

Abstract
Protein acetylation is an important and reversible post-translational modification (PTM), and it governs a variety of cellular dynamics and plasticity. Experimental identification of acetylation sites is labor-intensive and often limited by the availability reagents such as acetyl-specific antibodies and optimization of enzymatic reactions. Computational analyses may facilitate the identification of potential acetylation sites and provide insights into further experimentation. In this manuscript, we present a novel protein acetylation prediction program named PAIL, prediction of acetylation on internal lysines, implemented in a BDM (Bayesian Discriminant Method) algorithm. The accuracies of PAIL are 85.13%, 87.97% and 89.21% at low, medium and high thresholds, respectively. Both Jack-Knife validation and n-fold cross validation have been performed to show that PAIL is accurate and robust. Taken together, we propose that PAIL is a novel predictor for identification of protein acetylation sites and may serve as an important tool to study the function of protein acetylation. PAIL has been implemented in PHP and is freely available on a web server at: http://bioinformatics.lcd-ustc.org/pail.
Keywords: PAIL, Bayesian Discriminant Method, Nε-acetylation, post-translation modification, lysine, internal
Introduction
Protein acetylation is a widespread covalent modification in eukaryotes, transferring acetyl groups from acetyl coenzyme A (acetyl CoA) to either the α -amino (Nα ) group of amino-terminal residues or to the ε-amino group (Nε) of internal lysines at specific sites [1–5]. As one of the most ubiquitous protein modifications, approximately 85% of eukaryotic proteins are Nα -terminally acetylated in a co-translational manner on several types of residues such as serine, alainine, and so on [3, 4]. Although Nε-lysine acetylation is less common, its role is probably more important [1, 2, 4–12]. Nε-acetylation of proteins in internal lysine residues is an essential and highly reversible type of post-translational modification (PTM), and the Nε-acetylation orchestrates a variety of cellular processes, including transcription regulation [7, 9], DNA repair [10], apoptosis [8, 11], cytokine signaling [12], and nuclear import [6]. As a ‘loss-of-function’ mechanism proposed, Nε-acetylation greatly alters the electrostatic properties of a protein by neutralizing the positive charge of the lysine residues. The formation of hydrogen bonds on lysine side-chains are also disrupted [5, 13]. In addition, lysine acetylation also creates a new interface for protein binding, as a ‘gain-of-function’ mechanism [5, 13]. Thus, Nε-acetylation may modulate the protein function, such as of protein-protein interaction, DNA binding, enzymatic activity, stability and subcellular localization [1, 4–7, 9, 12, 13].
Early studies of histone acetylation have proposed that the modification regulates the gene expression and stabilizes the chromatin structure. In the past decades, numerous non-histone acetylated proteins have been identified to play diversified regulatory roles among eukaryotic [1, 2, 5], archaeal [14], bacterial [15] and viral [16] proteins. As a highly reversible reaction, the level of lysine acetylation in vivo is controlled by the antagonism of HATs (histone acetyltransferases) and HDACs (histone deacetylases). About 30 HATs have been discovered and divided into three classes such as Gcn5/PCAF, p300/CBP and MYST proteins [5]. In human, there are 18 distinct HDACs grouped into three groups including Class I, IIa/IIb and III [17]. Aberrant lysine acetylation has been implicated in the development of cancer and other diseases, such as prostate cancer [18], myeloid leukemia [5, 19], and inflammatory lung diseases [20]. Thus, both HATs and HDACs are potential molecular targets for biochemical therapy. Indeed, numerous HDAC inhibitors have been developed successfully as anticancer drugs, selectively inducing the tumor cells into apoptosis [21–23].
