

Abstract
The clinical use of doxorubicin, an anthracycline chemotherapeutic agent, is limited by cardiotoxicity, particularly when combined with herceptin, an antibody that blocks the HER2 receptor. Doxorubicin induces cyclooxygenase–2 (COX-2) activity in rat neonatal cardiomyocytes. This expression of COX-2 limits doxorubicin-induced cardiac cell injury, raising the possibility that the administration of a prostaglandin may protect the heart during the in vivo administration of doxorubicin. Doxorubicin (15 mg/kg) administered to adult male Sprague Dawley rats induced COX-2 expression and activity in cardiac tissue. Prostacyclin generation measured as the excretion of 2,3-dinor-6-keto-PGF1α also increased, and this was blocked by a COX-2 inhibitor, SC236. In contrast, administration of a COX-1 inhibitor SC560 at a dose that reduced serum thromboxane B2 by more than 80% did not prevent the doxorubicin-induced increase in prostacyclin generation. Doxorubicin increased cardiac injury, detected as a rise in plasma cardiac troponin T, serum lactate dehydrogenase, and cardiomyocyte apoptosis; this was aggravated by coadministration of SC236 but not SC560. The degree of injury in animals treated with a combination of doxorubicin and SC236 was attenuated by prior administration of the prostacyclin analogue iloprost. These data raise the possibility of protecting the heart during the administration of doxorubicin by prior administration of prostacyclin.
Introduction
Doxorubicin (DX) is an anthracycline that is a highly effective chemotherapeutic agent used largely in the treatment of solid tumors. However, the dose of DX is limited owing to a dose-related cardiac toxicity (1–3). In cardiac cells, DX is metabolized to the corresponding semiquinone free radical by flavin reductases (4, 5). This or a related species induces apoptosis in cardiomyocytes and is prevented by free radical-scavengers (6) or by chelating iron (7).
We have previously shown that DX induces cyclooxygenase (COX) activity in rat neonatal cardiomyocytes (8). COX catalyzes the first step in the conversion of arachidonic acid to prostaglandins (9). Two isoforms of COX have been identified that are the products of distinct genes. COX-1 is expressed in most cells and is the only isoform found in normal cardiomyocytes. COX-2 is largely absent from cells but is induced by a number of factors (10, 11) including presence of free radicals (8) and hypoxia (12). The differential expression of COX-1 and COX-2 in part reflects differences in the promoter structure of the two genes, with the COX-2 promoter containing the response elements of an inflammatory or acute-phase gene (13, 14). COX-2 expression is also regulated by modulation of mRNA stability (15, 16). In several types of cells, COX-2 expression has been shown to protect against apoptosis (17, 18). In rat neonatal cardiomyocytes, the induction of COX activity by DX is due to COX-2 gene expression (9). Inhibition of COX-2 aggravated the injury of the cells by DX, detected as the release of LDH or as apoptosis, and a prostaglandin prevented the injury. These findings raise the possibility of protecting the heart during treatment with DX by exogenous administration of a prostaglandin. Here, we examine whether DX induces cardiac injury in vivo in the rat and whether this is regulated by prostaglandins.
Methods
DX was from Pharmacia & Upjohn SpA (Milan, Italy). The 3,3′-diaminobenzidine tetrahydrochloride (DAB), Harris hematoxylin solution, Permount, and commonly used laboratory chemicals were obtained from Sigma Chemical Co. (St. Louis, Missouri, USA). Deuterated eicosanoid standards, NS-398, and arachidonic acid were obtained from Cayman Chemical Co. (Ann Arbor, Michigan, USA). Goat polyclonal anti-COX-1 antibody was from Oxford Biomedical Research Inc. (Rochester Hills, Missouri, USA). Monoclonal anti-mouse COX-2 antibody (R6), SC560, and SC236 were gifts from P. Isakson (Pharmacia Corp., Skokie, Illinois, USA). Immunohistochemical analysis kits containing secondary antibodies and reagents were from DAKO Corp. (Carpinteria, California, USA). Iloprost was obtained from Schering Aktiengesellschaff (Berlin, Germany). Enzyme immunoassays were obtained from R&D Systems Inc. (Minneapolis, Minnesota, USA).
Model.
Male Sprague Dawley rats (6–8 weeks old; 200–250 g) were studied. All experiments involving the use of rats were conducted in accordance with protocols approved by the institutional Biomedical Research Committee and with a license granted under the Cruelty to Animals Acts of 1876 by the Department of Health of Ireland. All study drugs were administered by intraperitoneal injection.
