frequency of mammalian respiratory tract cells in culture. Am Rev Respir Dis. 1989;139:432–440. doi: 10.1164/ajrccm/139.2.432. [DOI] [PubMed] [Google Scholar]
- 25.Sisson JH, Stoner JA, Ammons BA, Wyatt TA. All-digital image capture and whole-field analysis of ciliary beat frequency. J Microsc. 2003;211:103–111. doi: 10.1046/j.1365-2818.2003.01209.x. [DOI] [PubMed] [Google Scholar]
- 26.Jiang H, Colbran JL, Francis SH, Corbin JD. Direct evidence for cross-activation of cGMP-dependent protein kinase by cAMP in pig coronary arteries. J Biol Chem. 1992;267:1015–1019. [PubMed] [Google Scholar]
- 27.Roskoski R., Jr Assays of protein kinase. Methods Enzymol. 1983;99:3–6. doi: 10.1016/0076-6879(83)99034-1. [DOI] [PubMed] [Google Scholar]
- 28.Lorenz E, Stewart HL, Daniel JH, Nelson CV. The effects of breathing tobacco smoke on strain A mice. Cancer Res. 1943;3:123. [Google Scholar]
- 29.Fonseca-Aten M, Rios AM, Mejias A, Chavez-Bueno S, Katz K, Gomez AM, McCracken GH, Jr, Hardy RD. Mycoplasma pneumoniae induces host-dependent pulmonary inflammation and airway obstruction in mice. Am J Respir Cell Mol Biol. 2005;32:201–210. doi: 10.1165/rcmb.2004-0197OC. [DOI] [PubMed] [Google Scholar]
- 30.Preston JA, Beagley KW, Gibson PG, Hansbro PM. Genetic background affects susceptibility in nonfatal pneumococcal bronchopneumonia. Eur Respir J. 2004;23:224–231. doi: 10.1183/09031936.03.00081403. [DOI] [PubMed] [Google Scholar]
- 31.Singh B, Shinagawa K, Taube C, Gelfand EW, Pabst R. Strain-specific differences in perivascular inflammation in lungs in two murine models of allergic airway inflammation. Clin Exp Immunol. 2005;141:223–229. doi: 10.1111/j.1365-2249.2005.02841.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shinagawa K, Kojima M. Mouse model of airway remodeling: strain differences. Am J Respir Crit Care Med. 2003;168:959–967. doi: 10.1164/rccm.200210-1188OC. [DOI] [PubMed] [Google Scholar]
- 33.Gairola CG. Free lung cell response of mice and rats to mainstream cigarette smoke exposure. Toxicol Appl Pharmacol. 1986;84:567–575. doi: 10.1016/0041-008x(86)90262-0. [DOI] [PubMed] [Google Scholar]
- 34.Robbins CS, Pouladi MA, Fattouh R, Dawe DE, Vujicic N, Richards CD, Jordana M, Inman MD, Stampfli MR. Mainstream cigarette smoke exposure attenuates airway immune inflammatory responses to surrogate and common environmental allergens in mice, despite evidence of increased systemic sensitization. J Immunol. 2005;175:2834–2842. doi: 10.4049/jimmunol.175.5.2834. [DOI] [PubMed] [Google Scholar]
- 35.Coote K, Nicholls A, Atherton HC, Sugar R, Danahay H. Mucociliary clearance is enhanced in rat models of cigarette smoke and lipopolysaccharide-induced lung disease. Exp Lung Res. 2004;30:59–71. doi: 10.1080/01902140490252885. [DOI] [PubMed] [Google Scholar]
- 36.Lewis SJ, Cherry NM, Mc L, Niven R, Barber PV, Wilde K, Povey AC. Cotinine levels and self-reported smoking status in patients attending a bronchoscopy clinic. Biomarkers. 2003;8:218–228. doi: 10.1080/1354750031000120125. [DOI] [PubMed] [Google Scholar]
- 37.Gaworski CL, Dozier MM, Gerhart JM, Rajendran N, Brennecke LH, Aranyi C, Heck JD. 13-Week inhalation toxicity study of menthol cigarette smoke. Food Chem Toxicol. 1997;35:683–692. doi: 10.1016/s0278-6915(97)00033-1. [DOI] [PubMed] [Google Scholar]
- 38.Witschi H, Espiritu I, Uyeminami D, Suffia M, Pinkerton KE. Lung tumor response in strain a mice exposed to tobacco smoke: some dose-effect relationships. Inhal Toxicol. 2004;16:27–32. doi: 10.1080/08958370490258372. [DOI] [PubMed] [Google Scholar]
- 39.Kazmierowski JA, Gallin JI, Reynolds HY. Mechanism for the inflammatory response in primate lungs. Demonstration and partial characterization of an alveolar macrophage-derived chemotactic factor with preferential activity for polymorphonuclear leukocytes. J Clin Invest. 1977;59:273–281. doi: 10.1172/JCI108638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cohen AB, Batra GK. Bronchoscopy and lung lavage induced bilateral pulmonary neutrophil influx and blood leukocytosis in dogs and monkeys. Am Rev Respir Dis. 1980;122:239–247. doi: 10.1164/arrd.1980.122.2.239. [DOI] [PubMed] [Google Scholar]
- 41.Weiss RA, Chanana AD, Joel DD. Localized pulmonary neutrophil influx induced by lung lavage in sheep. Lung. 1983;161:369–374. doi: 10.1007/BF02713886. [DOI] [PubMed] [Google Scholar]
