

Abstract
Mitochondrial trifunctional protein (MTP) is a hetero-octamer of four α and four β subunits that catalyzes the final three steps of mitochondrial long chain fatty acid β-oxidation. Human MTP deficiency causes Reye-like syndrome, cardiomyopathy, or sudden unexpected death. We used gene targeting to generate an MTP α subunit null allele and to produce mice that lack MTP α and β subunits. The Mtpa–/– fetuses accumulate long chain fatty acid metabolites and have low birth weight compared with the Mtpa+/– and Mtpa+/+ littermates. Mtpa–/– mice suffer neonatal hypoglycemia and sudden death 6–36 hours after birth. Analysis of the histopathological changes in the Mtpa–/– pups revealed rapid development of hepatic steatosis after birth and, later, significant necrosis and acute degeneration of the cardiac and diaphragmatic myocytes. This mouse model documents that intact mitochondrial long chain fatty acid oxidation is essential for fetal development and for survival after birth. Deficiency of MTP causes fetal growth retardation, neonatal hypoglycemia, and sudden death.
Introduction
Mitochondrial β-oxidation of fatty acids is the major source of energy for skeletal muscle and the heart, and it plays an essential role in intermediary metabolism in the liver. The β-oxidation cycle is a repetitive process of four steps. Mitochondrial trifunctional protein (MTP) is a hetero-octamer of four α and four β subunits associated with the inner mitochondrial membrane that utilizes long chain fatty acids as substrate (1, 2). The MTP α subunit (MTPα) N-terminal domain contains the long chain 3-enoyl-CoA hydratase activity that catalyzes the second step, while long chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD) activity resides in the C-terminal domain and catalyzes the third step. The MTP β subunit (MTPβ) has the long chain 3-ketoacyl-CoA thiolase activity and catalyzes the fourth step. Human genes coding for MTPα (HADHA) and MTPβ (HADHB) are characterized and localized to chromosome 2 (3–6).
Human defects in the MTP complex are recessively inherited and can be classified into two biochemical subgroups (2, 5, 7–11). The first has isolated LCHAD deficiency with normal or slightly reduced thiolase and hydratase activities, while the second has complete MTP deficiency with markedly reduced activity of all three enzymes. In the United States, a defect in MTP causing complete MTP deficiency or isolated LCHAD deficiency occurs about once in 38,000 pregnancies (11). Recently, we reported HADHA mutations and phenotypes in 24 patients (11). Patients with the more common, isolated LCHAD deficiency present predominantly with a Reye-like syndrome and carry a prevalent mutation (G1528C, E474Q) on one or both HADHA alleles, whereas patients with complete MTP deficiency present predominantly with cardiomyopathy or neuromyopathy and carry mutations other than the prevalent G1528C mutation. Individuals with either isolated LCHAD deficiency or complete MTP deficiency may also present with sudden, initially unexplained death in infancy (2, 4, 11–14). Furthermore, fetal MTP defects cause a fetal-maternal interaction with the development of maternal liver disease. Many heterozygote women who carry fetuses with isolated LCHAD deficiency develop acute fatty liver of pregnancy or the HELLP (hemolysis, elevated liver enzymes, and low platelets) syndrome (4, 11, 14–17). In addition, fetal and perinatal outcome may be affected by the fetal defects in MTP, as higher frequencies of prematurity and intrauterine growth retardation (IUGR) have been documented in these individuals (ref. 16; and J.A. Ibdah, unpublished data).
Here, we report the generation and characterization of a knockout mouse model for complete MTP deficiency with biochemical changes identical to those of human deficiency. Homozygous deficient mice suffer intrauterine fetal growth retardation, hypoglycemia, and early neonatal death.
Methods
Generation of MTP-deficient mice.