A total of 15 mg/kg of DX or an equal volume of DMSO (Sigma Chemical Co.) was injected into the intraperitoneal space. Three hours after the injection, general anesthesia was induced and maintained by inhalation of halothane 1.5%–2% in an animal-specific anesthetic chamber. A transverse subdiaphragmatic incision was made, and a section of diaphragm (∼0.5 × 0.3 cm) was dissected and removed. The thoracic cage was immediately opened, and the heart was dissected from the thoracic cavity. The tissues were analyzed for prostaglandin generation or fixed in formalin saline solution (0.9% NaCl and 10% formaldehyde) for immunohistochemistry. In additional experiments, the animals were treated with the following: (a) Three milligrams per kilogram of the selective COX-2 inhibitor SC236 3, followed 1 hour later by 3 mg/kg of SC236 and 15 mg/kg of DX. (b) A total of 3–10 mg/kg of the selective COX-1 inhibitor SC560, followed 1 hour later by 3–10 mg/kg of SC560 and 15 mg/kg of DX. (c) Two milligrams per kilogram of indomethacin (a nonselective cyclooxygenase inhibitor), followed 1 hour later by 2 mg/kg of indomethacin and 15 mg/kg of DX. (d) A total of 9 μg/kg of iloprost in three divided doses at 12-hour intervals; after the third dose, the animals were treated as above.
Immunohistochemistry.
The tissue was processed and embedded in wax using a Raymond Lamb Blockmaster II. Sections (5–8 μm) were cut onto silane-coated slides. The slides were baked overnight at 37°C and then deparaffinized and hydrated as follows: xylene (three times for 5 minutes), 100% ethanol (3 minutes), 90% ethanol (3 minutes), 70% ethanol (3 minutes), 50% ethanol (3 minutes), 30% ethanol (5 minutes), Tris-MgCl2 (5 minutes), and PBS (5 minutes). The slides were washed in PBS for 5 minutes and incubated in 0.3% hydrogen peroxide (H2O2) in methanol for 30 minutes, to quench endogenous peroxidase activity. This was followed by three additional 5-minute washes in PBS. After blocking for 1 hour in normal serum from the species from which the secondary antibody was raised (mouse serum for Cox-1; goat for Cox-2), the slides were incubated in primary antibody (polyclonal anti-Cox-1 antibody 1:500 or a monoclonal anti Cox-2 1: 1500 antibody) for 1 hour and again washed three times for five minutes. All antibody incubations were performed in a moist chamber at room temperature for 1 hour. The slides were incubated in the secondary biotinylated antibody for 1 hour (ABC complex; Vectastain Elite Kit; Vector Laboratories Inc., Burlingame, California, USA), and the slides were washed in PBS. The immunocomplex was visualized using DAB + 0.01% H2O2 for less than 10 minutes at room temperature. The slides were counterstained with hematoxylin, “blued” in running tap water for 5 minutes, cleared in xylene, and mounted in DPX mountant (BDH Laboratory Supplies, Poole, United Kingdom). The slides were visualized by a light microscope with color video attachment for recording.
Prostacyclin formation by the cardiac tissue.
Three hours after intraperitoneal injection with 15 mg/kg of DX, or an equal volume of saline or DMSO, the thoracic cage was opened and the heart was removed under anesthesia as described above. Each heart was then sectioned into three segments. The first segment was placed into a test tube with Hanks’ HEPES (H/H) solution and 50 μM arachidonic acid; the second segment was placed into a test tube with H/H solution, arachidonic acid, and 1 μM of a specific COX-2 inhibitor, NS 398; the third segment was placed into a test tube with H/H solution, arachidonic acid, and 200 μM of aspirin (a nonselective COX inhibitor). After 15 minutes, the supernatant in each tube was aspirated and stored at –20°C. The samples were analyzed for 6-keto-prostaglandin F1α (6-keto-PGF1α), the stable hydrolysis product of PGI2 by enzyme immunoassay.
Urinary excretion of prostaglandin metabolites.