Although intensive research has been performed, the study of Nε-acetylation is still in its infancy. The full content of regulatory functions of lysine acetylation remains to be elucidated. Both HATs and HDACs have their substrate specificities, for example, peptide motif GKXXP as a potential recognition signal of GCN5 in yeast [2, 4, 13]. However, the general consensus sequences/motifs/profiles of substrates for HATs and HDACs targeting are still unclear. In this regard, dissection of acetylation and deacetylation on specific lysines of acetylated proteins will be a foundation of understanding the molecular mechanism and dynamics of Nε-acetylation. Besides the conventional experimental methods, such as mutagenesis of potential acetylation sites [12], acetylation-specific antibodies [6, 7] and mass-spectrometry [8, 14, 24] have also been employed. However, these experimental approaches are laborious and expensive. Therefore, the prediction of acetylation sites in silico is desirable. Previous computational studies only have focused on Nα -terminal acetylation [25, 26].
In this work, we present a novel online computational program for protein acetylation site prediction named PAIL, Prediction of Acetylation on Internal Lysines. We manually mined scientific literature to collect 249 experimentally verified acetylation sites of 92 distinct proteins. After redundant-clearing, there are 246 sites from 89 substrates reserved. Then the BDM (Bayesian Discriminant Method) algorithm [27] was employed. The window length of a potential acetylated peptide has been optimized as 13. The accuracy of PAIL is highly encouraging with, 85.13%, 87.97%, and 89.21% at low, medium and high thresholds, respectively. Both Jack-knife validation and n-fold (6-, 8-, and 10-fold) cross-validation have been employed. The accuracies of two validations fluctuate from 82.17% to 86.11%, and these results confirm that the PAIL is accurate and robust. In this regard, we propose that PAIL might be a useful in silico tool for further experimental consideration.
Materials & Methods
Data Preparation
Here we define the lysine (K) residues that undergo acetylated modification as positive data (+), while those non-acetylated lysine residues are regarded as negative data (−). Furthermore, we define a potential acetylated peptide (PAP) (denoted by x⃗ = (p1p2…pmKp1…pn)′, pi represents a residue, m ≥ 1, n ≥ 1) as a local peptide flanking a lysine residue. Then the window length of a PAP is m+n+1. In this work, m is equal to n and the windows with length of 9, 11 and 13 have been examined.
First, we searched PubMed with the key word “acetylation lysine”, and collected 249 unambiguously experimental verified acetylation sites of 92 distinct proteins from >1000 scientific articles. Although the acetylation-related literature is increasing rapidly, we only adopted the acetylation sites published online before Dec. 10th, 2005. Then we retrieved the primary sequences of these proteins from Swiss-Prot/TrEMBL database (http://cn.expasy.org). And the acetylated peptides with length of 9, 11 and 13 were parsed as positive (+) data, separately.
The positive data (+) set for training might contain several homologous sites from homologous proteins. If the training data are highly redundant with too many homologous sites, the prediction accuracy will be overestimated. To avoid the overestimation, we clustered the protein sequences from positive(+) data set with a threshold of 30% identity by BLASTCLUST, one program in the BLAST package [28]. If two proteins were similar with ≥30% identity, we re-aligned the proteins with BL2SEQ, another program in the BLAST package [28], and checked the results manually. If two acetylation sites from two homologous proteins were at the same position after sequence alignment, only one item was reserved while the other was discarded. Thus, we obtained non-redundant positive data (+) of high quality with 246 acetylation sites from 89 proteins. Only three acetylation sites from three proteins were truly redundant sites to be removed. As previously described [29, 30], the negative (−) sites were taken from non-annotated lysine sites in the same proteins from which (+) sites were chosen. The homology reducing process was also carried out on (−) data. If the identity between a PAP of (−) data and an acetylated peptide of (+) data was not less than 30%, the PAP of (−) data was removed as a redundant site. The final curated data set is available upon request.