Primers from human α-subunit cDNA were used to amplify a segment of mouse strain 129/SvJ heart mRNA encoding the α subunit. This fragment of mouse cDNA was used to initially screen a mouse heart cDNA library. A full-length cDNA for the mouse cDNA was isolated. Primers designed from the cDNA coding regions were then used to isolate a P1 mouse 129/SvJ genomic clone. A 15-kb genomic fragment containing exons 1, 2, and 3 of Mtpa (the mouse gene homologous to human HADHA) was subcloned and used to create a targeting construct by replacing an Mtpa 1.6-kb region containing exon 1 by a neomycin phosphotransferase gene (neo) driven by phosphoglycerate kinase (PGK) promoter. To create this construct (Figure 1), an Mtpa 900 bp fragment upstream of the 1.6 kb region containing exon 1 was isolated and subcloned in the EcoRI-KpnI site of a pPNT vector between the neo and herpes simplex virus thymidine kinase (HSV-TK) expression cassettes in an opposite orientation. To create the long arm of the targeting construct, an Mtpa 4.7 intronic fragment downstream of exon 1 was isolated and subcloned in the XhoI-NotI site of the pPNT vector, in an opposite orientation. RW4 embryonic stem (ES) cells from 129X1/SvJ strain (Genome Systems Inc., St. Louis, Missouri, USA) were then transfected with the replacement-targeting vector using electroporation. Positive/negative drug selection strategy was employed and 110 colonies were selected. Three colonies were identified by PCR and Southern blot analyses that successfully underwent homologous recombination and contained the neo mutation. Two hundred thirty-four C57BL/6J blastocysts were injected with the successfully targeted mycoplasma-free ES cells and implanted into pseudopregnant C57BL/6J female mice. Eighteen male chimeric offspring were mated with NIH Swiss black female mice to produce F1 progeny. The care of the animals was in accordance with Wake Forest University School of Medicine and Institutional Animal Care and Use Committee guidelines.
Figure 1.

Targeting of the mouse Mtpa gene. A schematic diagram of MTPα knockout construct. Restriction enzyme sites are indicated by E, EcoRI; P, PstI; Hc, HincI; N, NotI; K, KpnI; and X, XhoI. HSV-TK is the herpes simplex virus thymidine kinase cassette in the pPNT targeting vector.
Genotyping of mice.
To facilitate genotyping analysis of the large number of mice generated, we designed three oligonucleotide primers to distinguish the mutant allele from the wild-type allele by PCR (Figure 1). Separate PCR analyses were used to distinguish the PCR products (∼950 bp) from the wild-type and mutant alleles. All PCR analyses were confirmed in duplicates. We verified the initial PCR analyses by Southern blot analysis in 12–16 mice of each genotype. For Southern blot analysis, 10 μg genomic DNA was subjected to EcoRI and NotI digestion prior to transfer, then hybridized with a 32P-labeled probe. We used an approximately 750-bp PstI fragment from the Mtpa 5′ flanking region located outside of the targeting vector (Figure 1) as a probe to screen for correctly targeted ES cell clones and subsequent mutant mice. Replacement of the Mtpa exon 1 and the flanking regions by the PGK-neo cassette deleted a NotI restriction site upstream of exon 1 (Figure 1). Consequently, this probe detected a 2.2-kb fragment in the wild-type allele and a 4.1-kb fragment in the mutant allele containing the PGK-neo cassette.
Northern blot analysis.
Total RNA from various tissues was isolated using the guanidinium thiocyanate method (18). RNA samples were analyzed by formaldehyde gel electrophoresis and ethidium bromide staining. Northern blot analysis was performed using the mouse α-subunit 32P-cDNA probe. Staining of the transferred RNA with ethidium bromide was used to ensure uniform total cellular RNA recovery and transfer.
Western blot analysis.
This was performed following 10% SDS-PAGE according to Laemmli (19) with rabbit polyclonal antibodies raised against the mouse LCHAD domain of MTPα, the entire mouse MTPβ, and the entire mouse short chain 3-hydroxyacyl-CoA dehydrogenase (SCHAD) expressed in bacteria. For this, the corresponding cDNA was placed into the NdeI and SalI sites of the bacterial expression plasmid, pet21a (Novagen, Madison, Wisconsin, USA). After transformation into Escherichia coli and induction with isopropyl β-D-thiogalactoside for 4 hours, bacteria were lysed by sonication and centrifuged at 5,000 g. The supernatant was analyzed by SDS gel electrophoresis. The corresponding band was excised form the gel and injected into rabbits as the antigen. Antisera were diluted 1:2000.
Biochemical analyses.
All blood samples were obtained at the time of sacrifice by cardiac puncture. Sera were analyzed using an automated analyzer (Technicon CHEM I; Bayer Corp., Tarrytown, New York, USA) for measurement of glucose, alanine aminotransferase (ALT), bilirubin, gamma glutamyl transferase (GGT), and urea levels. Serum and tissue FFAs, serum acyl carnitines, and urine organic acids were analyzed by using gas chromatography–mass spectrometry as previously described (20). The activities of LCHAD and long chain 3-ketoacyl-CoA thiolase were measured in crude extracts of liver, skeletal muscle, or heart tissues, as previously described (21). Because the active sites of these two enzymes are in the α and β subunits of the MTP, respectively, a deficiency of both activities is always associated with a lack of activity of long chain 2,3 enoyl-CoA hydratase whose active site is in the α subunit.
Histopathological analysis.