After injection, urine was collected over 24 hours in a metabolic cage and stored at –20°C until analyzed for 2,3-dinor-6-keto-PGF1α, the principal enzymatic metabolite of prostacyclin (PGI2); for PGF2α; and for the isoprostane 8-iso-PGF2α by gas chromatography/mass spectrometry. Two milliliters of urine were spiked with deuterated internal standards and allowed to equilibrate for 15 minutes. The sample was deprotonated by lowering the pH to 3.0 using 10% formic acid (100 μl), and the samples were equilibrated for a further 30 minutes. The sample was applied to c-18 SPE cartridges, washed, and then eluted with 1.5 ml of 10% MeOH in ETOAc. The methoxylamine derivative was formed by adding 50 μl of 0.5% methoxylamine in pyridine and was then incubated overnight at room temperature. The sample was dried under vacuum, resuspended in 25 μl of MeOH, and separated by thin layer chromatography (TLC) with a developing solvent of 36 ml of ETOAc, 4 ml of MeOH, and 0.04 ml of concentrated acetic acid. The sample was derivatized as the pentaflourobenzyl ester, trimethylsilyl ether for 2,3-dinor-6-keto-PGF1α, or t-butyl ether for 8-iso-PGF2. The samples were redried under vacuum and finally resuspended in 20 μl of dodecane. Quantitation was performed on a Varian 3400 gas chromatograph linked to a Finnigan Incos XL mass spectrometer (Thermo Finnigan, Whythenshawe, United Kingdom) operated in the negative ion, chemical ionization mode. The results were corrected for urinary creatinine levels.
Serum thromboxane B2.
A dose response of SC560-induced inhibition of serum thromboxane B2, an assay of COX-1 activity, was established before the experiments. Animals were injected intraperitoneally with 3 mg/kg of SC560 (n = 3), 10 mg/kg of SC560 (n = 6), 3 mg/kg of SC236 (n = 3), or vehicle alone (n = 3). Four hours after injection, 1-ml aliquots of blood were sampled from the jugular vein into glass tubes and allowed to clot at 37°C for 1 hour. The plasma was separated by centrifugation (900 g for 10 minutes) and stored at –70°C until assayed for thromboxane B2 by enzyme immunoassay.
Serum cardiac troponin T.
Myocardial injury was detected by measuring cardiac troponin T (CTnT) and lactate dehydrogenase (LDH) levels in serum from venous blood samples. Blood from the jugular vein was sampled at 4 hours. The blood samples were centrifuged, and the serum was stored at –20°C until analyzed. CTnT levels were measured by enzyme immunoassay using an ES300 troponin T assay kit (Boehringer Mannheim Immunodiagnostics, Mannheim, Germany), with a lower limit of detection of 0.01 ng/ml. LDH levels were measured by ultraviolet photometric assay (Roche Diagnostics Ltd., Lewes, United Kingdom) with a lower detection point of 6 U/l.
TUNEL assay.
TUNEL assay was performed to detect apoptosis in cardiac sections. Briefly, after pretreatment with proteinase K (20 μg/ml) and 3.0% hydrogen peroxide, paraffin-fixed slides were treated with a digoxigenin-dNTP complex for 1 hour at 37°C (ApopTag; Intergen Co., Oxford, United Kingdom). This complex binds to free 3-OH termini of nucleosome-sized DNA fragments that are formed during apoptosis, and it is detected by anti-digoxigenin conjugate antibody. The bound complex stained with a DAB-based substrate. The slides were counterstained with 0.5% methyl green in 0.1 M sodium acetate, dehydrated in xylene, and mounted and examined by light microscopy. Examination of the slides was performed by two observers (one blinded) who counted the total number of apoptotic and normal cells over 25 high-powered fields for each slide using an ocular grid (0.25 mm2 area of field). The number of apoptotic cells was expressed as the percent of the total number of cells.
Statistical analysis.
All quantitative data are expressed as mean ± SEM. The data were analyzed by ANOVA and subsequently, where appropriate, by paired analysis.
Results
DX induces COX-2 isoform expression in the heart.
Immunohistochemistry showed constitutive expression of COX-1 in cardiac cells and in the myocytes of the diaphragm. COX-2 was absent from the normal heart but was induced 4 hours after the administration of DX (Figure 1). In contrast, COX-2 expression was absent in the diaphragm of DX-treated rats. These findings were confirmed by assay of COX activity, detected as the generation of PGI2, the main product in myocardial tissue. Myocardial biopsies from DX- and vehicle-treated rats were exposed to arachidonic acid ex vivo, and the levels of 6-keto-PGF1α, the hydrolysis product of PGI2, was measured in the supernatant. The concentration of 6-keto-PGF1α was higher in the DX-treated rats compared with vehicle-treated controls (P = 0.02; Figure 2). The increase in 6-keto-PGF1α formation was reduced in the presence of NS 398, the selective COX-2 inhibitor, or aspirin, the nonselective COX inhibitor. NS 398 had no effect on the generation of 6-keto-PGF1α by myocardial tissue from vehicle-treated animals.
Figure 1.