Algorithm Design
The standard Bayesian Discriminant Method (BDM) has been employed in PAIL. By this means, acetylated peptides from (+) data and PAPs from (−) data have been extracted from protein sequences. Thus, the assignment rule of candidate acetylation local peptides given by BDM can be described as:
| (1) |
Here P((+) ∣ x⃗) and P((−) ∣ x⃗) are the posterior probabilities of x⃗ for both (+) and (−) site, respectively. The b is the cut-off value to obtain the prediction performance. At the same time, by the Bayesian Role, the posterior probability for (+) sites can further be expressed as:
| (2) |
Here P(+) is the prior probability that is assumed to be a constant. And in this work, although there are more (−) sites than (+) sites in the data set, we regard the prior probabilities for both kinds of sites as equal, i.e., no prior information for prediction, which can avoid bias prediction results. At the same time, there are many ways to estimate the probability P(x⃗ ∣ +) and one simple way is to assume that all flanking residues are mutually independent. Thus, given the local peptides of PAPs with length m, it can be formulated as:
| (3) |
Here P(pi|+),i = 1,...,m are calculated by the occurrence of each residue in training data. So equation (2) can be further described as:
| (4) |
In the same way, we can describe the posterior probability for (−) sites as:
| (5) |
Thus, the final discriminant function can be stated as:
| (6) |
And is the final threshold for prediction.
Construction of the PAIL Web Server
We have implemented our PAIL as an easy-to-use web server, which can be accessed from http://bioinformatics.lcd-ustc.org/pail. The prediction page of PAIL is shown in Figure 1. Users can paste the protein sequence either in raw sequence or FASTA format (one or more sequences) into the text form and obtain the prediction result by clicking on the “Submit” button. In addition, the prediction result is downloadable in a tab-deliminated plain text by clicking on the word here in the sentence of “Download the TAB-deliminated data file from here”.
Figure 1.
The prediction page of PAIL.
Results
Functional analysis of Acetylated Proteins
To determine which types of proteins are acetylated, we have downloaded the GO annotation files for Uniprot from EBI-GOA (http://www.ebi.ac.uk/GOA/) for analyzing. In our non-redundant data set with 89 acetylated proteins, we observe 329 distinct GO categories. The Table 1 shows the top five Gene Ontology (GO) entries of biological processes, molecular functions and cellular components of acetylated proteins.
Table 1.
The top five GO categories of biological process, molecular function and cellular component of acetylated proteins.
| GO Symbol | Name of Gene Ontology | No. of Proteins |
|---|---|---|
| Top five biological process | ||
| GO:0006355 | regulation of transcription, DNA-dependent | 56 |
| GO:0006350 | transcription | 53 |
| GO:0045449 | regulation of transcription
regulation of transcription from RNA polymerase II |
16 |
| GO:0006357 | promoter | 10 |
| GO:0007165 | signal transduction | 10 |
| Top five molecular function | ||
| GO:0003677 | DNA binding | 59 |
| GO:0005515 | protein binding | 43 |
| GO:0003700 | transcription factor activity | 31 |
| GO:0008270 | zinc ion binding | 19 |
| GO:0046872 | metal ion binding | 19 |
| Top five cellular component | ||
| GO:0005634 | nucleus | 66 |
| GO:0005737 | cytoplasm | 11 |
| GO:0005739 | mitochondrion | 9 |
| GO:0016020 | membrane | 7 |
| GO:0000785 | chromatin | 6 |
The most frequent GO item of biological process in which acetylated proteins are involved in is “regulation of transcription, DNA-dependent” (56 proteins). The other four significantly biological processes are “transcription” (53 proteins), “regulation of transcription” (16 proteins), “regulation of transcription from RNA polymerase II promoter” (10 proteins) and “signal transduction” (10 proteins). The most enriched GO group of molecular function is “DNA binding” (59 proteins), while the other four highly-abundant molecular functions are “protein binding” (43 proteins), “transcription factor activity” (31 proteins), “zinc ion binding” (19 proteins) and “metal ion binding” (19 proteins). Again, the most abudant GO entry of cellular component is “nucleus” (66 proteins), and the other four highly-frequent cellular components are “cytoplasm” (11 proteins), “mitochondrion” (9 proteins), “membrane” (7 proteins) and “chromatin” (6 proteins).