Tissues were collected and fixed in 4% paraformaldehyde for histology or 2.5% glutaraldehyde for electron microscopy. Tissues for routine histology were trimmed to less than 3 mm in thickness, embedded in paraffin, sectioned at 4 μm, and stained with hematoxylin and eosin. Tissues used to detect the presence of fat were quick-frozen, cryostatically sectioned at 4 μm, and stained using Oil Red O (Hartman-Leddon Co., Philadelphia, Pennsylvania, USA). Tissues used for electron microscopy were postfixed in osmium tetroxide, embedded in Spurr’s resin, sectioned at 90 nm, and stained with uranyl acetate/lead citrate. All sections were reviewed by a board-certified veterinary pathologist who was blinded to the genotype of the animals. Standard histopathologic criteria were used for the assessment of tissue viability, necrosis, and degenerative changes at the light-microscopic and electron-microscopic levels.
Results
Generation and molecular characterization of MTP-deficient mice.
A targeting vector was used to replace Mtpa exon 1, a flanking 5′ upstream region, and an intron 1 region by a PGK-neomycin-resistance cassette (neo) using homologous recombination in 129X1/SvJ ES cells (Figure 1). Heterozygous F1 mice were then intercrossed to produce the three genotypes: wild-type (Mtpa+/+), heterozygotes (Mtpa+/–), and homozygotes (Mtpa–/–). All mice analyzed in this study were F2 and F3 generations.
Figure 2 shows a representative Southern blot (Figure 2a) using EcoRI and NotI digestion and the probe illustrated in Figure 1, a Northern blot using α subunit cDNA probe (Figure 2b), and a Western blot with β subunit antibodies as a surrogate for the MTP complex (Figure 2c) with DNA, RNA, and protein isolated from the livers and hearts of pups with different genotypes sacrificed at birth. Mtpa–/– mice have no detectable MTPα mRNA, whereas Mtpa+/– mice have intermediate levels. High expression is obvious in the cardiac muscle at birth in the Mtpa+/+ mice (Figure 2b). The MTP complex is absent in the Mtpa–/– mice and has intermediate expression in the Mtpa+/– mice for both α and β subunits (Figure 2c, data for the α subunit not shown). As a control, we used antibodies raised against the homologous SCHAD to determine SCHAD protein expression in pups with different genotypes. The results in Figure 2c show equal expression of SCHAD at birth in all three genotypes without compensatory increase in the MTP-deficient mice. Thus, Mtpa–/– mice lack MTP complex as determined by Western blot analysis and hence lack the activity of all MTP enzymes. The above analyses were confirmed in 12–16 pups of each genotype.
Figure 2.

Molecular characterization of MTP-deficient mice. Representative Southern blot (a), Northern blot (b), and Western blot (c) analyses using DNA, total RNA, and protein isolated from liver and heart tissues from pups sacrificed at birth with different genotypes: Mtpa+/+ (+/+), Mtpa+/– (+/–), and Mtpa–/– (–/–). Southern blot analysis was performed using EcoRI and NotI digestion. Mtpa+/+ mice had one band corresponding to the wild-type 2.2-kb fragment, Mtpa+/– mice had two bands corresponding to the wild-type 2.2-kb fragment and a mutant 4.1-kb fragment containing the neo cassette (see Figure 1), and Mtpa–/– mice had the mutant 4.1-kb band. An α-subunit cDNA probe was used in the Northern blot analysis, and antibodies raised against MTPβ or SCHAD were used in the Western blot analyses.
Sudden neonatal death of MTP-deficient mice.
All Mtpa–/– mice died within 6–36 hours after birth, most within 12–24 hours. Thirty-eight litters (345 pups) of crosses between Mtpa+/– parents and 27 control litters (244 pups) from Mtpa+/+ mice were observed closely for 48 hours after birth (at least four times a day by two different observers). Eighty-one pups died in the litters of the crosses between Mtpa+/– mice. Of these, 78 were Mtpa–/–, two were Mtpa+/–, and one was Mtpa+/+ (100%, 1.2%, and 1.2% of the Mtpa–/–, Mtpa+/–, and Mtpa+/+ pups, respectively). By comparison, four pups (1.6%) died in the litters from Mtpa+/+ mice. Most of the Mtpa–/– pups’ deaths were sudden. However, some Mtpa–/– pups became symptomatic (weak with lack of suckling activity, separated from the litter) for a brief period of time (1–3 hours) prior to death.