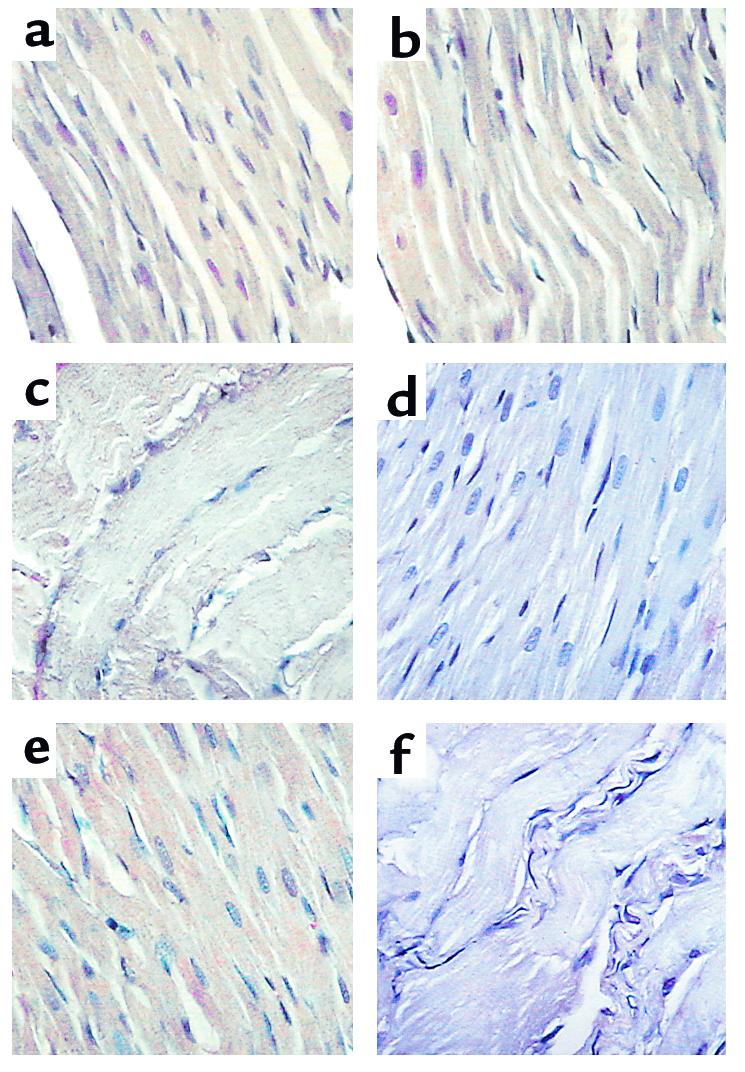
DX-induced COX-2 expression in cardiac tissue (representative of six to nine experiments for each treatment). COX-1 is constitutively expressed in cardiomyocytes in vehicle-treated control animals (a) and after DX injection (b), and in the myocytes of the diaphragm (c). COX-2 is not detected in hearts from vehicle-treated controls (d) but is induced 4 hours after administration of DX (e). COX-2 is not detected in the diaphragm after DX administration (f).
Figure 2.

Generation of prostacyclin-measured 6-keto-PGF1α in cardiac tissue after DX administration. The increase in 6-keto-PGF1α formation after DX administration was reduced by 1 μM of the selective COX-2 inhibitor NS 398 and by 200 nM of aspirin (ASA) (data are expressed as the mean ± SEM of six separate experiments per treatment).
Generation of prostaglandins in vivo.
To determine whether DX increased eicosanoid generation in vivo and identify the COX isoform responsible, we examined their urinary excretion of prostaglandins in the presence and absence of selective inhibitors SC236 (for COX-2) and SC560 (for COX-1). Given that SC560 is highly selective but not specific for COX-1 in some species, a dose response for SC560-induced inhibition of serum thromboxane B2, an assay of COX-1 activity, was established before the experiments (Figure 3). Administration of 10 mg/kg SC560 reduced the serum thromboxane B2 by more than 80% (290 ± 42 to 18.5 ± 9.8 ng/ml; P < 0.001), and this dose was used in subsequent experiments. In contrast, the COX-2 inhibitor SC236 had no effect on serum thromboxane B2 (290 ± 42 to 224 ± 90 ng/ml; P = NS). PGI2 production in vivo was quantified as the excretion of 2,3-dinor 6-keto-PGF1α, its major enzymatic metabolite. Urinary 2,3-dinor 6-keto-PGF1α increased after the administration of DX (Figure 4). The increase in 2,3-dinor 6-keto-PGF1α was abolished by the administration of the selective COX-2 inhibitor SC236. A similar effect was seen with the nonselective COX inhibitor indomethacin. The selective COX-1 inhibitor SC560 induced a small reduction in urinary 2,3-dinor 6-keto-PGF1α, probably reflecting inhibition of PGI2 generated by constitutively expressed COX-1. However, SC560 did not prevent the increase in PGI2 induced by DX (Figure 4). A similar trend was seen with urinary PGF2α, although none of the changes were statistically significant. There was a small, although not significant, increase in the excretion of the isoprostane 8-iso-PGF2α (from 2.02 ± 0.2 to 3.3 ± 2.1 ng/mg creatinine), a marker of gross free-radical–induced injury.