Taken together, the analyses propose that protein acetylation plays important roles in transcription regulation and signal transduction. Also, the functions of acetylated proteins are quite diverse. Thus, the data set is suitable for our prediction work as training data.
Performance evaluation
We have adopted four frequently considered measurements: accuracy (Ac), sensitivity (Sn), specificity (Sp) and Mathew correlation coefficient (MCC). Accuracy (Ac) illustrates the correct ratio between both positive (+) and negative (−) data sets, while sensitivity (Sn) and specificity (Sp) represent the correct prediction ratios of positive (+) and negative data (−) sets respectively. But when the number of positive data and negative data differ too much from each other, the Mathew correlation coefficient (MCC) should be included to evaluate the prediction performance. The value of MCC ranges from -1 to 1, and a larger MCC value stands for better prediction performance.
Among the data with positive hits by PAIL, the real positives are defined as true positives (TP), while the others are defined as false positives (FP). Among the data with negative predictions by PAIL, the real positives are defined as false negatives (FN), while the others are defined as true negatives (TN).
The performance measurements of sensitivity (Sn), specificity (Sp), accuracy (Ac), and Mathew correlation coefficient (CC) are all defined as below:
In addition to assess whether PAIL is unbiased and robust for prediction, we adopt the standard evaluations of Jack-Knife validation and n-fold (6-, 8- and 10-fold in this work) cross-validation. For Jack-Knife validation, one sample is removed from the training data set at a time and the Ac, Sn, Sp and MCC are re-calculated, respectively. The final results are the average of the all Ac, Sn, Sp and MCC of the Jack-Knife validation. As previously proposed [27], we have also taken an additional test with n-fold (6-, 8- and 10-fold in this work) cross-validation. The tests are repeated 20 times and the Ac, Sn, Sp and MCC are re-computed each time. The average Ac, Sn, Sp and MCC are adopted as the final value.
Prediction performance of PAIL
In this work, the PAPs with window length of 9, 11 and 13 were examined. Also, three cut-offs of high, medium and low thresholds were adopted in each condition. A specificity of ~95% was adopted for high stringency, while the medium and low stringencies denote the specificities of ~90% and ~85%, respectively. Then the prediction performances of self-consistency, Jack-Knife validation and n-fold (6-, 8- and 10-fold in this work) cross-validation have been calculated and shown in Table 2, 3 & 4, respectively.
Table 2.
The prediction performance of self-consistency, Jack-knife validation and n-fold validation of PAIL with window length of 9.
| Window length (9) | Threshold | Accuracy | Sensitivity | Specificity | MCC |
|---|---|---|---|---|---|
| Self-Consistency | High | 86.37% | 49.19% | 96.72% | 0.5584 |
| Medium | 85.75% | 64.63% | 91.63% | 0.5739 | |
| Low | 82.65% | 69.92% | 86.19% | 0.5277 | |
| Jack-Knife validation | High | 84.25% | 42.28% | 95.93% | 0.4785 |
| Medium | 81.42% | 53.25% | 89.25% | 0.4385 | |
| Low | 78.76% | 60.16% | 83.94% | 0.4167 | |
| 6-fold cross-validation | High | 83.27% | 40.63% | 95.13% | 0.4439 |
| Medium | 80.92% | 51.38% | 89.15% | 0.4207 | |
| Low | 79.71% | 55.33% | 86.50% | 0.4126 | |
| 8-fold cross-validation | High | 83.49% | 41.06% | 95.29% | 0.4518 |
| Medium | 81.39% | 52.22% | 89.51% | 0.4341 | |
| Low | 78.26% | 59.33% | 83.52% | 0.4042 | |
| 10-fold cross-validation | High | 83.74% | 41.40% | 95.53% | 0.4606 |
| Medium | 81.34% | 51.99% | 89.51% | 0.4320 | |
| Low | 78.52% | 60.00% | 83.68% | 0.4118 |
Table 3.