We measured serum levels of glucose, bilirubin, parenchymal and cholestatic hepatic enzyme markers (ALT and GGT, respectively), and serum urea in pups of different genotypes sacrificed within 36 hours after birth, including Mtpa–/– pups that were symptomatic (Table 1). The results show significant differences in the glucose levels between the Mtpa–/– pups and Mtpa+/+ or Mtpa+/– pups (P < 0.001 and P < 0.002, respectively), but the difference between the heterozygotes and wild-type was not significant (P = 0.212). There were mild but significant elevations of ALT in the Mtpa–/– pups compared to the Mtpa+/+ or Mtpa+/– pups (P < 0.05), with more significant elevations of ALT, GGT, and bilirubin in the symptomatic Mtpa–/– pups. The urea level was also significantly elevated in the Mtpa–/– pups, with a normal creatinine level, suggesting that Mtpa–/– pups were dehydrated.
Table 1.
Serum laboratory testsA and genotypes in MTP knockout mice

We performed frequent intraperitoneal glucose injections starting at birth in seven litters of crosses between Mtpa+/– mice (50 μl 5% dextrose, every 4 hours for 5 days). Litters were not separated from dams. Despite treatment, 16 Mtpa–/– pups died within 96 hours after birth: 13 within 36 hours, two within 48–72 hours, and one at 96 hours. None of the remaining 46 surviving pups was homozygous. Thus, treatment with glucose prolonged survival in some affected mice, but did not prevent death.
Biochemical analysis.
Characterization of the biochemical phenotype was based on the analysis of coded specimens of serum, liver, and urine of five Mtpa+/+, five Mtpa+/–, five asymptomatic Mtpa–/–, and eight symptomatic Mtpa–/– mice sacrificed within 24 hours of birth. In serum, quantitative analyses were performed of the following metabolites: carnitine, C2-C18 acylcarnitines and C12-C16 3-hydroxy acylcarnitines, C8-C18 total fatty acids, and C6-C16 free 3-hydroxy fatty acids. C6-C10 dicarboxylic acids and C6-C14 3-hydroxy dicarboxylic acids were also measured in urine. C8-C18 fatty acids and carnitine were analyzed in liver homogenates. Differences between Mtpa+/+ and Mtpa+/– mice were not statistically significant (P > 0.1). However, the Mtpa–/– mice clearly had abnormal biochemical features identical to those observed in human cases with MTP deficiency (22) including elevated C8-C14 urine dicarboxylic acids and C14-C18 serum acyl carnitines and fatty acids (P < 0.05). These biochemical changes in the Mtpa–/– mice were most significant in the symptomatic mice (Table 2). Serum free carnitine levels were significantly reduced in the asymptomatic and symptomatic Mtpa–/– mice (P < 0.01) compared with the Mtpa+/+ and Mtpa+/– mice. Serum free carnitine levels were 19.2 ± 14.2, 18 ± 9, 57.7 ± 20, and 46.4 ± 22.8 μmol/l for asymptomatic Mtpa–/–, symptomatic Mtpa–/–, Mtpa+/+, and Mtpa+/– mice, respectively.
Table 2.
Representative mean values of biochemical metabolites in Mtpa+/+ and symptomatic Mtpa–/– mice

Enzymatic measurements of LCHAD and long chain thiolase activities in samples of liver, heart, and muscle homogenates of five Mtpa+/+, five Mtpa+/–, and five Mtpa–/– mice sacrificed at birth revealed marked reductions similar to those reported in human MTP deficiency (2, 5, 6, 9, 11, 21). Table 3 shows the activities of cardiac LCHAD and long chain 3-ketothiolase in comparison with activities of SCHAD and short chain 3-ketothiolase. Similar results were obtained from enzymatic measurements in the other tissues (data not shown).
Table 3.
Heart enzymatic activitiesA in MTP knockout mice

Histopathological analysis.
We studied the histopathological changes in 60 pups from seven litters of crosses between Mtpa+/– mice sacrificed at 0, 6, 12, and 24 hours after birth. In these litters, five pups died within 6–24 hours after birth, and another four pups became symptomatic and were sacrificed. All symptomatic and dead pups were Mtpa–/– by PCR and Southern blot analysis. Gross findings included diffuse hepatic enlargement and yellow hepatic discoloration in the symptomatic and dead MTP-deficient pups. No gross abnormalities were noted in other organs. Table 4 shows the frequency and severity of the histological lesions in pups with different genotypes. Representative histological changes in the dead Mtpa–/– pups compared with Mtpa+/+ pups are illustrated in Figure 3. All dead and symptomatic Mtpa–/– pups and three-fifths of asymptomatic Mtpa–/– pups showed moderate to severe hepatic lipidosis (Figure 3b). Oil Red O–stain on frozen sections showed micro- and macrovesicular fatty infiltration pattern (data not shown). All dead Mtpa–/– pups had moderate to severe necrosis and vacuolation, with acute degenerative changes of the cardiac and diaphragmatic myocytes (Figure 3, d and f). All symptomatic Mtpa–/– pups had moderate to severe cardiomyocyte degeneration and necrosis, but none had acute degenerative changes in the diaphragm. All Mtpa–/– pups sacrificed immediately at birth showed no histological lesions. Thus, Mtpa–/– pups develop hepatic lipidosis very rapidly after birth and, later, cardiac and diaphragmatic lesions.