Figure 3.

Dose-dependent inhibition of serum thromboxane B2 by SC560 (the data are expressed as the mean ± SEM of n = 3 for each treatment).
Figure 4.

Urinary excretion of 2,3-dinor 6-keto-PGF1α, 8-iso-PGF2α, and PGF2α, after the administration of DX. The increase in 2,3-dinor 6-keto-PGF1α was inhibited by the administration of the selective COX-2 inhibitor SC236 and by indomethacin (Indo), whereas the selective COX-1 inhibitor SC560 had little effect. A similar trend was seen with urinary PGF2α, although none of the changes were statistically significant. (The results are the mean ± SEM of five separate experiments for each treatment.)
Cardiac injury.
Myocardial injury was evaluated by measuring CTnT in venous blood. No measurable CTnT was detected in animals treated with vehicle, SC236, or SC560 alone. CTnT levels increased in the DX-treated rats, and the levels increased further when DX was combined with the COX-2 inhibitor SC236 (Figure 5). Similar results were seen with plasma LDH, which increased with DX (578 ± 227 U/l; n = 6) and was further increased when DX was combined with SC236 (1,552 ± 58 U/l; n = 3, P = 0.02), versus 156 ± 28 U/l (n = 6) in untreated controls. The CTnT levels were not further increased when DX was combined with the COX-1 inhibitor SC560. We examined whether the enhanced injury seen when DX was combined with SC236 could be offset by PGI2, administered as its stable analogue iloprost. No measurable concentrations of CTnT were detected in seven of 12 rats injected with iloprost combined with DX (Figure 5). DX also increased the percentage of apoptotic cells detected as TUNEL-positive cells (Figure 6). A further increase was seen when DX was combined with SC236, and the effect of the combination was attenuated by prior treatment with iloprost.
Figure 5.

Cardiac injury detected as an increase in CTnT of more than 0.01 ng/ml, after DX administration alone and combined with the selective COX-2 inhibitor SC236. DX induced cardiac injury, and this was aggravated by SC236 but not SC560. The increase in injury seen with the combination of DX and SC236 was attenuated by prior administration of iloprost, a stable analogue of PGI2.
Figure 6.

Cardiac injury detected as apoptosis (TUNEL-positive cells) after DX treatment alone and combined with the selective COX-2 inhibitor SC236. DX increased the percentage of cells that were apoptotic at 4 hours, and this was further aggravated by SC236. The increase in apoptosis seen with the combination of DX and SC236 was attenuated by prior administration of iloprost. Similar results were seen at 12 hours after DX administration (data not shown). The data are the mean (± SEM) of 25 high-power fields from three to four experiments per treatment.
Discussion
COX-2 expression in several cells, but particularly cancer cell lines, appears to protect against apoptosis and to promote cell growth (17, 18). Thus, COX-2 inhibition suppresses the growth of tumor cells in vitro (19) and inhibits tumor formation in the APC-knockout mouse model of colon cancer (20) and in human familial polyposis coli (21). COX-2 is expressed in areas of infarction in the human heart (22), and myocardial COX-2 expression has been reported in the rat heart during cardiac transplant rejection (23). In addition, cardiac fibrosis has been reported in the COX-2–knockout mouse (24). Together, these findings prompted us to examine whether COX-2 was protective in cardiomyocytes in vitro. Cardiomyocytes only express COX-1 under normal circumstances, but COX-2 was induced on exposure to free radicals or DX (9). DX also provoked the release of LDH and increased the rate of apoptosis (S.R. Adderley and D.J. Fitzgerald, unpublished data), and this was aggravated by a COX-2 inhibitor.