The prediction performance of self-consistency, Jack-knife validation and n-fold validation of PAIL with window length of 11.
| Window length (11) | Threshold | Accuracy | Sensitivity | Specificity | MCC |
|---|---|---|---|---|---|
| Self-Consistency | High | 88.14% | 56.50% | 96.95% | 0.6232 |
| Medium | 87.08% | 72.36% | 91.18% | 0.6264 | |
| Low | 84.60% | 78.46% | 86.31% | 0.5967 | |
| Jack-Knife validation | High | 84.96% | 43.90% | 96.38% | 0.5046 |
| Medium | 83.19% | 53.66% | 91.40% | 0.4799 | |
| Low | 80.00% | 60.98% | 85.29% | 0.4423 | |
| 6-fold cross-validation | High | 84.23% | 43.66% | 95.52% | 0.4805 |
| Medium | 82.50% | 52.93% | 90.73% | 0.4614 | |
| Low | 80.12% | 59.78% | 85.78% | 0.4393 | |
| 8-fold cross-validation | High | 84.54% | 44.13% | 95.79% | 0.4915 |
| Medium | 82.59% | 53.13% | 90.79% | 0.4641 | |
| Low | 80.27% | 59.80% | 85.97% | 0.4422 | |
| 10-fold cross-validation | High | 84.65% | 44.27% | 95.88% | 0.4950 |
| Medium | 82.72% | 53.21% | 90.93% | 0.4674 | |
| Low | 80.34% | 60.14% | 85.97% | 0.4451 |
Table 4.
The prediction performance of self-consistency, Jack-knife validation and n-fold validation of PAIL with window length of 13.
| Window length (13) | Threshold | Accuracy | Sensitivity | Specificity | MCC |
|---|---|---|---|---|---|
| Self-Consistency | High | 89.21% | 61.38% | 96.95% | 0.6608 |
| Medium | 87.97% | 73.58% | 91.97% | 0.6499 | |
| Low | 85.13% | 79.68% | 86.65% | 0.6111 | |
| Jack-Knife validation | High | 86.11% | 52.85% | 95.36% | 0.5551 |
| Medium | 84.42% | 61.79% | 90.72% | 0.5348 | |
| Low | 82.92% | 63.82% | 88.24% | 0.5097 | |
| 6-fold cross-validation | High | 85.42% | 48.31% | 95.75% | 0.5266 |
| Medium | 83.58% | 59.76% | 90.20% | 0.5091 | |
| Low | 82.17% | 62.20% | 87.73% | 0.4886 | |
| 8-fold cross-validation | High | 85.53% | 51.40% | 95.02% | 0.5353 |
| Medium | 83.81% | 60.26% | 90.36% | 0.5160 | |
| Low | 82.34% | 62.52% | 87.85% | 0.4931 | |
| 10-fold cross-validation | High | 85.66% | 51.59% | 95.14% | 0.5396 |
| Medium | 83.69% | 60.29% | 90.21% | 0.5136 | |
| Low | 82.17% | 62.72% | 87.58% | 0.4905 |
With window length of 9, the accuracies of three thresholds are 86.37%, 85.75% and 82.65%, respectively (see in table 2). The sensitivity (Sn), specificity (Sp) and MCC are 49.19%~69.92%, 96.72%~86.19%, and 0.5584~0.5277. Also, the results of Jack-Knife validation and n-fold (6-, 8-, 10-fold) cross-validation proposes our prediction is robust. In table 3, the accuracy fluctuates from 88.14% to 84.60%, with the window length of 11. When the PAPs are chosen with length of 13, the accuracy is 89.21%~85.13% (see in Table 4). And MCC fluctuates from 0.6608 to 0.6111. Again, the validation results suggest that the prediction is accurate and robust. In this condition, the sensitivity (Sn) and specificity (Sp) are 61.38%~79.68% and 96.95%~86.65%, respectively.
Furthermore, to compare the prediction performance of PAPs with different window lengths, we also diagram their ROC (Receiver Operating Characteristic) curves (sensitivity vs. 1-specificity) shown in Figure 2. Three curves are quite similar. However, when the specificity is greater than 80% (that is to see, the value of 1-speficity is <0.2), the performance of PAPs with window length of 13 is better than others. In this regard, the PAPs with window length of 13 have been employed in current PAIL system.