Table 4.
Comparison of histological changes among various genotypes in MTP knockout mice

Figure 3.

Histopathological analyses of MTP-deficient mice. Representative sections from liver, diaphragm, and heart obtained from approximately 12-hour-old Mtpa+/+ (a, c, and e, respectively) and dead Mtpa–/– mice (b, d, and f, respectively) stained with hematoxylin and eosin (×40).
Three dead and three symptomatic pups were also examined for ultrastructural changes by electron microscopy. Figure 4 shows the ultrastructural changes in the hepatocytes of a symptomatic pup with significant fatty vacuolation and swollen, distorted mitochondria.
Figure 4.

Hepatic ultrastructural changes in MTP-deficient mice. Representative electron microscopy of a liver section from a symptomatic Mtpa–/– mouse 12 hours after birth showing extensive lipidosis, swelling, and distortion of mitochondria. L, lipid; Mt, mitochondria; N, nucleus.
Gestational complications.
We investigated the possibility of intrauterine fetal death of the Mtpa–/– fetuses by performing cesarean section and examining the fetuses at 9, 12, 15, and 19 days post coitus (dpc) in 27 litters of crosses between Mtpa+/– mice and 11 litters from Mtpa+/+ mice for comparison. There was no statistically significant difference in the litter size (9.1 ± 3.1 and 9.7 ± 2.3, respectively) or genotype ratios (∼1:2:1, Mtpa+/+:Mtpa+/–:Mtpa–/–, respectively) at different embryonic stages. Similar results were obtained by sacrificing 28 litters of crosses between Mtpa+/– mice and 27 litters from Mtpa+/+ mice for comparison. Thus, MTP-deficient mice do not suffer intrauterine fetal death to a significant extent.
However, Mtpa–/– fetuses suffer significant IUGR. We measured birth weights in 28 litters of crosses between Mtpa+/– mice (67 Mtpa+/+, 132 Mtpa+/–, and 70 Mtpa–/– pups) sacrificed at birth. The mean birth weight values (±SD) for Mtpa+/+, Mtpa+/–, and Mtpa–/– pups were 1.69 ± 0.2, 1.64 ± 0.26, and 1.42 ± 0.16, respectively. The low birth weight of the Mtpa–/– pups was statistically significant (P < 0.001) when compared with the Mtpa+/+ or Mtpa+/– pups. The difference in birth weight between Mtpa+/+ and Mtpa+/– pups was not statistically significant (P = 0.169). Our analysis of birth weights in the 19 dpc fetuses confirms this observation (data not shown).
We evaluated the possibility that maternal complications or placental abnormalities cause the IUGR. Gross clinical evaluation of the pregnant Mtpa+/– dams mated with Mtpa+/– males and histopathological evaluation of the liver, heart, and muscle at different stages during pregnancy (9, 12, 15, and 19 dpc, n = 5, 5, 5, and 12, respectively) revealed no abnormalities. Similarly, histopathological examination of the placentae with different fetal genotypes did not reveal any abnormalities (n = 12 of each fetal genotype). Maternal laboratory data including liver function tests and glucose levels were not statistically significant at P ≤ 0.05 when compared with Mtpa+/+ pregnant dams carrying Mtpa+/+ fetuses (n = 27 Mtpa+/– and 11 Mtpa+/+ dams).
We measured serum acylcarnitines and total fatty acids in 19 dpc fetuses in eight litters of crosses between Mtpa+/– mice (19 Mtpa–/–, 31 Mtpa+/–, and 15 Mtpa+/+ fetuses). Mtpa–/– fetuses had significantly higher levels of long chain acylcarnitines (Table 5) and long chain fatty acids (data not shown) with particular elevations of unsaturated species (C16:1, C18:1, C18:2). These results document that MTP-deficient fetuses accumulate long chain fatty acids and metabolites secondary to impaired utilization of long chain fatty acids in the Mtpa–/– fetus and/or placenta.
Table 5.
Representative mean values of serum acylcarnitines (μmol/l) in 19-dpc fetuses

Developmental stage–specific MTP expression.