These in vitro data raise the possibility of protecting the heart in patients during the administration of DX by coadministration of a prostaglandin. Here, we show that DX induces COX-2 in the adult rat heart when administered exogenously and enhances the generation of PGI2, the principal product generated in cardiac cells. The isoform responsible for the increased product formation was assessed using specific inhibitors of COX isoforms (25). We have confirmed the selectivity of the compounds using COS cells expressing either COX-1 or COX-2 (data not shown). In addition, at the dose used SC236 had no effect on serum thromboxane B2, an assay of COX-1 activity. In contrast, SC560 induced a greater than 80% inhibition of serum thromboxane B2. SC236 prevented the rise in urinary 2,3-dinor-6-keto-PGF1α seen with DX, demonstrating a role for COX-2 in the enhanced generation of PGI2 after administration of the anthracycline. In contrast, SC560 had very little effect.
DX administered alone induced cardiac injury measured as the increase in serum CTnT and by the number of apoptotic cells detected on TUNEL assay, as reported previously (26, 27). Coadministration of SC236 aggravated the cardiac injury, whereas the COX-1 inhibitor did not. There is evidence that COX-2 inhibitors enhance apoptosis in some cell lines in a prostaglandin-independent manner (28). Moreover, some NSAIDs (although not COX-2 inhibitors) inhibit a multidrug-resistance protein responsible for transporting DX out of cells (29). However, administration of an analogue of PGI2, the major product of the cardiomyocytes, prevented the injury. Further, in five of the 12 animals, there was still evidence of cardiac injury despite treatment with iloprost. This may reflect additional mechanisms of cell death induced by DX that are not reversed by iloprost, although, in the absence of dose-response data, the mechanism is unclear. Mass spectrometry of cell supernatants show that cardiomyocytes largely generate PGI2 in addition to PGF2α and PGE2. In vitro, only PGI2 protected against DX-mediated injury (8), suggesting that this may be the product responsible for the cytoprotective effect of COX-2 expression. Prostaglandins, however, can have the opposite effect. Thus, expression of COX-2 enhances apoptosis in neuronal cells (30) and the PGD2 metabolite 15-deoxy PGJ2 provokes endothelial cell death (31). Thus, the effect may be specific to prostacyclin or its analogues.
Prostaglandins act on two classes of receptors (a) surface transmembrane-spanning, G-protein–coupled receptors and (b) PPARs, which are nuclear membrane receptors. Prostaglandins and their precursors are ligands for several PPARs including PPARα (polyunsaturated fatty acids), PPARδ (PGI2), and PPARγ (PGD2, 15-deoxy PGJ2) (31). PPARα and PPARδ are widely expressed in tissues including the heart, whereas PPARγ is found particularly in adipocytes (32). PPARs heterodimerize with other proteins, such as the retinoid X receptor and ARA70, to act as transcription factor complexes for a wide number of genes, particularly those involved in lipid metabolism (33, 34). Alternatively, the effects of iloprost may be mediated through surface membrane receptors, transcripts for which have been identified in the adult human heart (35). Moreover, we cannot exclude the possibility that a vasodilator response to iloprost may have provided some degree of protection. However, the dose used was low relative to that reported to reduce blood pressure and increase heart rate in the rat (36).
In the rat model of DX-induced cardiotoxicity, COX-2 expression may provide some degree of protection against the injury induced by the anthracycline. Our experiments suggest that administration of a prostacyclin analogue may afford cardiac protection in this setting.
Acknowledgments
We thank P. Isakson for the generous gift of the monoclonal anti-mouse COX-2 antibody (R6) and Elaine Kay and Mohommad Mabruk for their support on TUNEL assays and immunohistochemistry. This work was supported by grants from the Health Research Board and Higher Education Authority of Ireland.