Figure 2.
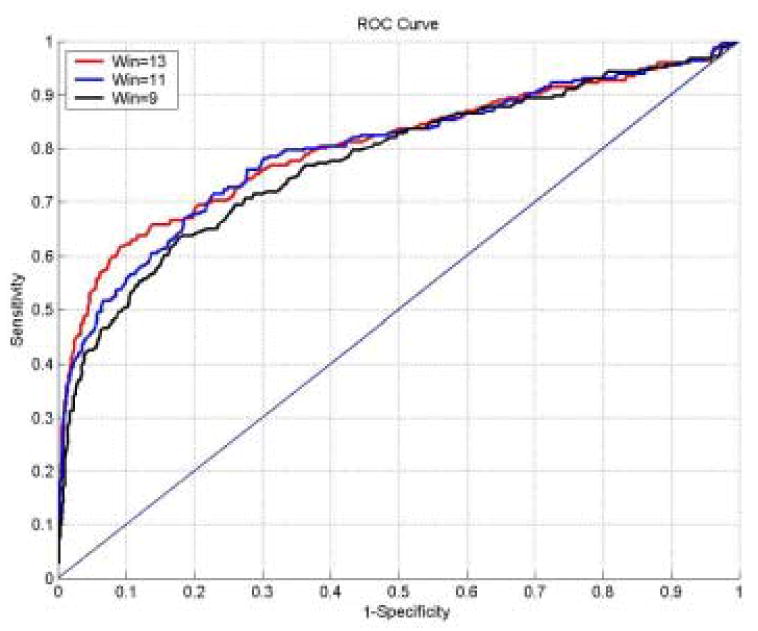
The Receiver Operating Characteristic (ROC) curve to diagram the prediction performances of PAIL with window length of 9, 11 and 13.
Discussion
PAIL is a novel in silico acetylation site prediction system with high-performance and may provide valuable insight into further experimentation. The study of protein acetylation is still in its infancy, and many problems remain to be resolved. For example, the prediction performance of PAIL is limited by the lack of a large amount of data sets as the known protein acetylation sites are still far fewer than those of phosphorylation [29, 30]. As large-scale screening strategies have been applied to identify the protein acetylation sites systematically [8, 14, 24], more and more bona fide data can be generated and integrated into the PAIL system to optimize its computing power. In addition, there have been ~30 HATs (histone acetyltransferases) and >18 HDACs (histone deacetylases) discovered [5, 17]. Thus, a more rigorous predictor in a HAT-specific mode is also desirable. However, due to the limit amount of data, such a computational tool currently is not available. In addition, some other computational approaches could be applied, i.e., Group-based Prediction and Scoring algorithm (GPS) [29, 30] and Support Vector Machines (SVMs) [31] These methods could be employed separately or combined together to obtain potentially better performance. Nevertheless, with high-accuracy PAIL provides the first computational tool for identifying protein acetylation sites in silico.