We determined MTP α and β subunit expression in Mtpa+/+ fetuses (livers and whole homogenized mice), in placenta at 9, 15, and 19 dpc, and in livers of Mtpa+/+ mice at birth, and at 1 day, 2 days, 1 week, 2 weeks, 3 weeks, 2 months, 4 months, 8 months, and 16 months of age. We detected low but significant fetal and placental expression in pregnancy starting at 9 dpc. A striking increase in neonatal hepatic MTP expression occurs after birth, reaching levels at 48 hours close to those observed in adult mice. Figure 5 shows representative Western blot analyses using antibodies raised against MTPβ in a 19 dpc Mtpa+/+ placenta, and in livers from 19-dpc Mtpa+/+ fetus, 1-day, 2-day, and 8-month-old Mtpa+/+ mice. These results were confirmed in three different animals of each developmental stage.
Figure 5.
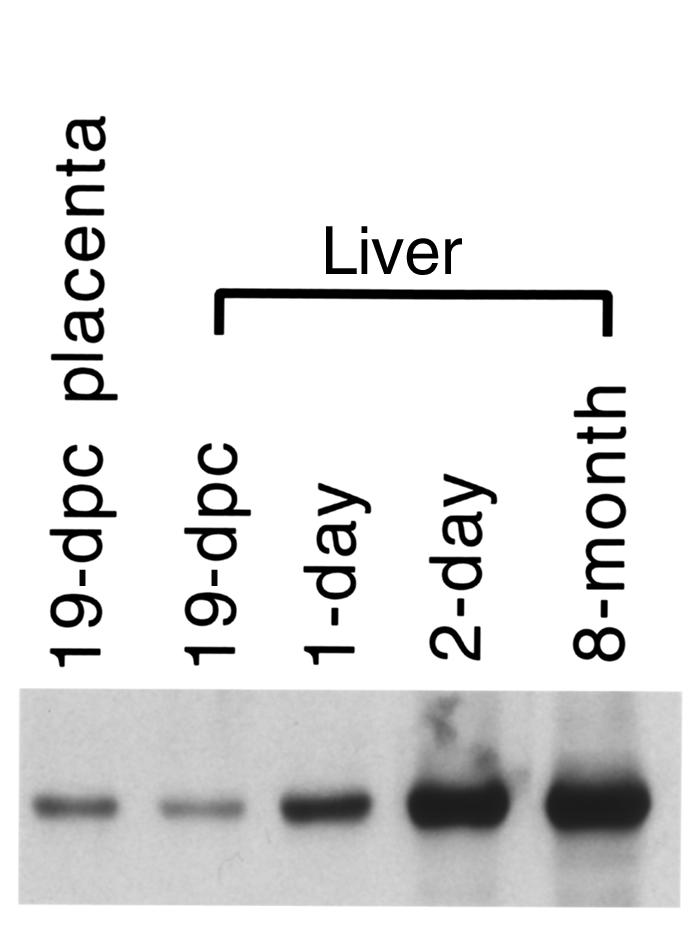
Developmental stage–specific expression of MTP. Representative Western blot analyses using protein isolated from 19-dpc fetal Mtpa+/+ placentae and livers from 19-dpc Mtpa+/+ fetus, 1-day, 2-day, and 8-month-old Mtpa+/+ mice. Antibodies raised against MTPβ were used in the Western blot analyses.
Discussion
Our results demonstrate that the MTP-deficient knockout mouse is a valid model for human MTP deficiency and that it has important implications for human disease. We have proven that intact mitochondrial long chain fatty acid oxidation is essential for fetal development and for survival after birth. Defective MTP in mice causes IUGR, neonatal hypoglycemia, and sudden neonatal death. All are serious and common human disorders.
Human studies (ref. 16; and J.A. Ibdah, unpublished data) document IUGR in fetuses with mitochondrial MTP enzymatic defects. Because most of the reported human cases were isolated LCHAD deficiency with associated maternal complications, it is difficult to determine whether the observed IUGR is secondary to fetal or maternal factors. Our current study in mice demonstrates that fetal MTP deficiency causes IUGR without associated maternal complications. Our data document an early placental and fetal expression of MTP in mice (Figure 5) suggesting a reliance of the placenta and/or the fetus on long chain fatty acid oxidation as a source of energy. This is consistent with our observation of accumulation of long chain FFAs and acylcarnitines in the Mtpa–/– fetuses (Table 5). Thus, intact mitochondrial long chain fatty acid oxidation is important for fetal development, and its impairment causes restriction of fetal growth.
The lack of the maternal phenotype in the MTP-deficient mouse model is consistent with our earlier observation in families with MTP mutations. Human maternal complications developed only in association with isolated fetal LCHAD deficiency, presumably secondary to accumulation of hepatotoxic fatty acid metabolites in the maternal circulation (11). However, it should be noted that the presence of multiple fetal genotypes in these multiparous mice may have obscured the development of the maternal phenotype.