References
- 1.Singal PK, Iliskovic N. Doxorubicin-induced cardiomyopathy. N Engl J Med. 1998;339:900–905. doi: 10.1056/NEJM199809243391307. [DOI] [PubMed] [Google Scholar]
- 2.Feldman AM, Lorell BH, Reis SE. Trastuzumab in the treatment of metastatic breast cancer: anticancer therapy versus cardiotoxicity. Circulation. 2000;102:272–274. doi: 10.1161/01.cir.102.3.272. [DOI] [PubMed] [Google Scholar]
- 3.Slamon DJ, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344:783–792. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- 4.Boveris A. Mitochondrial production of superoxide radical and hydrogen peroxide. Adv Exp Med Biol. 1977;78:67–82. doi: 10.1007/978-1-4615-9035-4_5. [DOI] [PubMed] [Google Scholar]
- 5.Bachur NR, Gordon SC, Gee MV. Anthracycline antibiotic augmentation of microsomal electron transport and free radical formation. Mol Pharmacol. 1977;13:901–910. [PubMed] [Google Scholar]
- 6.Della-Torre P. Long-lasting effect of dexrazoxone against anthracycline cardiotoxicity in rats. Toxicol Pathol. 1996;24:398–402. doi: 10.1177/019262339602400402. [DOI] [PubMed] [Google Scholar]
- 7.Sparano JA. Use of dexrazoxane and other strategies to prevent cardiomyopathy associated with doxorubicin-taxane combinations. Semin Oncol. 1998;25(Suppl. 10):66–71. [PubMed] [Google Scholar]
- 8.Adderley S, Fitzgerald D. Oxidative damage of cardiomyocytes is limited by extracellular regulated kinases 1/2-mediated induction of cyclooxygenase 2. J Biol Chem. 1999;27:5038–5046. doi: 10.1074/jbc.274.8.5038. [DOI] [PubMed] [Google Scholar]
- 9.Hemler M, Lands WEM, Smith WL. Purification of cyclooxygenase that forms prostaglandins. J Biol Chem. 1976;251:5575–5579. [PubMed] [Google Scholar]
- 10.Hla T, Neilson K. Human cyclooxygenase-2 cDNA. Proc Natl Acad Sci USA. 1992;89:7384–7388. doi: 10.1073/pnas.89.16.7384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kujubu DA, Herschman HR. Dexamethasone inhibits mitogen induction of the TIS10 prostaglandin synthase/cyclooxygenase gene. J Biol Chem. 1992;267:7991–7994. [PubMed] [Google Scholar]
- 12.Schmedtje JF, Jr, Ji YS, Liu WL, DuBois RN, Runge MSJ. Hypoxia induces cyclooxygenase-2 via the NF-kappaB p65 transcription factor in human vascular endothelial cells. J Biol Chem. 1997;272:601–608. doi: 10.1074/jbc.272.1.601. [DOI] [PubMed] [Google Scholar]
- 13.Xie W, Herschman HR. v-src induces prostaglandin synthase 2 gene expression by activation of the c-Jun N-terminal kinase and the c-Jun transcription factor. J Biol Chem. 1995;270:27622–27628. doi: 10.1074/jbc.270.46.27622. [DOI] [PubMed] [Google Scholar]
- 14.Xie W, Fletcher BS, Andersen RD, Herschman HR. Mol v-src induction of the TIS10/PGS2 prostaglandin synthase gene is mediated by an ATF/CRE transcription response element. Cell Biol. 1994;14:6531–6539. doi: 10.1128/mcb.14.10.6531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dixon DA, Kaplan CD, McIntyre TM, Zimmerman GA, Prescott SM. Post-transcriptional control of cyclooxygenase-2 gene expression. The role of the 3′-untranslated region. J Biol Chem. 2000;275:11750–11757. doi: 10.1074/jbc.275.16.11750. [DOI] [PubMed] [Google Scholar]
- 16.Sheng H, et al. Transforming growth factor-beta1 enhances Ha-ras-induced expression of cyclooxygenase-2 in intestinal epithelial cells via stabilization of mRNA. J Biol Chem. 2000;275:6628–6635. doi: 10.1074/jbc.275.9.6628. [DOI] [PubMed] [Google Scholar]
- 17.Kinoshita T, et al. Growth stimulation and induction of epidermal growth factor receptor by over expression of cyclooxygenases 1 and 2 in human colon carcinoma cells. . Biochim Biophys Acta . 1999;1438:120–130. doi: 10.1016/s1388-1981(99)00034-7. [DOI] [PubMed] [Google Scholar]
- 18.Kawamura T, et al. Prostaglandin E1 transported into cells blocks the apoptotic signals induced by nerve growth factor deprivation. J Neurochem. 1999;72:1907–1914. doi: 10.1046/j.1471-4159.1999.0721907.x. [DOI] [PubMed] [Google Scholar]
- 19.Liu XH, Yao S, Kirschenbaum A, Levine AC. NS398, a selective cyclooxygenase-2 inhibitor, induces apoptosis and down-regulates bcl-2 expression in LNCaP cells. Cancer Res. 1998;58:4245–4249. [PubMed] [Google Scholar]