Acknowledgments
The work is supported by Chinese Natural Science Foundation (39925018, 20270293, and 90508002), Chinese Academy of Science (KSCX2-2-01), Chinese 973 project (2002CB713700), Chinese 863 project (2001AA215331), and Chinese Minister of Education (20020358051) to X. Yao. X. Yao is a Cheung Kong Scholar.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Glozak MA, Sengupta N, Zhang X, Seto E. Acetylation and deacetylation of non-histone proteins. Gene. 2005;363:15–23. doi: 10.1016/j.gene.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 2.Kouzarides T. Acetylation: a regulatory modification to rival phosphorylation? Embo J. 2000;19:1176–1179. doi: 10.1093/emboj/19.6.1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Polevoda B, Sherman F. Nalpha -terminal acetylation of eukaryotic proteins. J Biol Chem. 2000;275:36479–36482. doi: 10.1074/jbc.R000023200. [DOI] [PubMed] [Google Scholar]
- 4.Polevoda B, Sherman F. The diversity of acetylated proteins. Genome Biol. 2002;3:reviews0006. doi: 10.1186/gb-2002-3-5-reviews0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang XJ. The diverse superfamily of lysine acetyltransferases and their roles in leukemia and other diseases. Nucleic Acids Res. 2004;32:959–976. doi: 10.1093/nar/gkh252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bannister AJ, Miska EA, Gorlich D, Kouzarides T. Acetylation of importin-alpha nuclear import factors by CBP/p300. Curr Biol. 2000;10:467–470. doi: 10.1016/s0960-9822(00)00445-0. [DOI] [PubMed] [Google Scholar]
- 7.Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, Hu LS, Cheng HL, Jedrychowski MP, Gygi SP, Sinclair DA, Alt FW, Greenberg ME. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- 8.Cohen HY, Lavu S, Bitterman KJ, Hekking B, Imahiyerobo TA, Miller C, Frye R, Ploegh H, Kessler BM, Sinclair DA. Acetylation of the C terminus of Ku70 by CBP and PCAF controls Bax-mediated apoptosis. Mol Cell. 2004;13:627–638. doi: 10.1016/s1097-2765(04)00094-2. [DOI] [PubMed] [Google Scholar]
- 9.Faiola F, Liu X, Lo S, Pan S, Zhang K, Lymar E, Farina A, Martinez E. Dual regulation of c-Myc by p300 via acetylation-dependent control of Myc protein turnover and coactivation of Myc-induced transcription. Mol Cell Biol. 2005;25:10220–10234. doi: 10.1128/MCB.25.23.10220-10234.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Murr R, Loizou JI, Yang YG, Cuenin C, Li H, Wang ZQ, Herceg Z. Histone acetylation by Trrap-Tip60 modulates loading of repair proteins and repair of DNA double-strand breaks. Nat Cell Biol. 2006;8:91–99. doi: 10.1038/ncb1343. [DOI] [PubMed] [Google Scholar]
- 11.Subramanian C, Opipari AW, Jr, Bian X, Castle VP, Kwok RP. Ku70 acetylation mediates neuroblastoma cell death induced by histone deacetylase inhibitors. Proc Natl Acad Sci U S A. 2005;102:4842–4847. doi: 10.1073/pnas.0408351102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yuan ZL, Guan YJ, Chatterjee D, Chin YE. Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science. 2005;307:269–273. doi: 10.1126/science.1105166. [DOI] [PubMed] [Google Scholar]
- 13.Yang XJ. Lysine acetylation and the bromodomain: a new partnership for signaling. Bioessays. 2004;26:1076–1087. doi: 10.1002/bies.20104. [DOI] [PubMed] [Google Scholar]
- 14.Marsh VL, Peak-Chew SY, Bell SD. Sir2 and the acetyltransferase, Pat, regulate the archaeal chromatin protein, Alba. J Biol Chem. 2005;280:21122–21128. doi: 10.1074/jbc.M501280200. [DOI] [PubMed] [Google Scholar]
- 15.Starai VJ, Gardner JG, Escalante-Semerena JC. Residue Leu-641 of Acetyl-CoA synthetase is critical for the acetylation of residue Lys-609 by the Protein acetyltransferase enzyme of Salmonella enterica. J Biol Chem. 2005;280:26200–26205. doi: 10.1074/jbc.M504863200. [DOI] [PubMed] [Google Scholar]
