

Abstract
In normal epidermis, β1 integrin expression is confined to the basal layer, whereas in hyperproliferative epidermis, integrins are also expressed in the suprabasal layers. Transgenic mice in which integrins are expressed suprabasally via the involucrin promoter have a sporadic psoriatic phenotype; however, the mechanism by which integrins contribute to the pathogenesis of psoriasis is unknown. We observed activation of mitogen-activated protein kinase (MAPK) in basal and suprabasal keratinocytes of human and transgenic mouse psoriatic lesions and healing mouse skin wounds, correlating in each case with suprabasal integrin expression. Phenotypically normal human and transgenic mouse epidermis did not contain activated MAPK. Transgene-positive keratinocytes produced more IL-1α than controls did, and keratinocyte MAPK could be activated by ligation of suprabasal integrins or treatment with IL-1α. Constitutive activation of MAPK increased the growth rate of human keratinocytes and delayed the onset of terminal differentiation, recreating many of the histological features of psoriatic epidermis. We propose that activation of MAPK by integrins, either directly or through increased IL-1α production, is responsible for epidermal hyperproliferation in psoriasis and wound healing, and that the sporadic phenotype of the transgenic mice may reflect the complex mechanisms by which IL-1 release and responsiveness are controlled in skin.
Introduction
The unique structure of the epidermis is maintained by a tightly regulated balance between keratinocyte proliferation and terminal differentiation. Stem cells within the epidermal basal layer self-renew and also produce nonstem daughters, known as transit-amplifying cells (1). Transit-amplifying cells divide a small number of times before commitment to terminal differentiation. Committed cells withdraw from the cell cycle, detach from the epidermal basement membrane, and move into the suprabasal layers of the epidermis. As they migrate through the suprabasal layers toward the skin surface, keratinocytes undergo striking changes in morphology and gene expression (2). In the first suprabasal layers, they enlarge; they express novel keratins and several proteins, including involucrin and cornifin, that are precursors of a structure known as the cornified envelope. Once they have entered the outermost layers, the cells flatten and lose their nucleus, and the envelope precursors become enzymatically crosslinked by transglutaminase 1, resulting in envelope assembly. The cornified cells are continually shed from the surface of the skin and are replenished through proliferation of stem cells in the basal layer.
Attachment of basal keratinocytes to the basement membrane is mediated by integrin receptors, including the α6β4 integrin, which is a receptor for laminin, and several β1 integrins, the most abundant of which are α2β1 (collagen receptor) and α3β1 (laminin receptor). In addition to their adhesive role, β1 integrins regulate keratinocyte differentiation. Human epidermal stem cells have higher levels of β1 integrins and are more adhesive to ECM proteins than are transit-amplifying cells (1, 3), and when a dominant negative β1 integrin mutant is introduced into human keratinocytes in culture the proportion of transit-amplifying cells is markedly increased (4). When keratinocytes are deprived of contact with the ECM, both stem and transit-amplifying cells differentiate without further rounds of division (3); suspension-induced terminal differentiation can be inhibited with ECM proteins or antibodies to β1 integrins, suggesting that integrin ligation is a negative regulator of terminal differentiation (5).
Given the role of integrins in regulating keratinocyte adhesion and differentiation, it is not surprising that integrin expression is altered in both benign and neoplastic keratinocyte disorders (6). In particular, although integrin expression is normally confined to the basal layer of the epidermis, suprabasal integrin expression is a feature of hyperproliferative epidermis, as found, for example, after wound closure or in lesions of the benign human skin disorder psoriasis (6). That suprabasal integrin expression can play a causal role in the onset of psoriasis has been demonstrated by creating transgenic mice in which various integrin subunits are expressed under the control of the involucrin promoter (7, 8). In these mice, sporadic epidermal hyperproliferation with accompanying histological features of psoriasis, including a lymphocytic infiltrate, is observed.
There are two alternative hypotheses for how suprabasal integrin expression could trigger epidermal hyperproliferation: either the presence of integrins on the surface of suprabasal cells provides a signal that the underlying basal cells respond to, or the integrins signal to the suprabasal cells themselves, possibly leading to unscheduled synthesis of growth factors or cytokines (7, 8). Basal keratinocytes are not stimulated to proliferate when exposed to integrin-positive suprabasal keratinocytes in vitro (8); therefore, there is currently no evidence in favor of the first hypothesis. This has led us to consider the alternative hypothesis and to examine signal transduction by suprabasal integrins.
The classic mitogen-activated protein kinase (MAPK) cascade has previously been implicated in integrin regulation of keratinocyte terminal differentiation (4). The cascade consists of a module of three protein kinases acting in a hierarchical order: the MAP kinases ERK (extracellularly regulated kinase) 1 and 2, which are activated by different isoforms of MAP kinase kinases (MEK) through phosphorylation of conserved threonines and tyrosines within a TXY motif (9), and the MAP kinase kinase kinases, including the Raf family, which regulate MAP kinase kinase (MAPKK) activity by phosphorylation of serine residues, such as serines 217 and 221 in MAPK kinase 1 (10). Expression of constitutively activated MAPKK (with glutamic acid substitutions at serines 217 and 221; known as MAPKK1; ref. 11) can overcome the effect of a dominant negative β1 integrin mutant on human keratinocytes, thereby restoring the size of the stem cell compartment in vitro (4). In addition, a dominant negative MAPKK mutant (with an alanine substitution at serine 221; known as MANA; ref. 11) has the same effect as the dominant negative integrin, reducing keratinocyte adhesiveness and β1 integrin levels and causing cells to leave the stem cell compartment (4). Given the role of β1 integrin signaling via MAPK in the stem to transit-amplifying cell transition, we decided to investigate whether suprabasal integrins contribute to epidermal hyperproliferation by activating MAPK.