MTP deficiency in mice causes the severe phenotype of early sudden death. Sudden death in the MTP-deficient mice occurs shortly after birth; this coincides with the neonatal metabolic switch in nutritional fuel from the maternal glucose to breast milk, which is high in fat. Our expression studies (Figure 5) reveal a major increase in the MTP expression within 48 hours after birth. Thus, there is high demand for long chain fatty acid β-oxidation after birth that is essential for survival consistent with reliance upon maternal milk with high fat content as the nutritional energy source. The high expression of the MTP in the neonatal heart (Figure 2) suggests an early reliance of the heart compared with other tissues on mitochondrial β-oxidation as a source of energy. Hence, the neonatal heart is probably more susceptible than other tissues to the toxic effect of long chain FFAs and acylcarnitines. Our biochemical and histopathological analyses in the Mtpa–/– mice after birth offer insight into the likely mechanism of sudden death. The histological analyses suggest that the underlying etiology for sudden death is the cardiac and diaphragmatic lesions (Figure 3, d and f). We suggest that the cardiac lesions may have caused cardiac arrhythmias, as found in newborns with documented long chain fatty acid oxidation disorders (23). We also suggest that the diaphragmatic lesions may have caused dysfunction of the diaphragm and subsequent respiratory insufficiency.
The phenotypes in the Mtpa–/– mice are strikingly severe compared with those observed in mice deficient in long chain acyl-CoA dehydrogenase (LCAD) (24). In humans, LCAD apparently has a limited role in mitochondrial long chain fatty acid oxidation, and to date, there are no reports of its deficiency. Significant fetal loss was reported in LCAD–/– mice, but those achieving birth appeared normal under nonfasting conditions, although about 5% died suddenly 4–14 weeks after birth. Thus, both Mtpa–/– and LCAD–/– mouse models suggest an important role for long chain fatty acid oxidation in fetal development. However, our mouse model proves that intact MTP, and, hence, intact long chain fatty acid oxidation, is essential for survival after birth.
In summary, the generation of this MTP-deficient mouse model has documented a pivotal role of long chain fatty acid β-oxidation in fetal development and has proven that intact long chain fatty acid oxidation is essential for survival. Mitochondrial long chain fatty acid oxidation disorders should be considered a significant cause for IUGR, neonatal hypoglycemia, and sudden death.
Acknowledgments
This study was supported by NIH grants DK-02574 (J.A. Ibdah) and AM-20407 (A.W. Strauss). Special thanks go to Hermina Borgerink for her expert assistance in the histopathological analysis. We also thank Laurie O’Brien and Beverly Gibson for their extensive efforts in preparing the antibodies to SCHAD and MTP subunits, and Harold Sims for his expert advice in mouse ES cell culture.
References
- 1.Uchida Y, Izai K, Orii T, Hashimoto T. Novel fatty acid β-oxidation enzymes in rat liver mitochondria. II. Purification and properties of enoyl-coenzyme A (CoA) hydratase/3-hydroxyacyl-CoA dehydrogenase/3-ketoacyl-CoA thiolase trifunctional protein. J Biol Chem. 1992;267:1034–1041. [PubMed] [Google Scholar]
- 2.Jackson S, et al. Combined enzyme defect of mitochondrial fatty acid oxidation. J Clin Invest. 1992;90:1219–1225. doi: 10.1172/JCI115983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kamijo T, Aoyama T, Komiyama A, Hashimoto T. Structural analysis of cDNAs for subunits of human mitochondrial fatty acid β-oxidation trifunctional protein. Biochem Biophys Res Commun. 1994;199:818–825. doi: 10.1006/bbrc.1994.1302. [DOI] [PubMed] [Google Scholar]
- 4.Sims HF, et al. The molecular basis of pediatric long chain 3-hydroxyacyl-CoA dehydrogenase deficiency associated with maternal acute fatty liver of pregnancy. Proc Natl Acad Sci USA. 1995;92:841–845. doi: 10.1073/pnas.92.3.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ushikubo S, et al. Molecular characterization of mitochondrial trifunctional protein deficiency: formation of the enzyme complex is important for stabilization of both α- and β-subunits. Am J Hum Genet. 1996;58:979–988. [PMC free article] [PubMed] [Google Scholar]