- 42.Von Essen SG, Robbins RA, Spurzem JR, Thompson AB, McGranaghan SS, Rennard SI. Bronchoscopy with bronchoalveolar lavage causes neutrophil recruitment to the lower respiratory tract. Am Rev Respir Dis. 1991;144:848–854. doi: 10.1164/ajrccm/144.4.848. [DOI] [PubMed] [Google Scholar]
- 43.Hahn HL, Kleinschrot D, Hansen D. Nicotine increases ciliary beat frequency by a direct effect on respiratory cilia. Clin Investig. 1992;70:244–251. doi: 10.1007/BF00184658. [DOI] [PubMed] [Google Scholar]
- 44.Strieter RM, Kunkel SL. Chemokines. In: Crystal R, et al., editors. The Lung: Scientific Foundations. 2. Philadelphia: Lippincott-Raven Publishers; 1997. [Google Scholar]
- 45.Rhee CS, Hong SK, Min YG, Lee CH, Lee KS, Ahn SH, Park KS, Yi WJ. Effects of IL-1 beta, TNF-alpha, and TGF-beta on ciliary beat frequency of human nasal ciliated epithelial cells in vitro. Am J Rhinol. 1999;13:27–30. doi: 10.2500/105065899781389920. [DOI] [PubMed] [Google Scholar]
- 46.Chen JH, Takeno S, Osada R, Ueda T, Yajin K. Modulation of ciliary activity by tumor necrosis factor-alpha in cultured sinus epithelial cells. Possible roles of nitric oxide. Hiroshima J Med Sci. 2000;49:49–55. [PubMed] [Google Scholar]
- 47.Jain B, Rubinstein I, Robbins RA, Sisson JH. TNF-alpha and IL-1 beta upregulate nitric oxide-dependent ciliary motility in bovine airway epithelium. Am J Physiol. 1995;268:L911–L917. doi: 10.1152/ajplung.1995.268.6.L911. [DOI] [PubMed] [Google Scholar]
- 48.Yanaura S, Imamura N, Misawa M. Effects of beta-adrenoceptor stimulants on the canine tracheal ciliated cells. Jpn J Pharmacol. 1981;31:951–956. doi: 10.1254/jjp.31.951. [DOI] [PubMed] [Google Scholar]
- 49.Verdugo P, Johnson NT, Tam PY. β-Adrenergic stimulation of respiratory ciliary activity. J Appl Physiol. 1980;48:868–871. doi: 10.1152/jappl.1980.48.5.868. [DOI] [PubMed] [Google Scholar]
- 50.Wong LB, Yeates DB. Dynamics of epithelial function. Comments Theor Biol. 1997;4:183. [Google Scholar]
- 51.Verdugo P. Ca2+-dependent hormonal stimulation of ciliary activity. Nature. 1980;283:764–745. doi: 10.1038/283764a0. [DOI] [PubMed] [Google Scholar]
- 52.Uzlaner N, Priel Z. Interplay between the NO pathway and elevated [Ca2+]i enhances ciliary activity in rabbit trachea. J Physiol (Lond) 1999;516:179–190. doi: 10.1111/j.1469-7793.1999.179aa.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Feschenko MS, Stevenson E, Sweadner KJ. Interaction of protein kinase C and cAMP-dependent pathways in the phosphorylation of the Na,K-ATPase. J Biol Chem. 2000;275:34693–34700. doi: 10.1074/jbc.M005869200. [DOI] [PubMed] [Google Scholar]
- 54.Li TF, Zuscik MJ, Ionescu AM, Zhang X, Rosier RN, Schwarz EM, Drissi H, O’Keefe RJ. PGE2 inhibits chondrocyte differentiation through PKA and PKC signaling. Exp Cell Res. 2004;300:159–169. doi: 10.1016/j.yexcr.2004.06.019. [DOI] [PubMed] [Google Scholar]
- 55.Yuan N, Friedman J, Whaley BS, Clark RB. cAMP-dependent protein kinase and protein kinase C consensus site mutations of the beta-adrenergic receptor. Effect on desensitization and stimulation of adenylylcyclase. J Biol Chem. 1994;269:23032–23038. [PubMed] [Google Scholar]
- 56.Zhang XF, Guo SZ, Lu KH, Li HY, Li XD, Zhang LX, Yang L. Different roles of PKC and PKA in effect of interferon-gamma on proliferation and collagen synthesis of fibroblasts. Acta Pharmacol Sin. 2004;25:1320–1326. [PubMed] [Google Scholar]
- 57.Sugita S, Baxter DA, Byrne JH. Modulation of a cAMP/protein kinase A cascade by protein kinase C in sensory neurons of Aplysia. J Neurosci. 1997;17:7237–7244. doi: 10.1523/JNEUROSCI.17-19-07237.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Salathe M, Pratt MM, Wanner A. Cyclic AMP-dependent phosphorylation of a 26 kD axonemal protein in ovine cilia isolated from small tissue pieces. Am J Respir Cell Mol Biol. 1993;9:306–314. doi: 10.1165/ajrcmb/9.3.306. [DOI] [PubMed] [Google Scholar]
- 59.DiPilato LM, Cheng X, Zhang J. Fluorescent indicators of cAMP and Epac activation reveal differential dynamics of cAMP signaling within discrete subcellular compartments. Proc Natl Acad Sci U S A. 2004;101:16513–16518. doi: 10.1073/pnas.0405973101. [DOI] [PMC free article] [PubMed] [Google Scholar]