- 20.Oshima M, et al. Suppression of intestinal polyposis in Apc delta716 knockout mice by inhibition of cyclooxygenase 2 (COX-2) Cell. 1996;87:803–809. doi: 10.1016/s0092-8674(00)81988-1. [DOI] [PubMed] [Google Scholar]
- 21.Steinbach G, et al. The effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenomatous polyposis. N Engl J Med. 2000;342:1946–1952. doi: 10.1056/NEJM200006293422603. [DOI] [PubMed] [Google Scholar]
- 22.Wong SCY, Mitsumasa F, Melnyk P, Rodger I, Giaid A. Induction of cyclooxygenase-2 and activation of nuclear factor-kB in myocardium of patients with congestive heart failure. Circulation. 1998;98:100–103. doi: 10.1161/01.cir.98.2.100. [DOI] [PubMed] [Google Scholar]
- 23.Yang X, et al. Upregulation of COX-2 during cardiac allograft rejection. Circulation. 2000;101:430–438. doi: 10.1161/01.cir.101.4.430. [DOI] [PubMed] [Google Scholar]
- 24.Dinchuk JE, et al. Renal abnormalities and an altered inflammatory response in mice lacking cyclooxygenase II. Nature. 1995;378:406–409. doi: 10.1038/378406a0. [DOI] [PubMed] [Google Scholar]
- 25.Smith CJ, et al. Pharmacological analysis of cyclooxygenase-1 in inflammation. Proc Natl Acad Sci USA. 1998;95:13313–13318. doi: 10.1073/pnas.95.22.13313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Herman EH, et al. Use of cardiac troponin T levels as an indicator of doxorubicin-induced cardiotoxicity. Cancer Res. 1998;58:195–197. [PubMed] [Google Scholar]
- 27.Kang YJ, Zhou ZX, Wang GW, Buridi A, Klein JB. Suppression by metallothionein of doxorubicin-induced cardiomyocyte apoptosis through inhibition of p38 mitogen-activated protein kinases. J Biol Chem. 2000;275:13690–13698. doi: 10.1074/jbc.275.18.13690. [DOI] [PubMed] [Google Scholar]
- 28.Hsu AL, et al. The cyclooxygenase-2 inhibitor celecoxib induces apoptosis by blocking Akt activation in human prostate cancer cells independently of Bcl-2. J Biol Chem. 2000;275:11397–11403. doi: 10.1074/jbc.275.15.11397. [DOI] [PubMed] [Google Scholar]
- 29.Duffy CP, et al. Enhancement of chemotherapeutic drug toxicity to human tumour cells in vitro by a subset of non-steroidal anti-inflammatory drugs (NSAIDs) Eur J Cancer. 1998;34:1250–1259. doi: 10.1016/s0959-8049(98)00045-8. [DOI] [PubMed] [Google Scholar]
- 30.Hewett SJ, Uliasz TF, Vidwans AS, Hewett JA. Cyclooxygenase-2 contributes to N-methyl-D-aspartate-mediated neuronal cell death in primary cortical cell culture. J Pharmacol Exp Ther. 2000;293:417–425. [PubMed] [Google Scholar]
- 31.Bishop-Bailey D, Hla T. Endothelial cell apoptosis induced by the peroxisome proliferator-activated receptor (PPAR) ligand 15-deoxy-Delta12, 14-prostaglandin J2. J Biol Chem. 1999;274:17042–17048. doi: 10.1074/jbc.274.24.17042. [DOI] [PubMed] [Google Scholar]
- 32.Forman BM, Chen J, Evans RM. Hypolipidemic drugs, polyunsaturated fatty acids, and eicosanoids are ligands for peroxisome proliferator-activated receptors alpha and delta. Proc Natl Acad Sci USA. 1997;94:4312–4317. doi: 10.1073/pnas.94.9.4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Braissant O, Foufelle F, Scotto C, Dauca M, Wahli W. Differential expression of peroxisome proliferator-activated receptors (PPARs): tissue distribution of PPAR-alpha, -beta, and -gamma in the adult rat. Endocrinology. 1996;137:354–366. doi: 10.1210/endo.137.1.8536636. [DOI] [PubMed] [Google Scholar]
- 34.Heinlein CA, Ting HJ, Yeh S, Chang C. Identification of ARA70 as a ligand-enhanced coactivator for the peroxisome proliferator-activated receptor gamma. J Biol Chem. 1999;274:16147–16152. doi: 10.1074/jbc.274.23.16147. [DOI] [PubMed] [Google Scholar]
- 35.Nakagawa O, et al. Molecular cloning of human prostacyclin receptor cDNA and its gene expression in the cardiovascular system. Circulation. 1994;90:1643–1647. doi: 10.1161/01.cir.90.4.1643. [DOI] [PubMed] [Google Scholar]
- 36.Steinberg H, Medvedev OS, Luft FC, Unger T. Effect of a prostacyclin derivative (iloprost) on regional blood flow, sympathetic nerve activity, and baroreceptor reflex in the conscious rat. J Cardiovasc Pharmacol. 1988;11:84–89. doi: 10.1097/00005344-198801000-00013. [DOI] [PubMed] [Google Scholar]