- 16.Cereseto A, Manganaro L, Gutierrez MI, Terreni M, Fittipaldi A, Lusic M, Marcello A, Giacca M. Acetylation of HIV-1 integrase by p300 regulates viral integration. Embo J. 2005;24:3070–3081. doi: 10.1038/sj.emboj.7600770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Verdin E, Dequiedt F, Kasler HG. Class II histone deacetylases: versatile regulators. Trends Genet. 2003;19:286–293. doi: 10.1016/S0168-9525(03)00073-8. [DOI] [PubMed] [Google Scholar]
- 18.Li D, Yea S, Dolios G, Martignetti JA, Narla G, Wang R, Walsh MJ, Friedman SL. Regulation of Kruppel-like factor 6 tumor suppressor activity by acetylation. Cancer Res. 2005;65:9216–9225. doi: 10.1158/0008-5472.CAN-05-1040. [DOI] [PubMed] [Google Scholar]
- 19.Bae SC, Lee YH. Phosphorylation, acetylation and ubiquitination: The molecular basis of RUNX regulation. Gene. 2005 doi: 10.1016/j.gene.2005.10.017. [DOI] [PubMed] [Google Scholar]
- 20.Barnes PJ, Adcock IM, Ito K. Histone acetylation and deacetylation: importance in inflammatory lung diseases. Eur Respir J. 2005;25:552–563. doi: 10.1183/09031936.05.00117504. [DOI] [PubMed] [Google Scholar]
- 21.Iwabata H, Yoshida M, Komatsu Y. Proteomic analysis of organ-specific post-translational lysine-acetylation and -methylation in mice by use of anti-acetyllysine and -methyllysine mouse monoclonal antibodies. Proteomics. 2005;5:4653–4664. doi: 10.1002/pmic.200500042. [DOI] [PubMed] [Google Scholar]
- 22.Kelly WK, Marks PA. Drug insight: Histone deacetylase inhibitors--development of the new targeted anticancer agent suberoylanilide hydroxamic acid. Nat Clin Pract Oncol. 2005;2:150–157. doi: 10.1038/ncponc0106. [DOI] [PubMed] [Google Scholar]
- 23.Tabe Y, Konopleva M, Contractor R, Munsell M, Schober WD, Jin L, Tsutsumi-Ishii Y, Nagaoka I, Igari J, Andreeff M. Upregulation of MDR1 and induction of doxorubicin resistance by histone deacetylase inhibitor depsipeptide (FK228) and ATRA in acute promyelocytic leukemia cells. Blood. 2005 doi: 10.1182/blood-2004-10-4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dormeyer W, Ott M, Schnolzer M. Probing lysine acetylation in proteins: strategies, limitations, and pitfalls of in vitro acetyltransferase assays. Mol Cell Proteomics. 2005;4:1226–1239. doi: 10.1074/mcp.M500047-MCP200. [DOI] [PubMed] [Google Scholar]
- 25.Kiemer L, Bendtsen JD, Blom N. NetAcet: prediction of N-terminal acetylation sites. Bioinformatics. 2005;21:1269–1270. doi: 10.1093/bioinformatics/bti130. [DOI] [PubMed] [Google Scholar]
- 26.Liu Y, Lin Y. A novel method for N-terminal acetylation prediction. Genomics Proteomics Bioinformatics. 2004;2:253–255. doi: 10.1016/S1672-0229(04)02032-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xue Y, Li A, Wang L, Xu W, Feng H, Yao X. PPSP: Prediction of PK-Specific phosphorylation site with Bayesian Decision Theory. BMC Bioinformatics. 2006;7:163. doi: 10.1186/1471-2105-7-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xue Y, Zhou F, Zhu M, Ahmed K, Chen G, Yao X. GPS: a comprehensive www server for phosphorylation sites prediction. Nucleic Acids Res. 2005;33:W184–187. doi: 10.1093/nar/gki393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou FF, Xue Y, Chen GL, Yao X. GPS: a novel group-based phosphorylation predicting and scoring method. Biochem Biophys Res Commun. 2004;325:1443–1448. doi: 10.1016/j.bbrc.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 31.Xie D, Li A, Wang M, Fan Z, Feng H. LOCSVMPSI: a web server for subcellular localization of eukaryotic proteins using SVM and profile of PSI-BLAST. Nucleic Acids Res. 2005;33:W105–110. doi: 10.1093/nar/gki359. [DOI] [PMC free article] [PubMed] [Google Scholar]