- 6.Ijlst L, Ruiter JPN, Hoovers JMN, Jakobs ME, Wanders RJA. Common missense mutation G1528C in long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. Characterization and expression of the mutant protein, mutation analysis on genomic DNA and chromosomal localization of the mitochondrial trifunctional protein alpha subunit gene. J Clin Invest. 1996;98:1028–1033. doi: 10.1172/JCI118863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kamijo T, et al. Mitochondrial trifunctional protein deficiency. Catalytic heterogeneity of the mutant enzyme in two patients. J Clin Invest. 1994;93:1740–1747. doi: 10.1172/JCI117158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hagenfeldt L, Venizelos N, von Dobeln U. Clinical and biochemical presentation of long chain 3-hydroxyacyl-CoA dehydrogenase deficiency. J Inherit Metab Dis. 1995;18:245–248. doi: 10.1007/BF00711779. [DOI] [PubMed] [Google Scholar]
- 9.Brackett JC, et al. Two α-subunit donor splice site mutations cause human trifunctional protein deficiency. J Clin Invest. 1995;95:2076–2082. doi: 10.1172/JCI117894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ibdah JA, et al. Mild trifunctional protein deficiency is associated with progressive neuropathy and myopathy and suggests a novel genotype-phenotype correlation. J Clin Invest. 1998;102:1193–1199. doi: 10.1172/JCI2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ibdah JA, et al. A fetal fatty-acid oxidation disorder as a cause of liver disease in pregnant women. N Engl J Med. 1999;340:1723–1731. doi: 10.1056/NEJM199906033402204. [DOI] [PubMed] [Google Scholar]
- 12.Wanders RJA, et al. Sudden infant death and long-chain 3-hydroxyacyl-CoA dehydrogenase. Lancet. 1989;2:52–53. doi: 10.1016/s0140-6736(89)90300-0. [DOI] [PubMed] [Google Scholar]
- 13.Saudubray JM, et al. Recognition and management of fatty acid oxidation defects: a series of 107 patients. J Inherit Metab Dis. 1999;22:488–502. doi: 10.1023/a:1005556207210. [DOI] [PubMed] [Google Scholar]
- 14.Ibdah JA, Dasouki MJ, Strauss AW. Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: variable expressivity of maternal illness during pregnancy and unusual presentation with infantile cholestasis and hypocalcaemia. J Inherit Metab Dis. 1999;22:811–814. doi: 10.1023/a:1005506024055. [DOI] [PubMed] [Google Scholar]
- 15.Wilcken B, Leung KC, Hammond J, Kamath R, Leonard JV. Pregnancy and fetal long-chain 3-hydroxyacyl coenzyme A dehydrogenase deficiency. Lancet. 1993;341:407–408. doi: 10.1016/0140-6736(93)92993-4. [DOI] [PubMed] [Google Scholar]
- 16.Tyni T, Ekholm E, Pihko H. Pregnancy complications are frequent in long-chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency. Am J Obstet Gynecol. 1998;178:603–608. doi: 10.1016/s0002-9378(98)70446-6. [DOI] [PubMed] [Google Scholar]
- 17.Ibdah JA, Yang Z, Bennett MJ. Liver disease in pregnancy and fetal fatty acid oxidation defects. Mol Genet Metab. 2000;71:182–189. doi: 10.1006/mgme.2000.3065. [DOI] [PubMed] [Google Scholar]
- 18.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 19.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 20.Boles RG, Martin SK, Blitzer MG, Rinaldo P. Biochemical diagnosis of fatty acid oxidation disorders by metabolite analysis of postmortem liver. Hum Pathol. 1994;25:735–741. doi: 10.1016/0046-8177(94)90240-2. [DOI] [PubMed] [Google Scholar]
- 21.Wanders RJA, et al. Human trifunctional protein deficiency: a new disorder of mitochondrial fatty acid beta-oxidation. Biochem Biophys Res Commun. 1992;188:1139–1145. doi: 10.1016/0006-291x(92)91350-y. [DOI] [PubMed] [Google Scholar]
- 22.Wanders RJA, et al. Disorders of mitochondrial fatty acyl-CoA β-oxidation. J Inherit Metab Dis. 1999;22:442–487. doi: 10.1023/a:1005504223140. [DOI] [PubMed] [Google Scholar]
- 23.Bonnet D, et al. Arrhythmias and conduction defects as presenting symptoms of fatty acid oxidation disorders in children. Circulation. 1999;100:2248–2253. doi: 10.1161/01.cir.100.22.2248. [DOI] [PubMed] [Google Scholar]
- 24.Kurtz DM, et al. Targeted disruption of mouse long-chain acyl-CoA dehydrogenase gene reveals crucial roles for fatty acid oxidation. Proc Natl Acad Sci USA. 1998;95:15592–15597. doi: 10.1073/pnas.95.26.15592. [DOI] [PMC free article] [PubMed] [Google Scholar]