Methods
Source of tissue.
Psoriatic lesions from the upper leg of nine untreated patients, and samples of normal human skin from the upper leg, back, and cheek of four patients who underwent tumor excisions, were kindly provided by K. Hartmann and M. Haitas-Haase (University of Cologne, Germany). Normal scalp skin and neonatal foreskin (one sample of each) were also examined. Skin samples were taken from the backs of transgenic mice expressing the human β1 integrin subunit or human α6β1 under the control of the involucrin promoter (founder lines 0840 and 1137B; CBA X C57BL/10F1; refs. 7, 8) and from littermate controls. In some experiments, 3-mm-diameter full thickness wounds were made in the backs of mice expressing the β1 transgene or wild-type controls; 6–7 days later, the animals were sacrificed and the wound sites collected for histological analysis.
Keratinocyte culture and retroviral infection.
Normal human epidermal keratinocytes from neonatal foreskins (strains kq, km, and kn; passages 2–8) were cultured in FAD medium (one part Ham’s F12 medium plus three parts DMEM and 0.18 mM adenine) supplemented with 10% FCS and a cocktail of 0.5 μg/ml hydrocortisone, 5 μg/ml insulin, 10–10 M cholera toxin, and 10 ng/ml epidermal growth factor (HICE) on a J2-3T3 feeder layer as described previously (3). To determine the proportion of S-phase cells, keratinocytes were pulse-labeled with 10 μg/ml BrdU for 1 hour and were harvested and immunolabeled as described previously (12). Organotypic cultures on de-epidermized dermis were carried out as described previously (13).
The constitutively active MAPK kinase 1 mutant, MAPKK1, with glutamic acid substitutions at serines 217 and 221, and the dominant negative mutant MANA, with an alanine substitution at serine 221 (11), cloned into pBabe puro, were generous gifts of C. Marshall (Institute of Cancer Research, London, United Kingdom). Keratinocytes were infected by coculture with retroviral producer cells as described previously (4).
Lines of murine keratinocytes derived from the skin of a transgenic mouse expressing human α2β1 or the human β1 integrin subunit under the control of the involucrin promoter (7, 8) (passages 19–24) were cultured at 32°C on a J2-3T3 feeder layer in the same medium as used to culture human keratinocytes.
Immunostaining of cells and tissue sections.
The following antibodies were used for immunohistochemistry: LH001 (monoclonal anti-human keratin 14; kindly provided by I. Leigh, London Hospital, London, United Kingdom), DH1 (rabbit anti-human involucrin), rabbit anti–Ki 67 (Novacastra, Newcastle, United Kingdom), rabbit antisera to doubly phosphorylated MAPK (NEB, Hitchin, United Kingdom, or MAPK-YT from Sigma Chemical Co., St. Louis, Missouri, USA), monoclonal anti-ERK2 (sc-1647 to stain human skin; Santa Cruz Biotechnology Inc., Santa Cruz, California, USA), rabbit anti-ERK2 (sc-153 to stain mouse skin; Santa Cruz Biotechnology Inc.), mAb13 (monoclonal anti-β1 integrin; Becton Dickinson, Lincoln Park, New Jersey, USA). Secondary antibodies labeled with Alexa 488 (green) and 594 (red) (Molecular Probes Inc., Eugene, Oregon, USA) were used. In some experiments, cell nuclei were stained using a DNA-specific dye, ToProIII (Molecular Probes Inc.) for 5–30 minutes at a concentration of 1 μM (applied after incubation with secondary antibody). Controls were to stain with secondary antibody alone or to preincubate MAPK antibodies with blocking peptides.
Before staining, cultured cells and frozen sections of skin were fixed in 4% paraformaldehyde for 10 minutes at room temperature; cultured cells were then permeabilized with 0.4% Triton X 100 in PBS for 4 minutes at room temperature. Incubations with primary and secondary antibodies were for approximately 1 hour at room temperature with subsequent PBS washes, except in the case of anti–doubly phosphorylated MAPK antibodies, when the manufacturers’ protocols were followed. Sections of paraformaldehyde-fixed, paraffin-embedded day 11 organotypic cultures were stained with anti–Ki 67, and the average number (± SD) of positive cells in 12 high-power fields per section was determined.
ELISAs of cytokine production.
A line of murine keratinocytes expressing human α2β1 under the control of the involucrin promoter and a transgene negative control line (8) were grown to near confluence on a J2-3T3 feeder layer. Medium conditioned by the cultures overnight was collected, and then the feeder cells were removed and the keratinocytes harvested. Keratinocytes were plated for 8 or 24 hours onto agarose-coated dishes in FAD + fibronectin-free FCS + HICE to prevent cell attachment. After 8 hours in suspension, a fraction of the cells was collected and allowed to attach to dishes coated with 10 μg/ml type IV collagen for a further 16 hours.
Both intracellular and secreted levels of cytokines were measured. To measure intracellular cytokines, cells were washed in PBS and lysed in 1% NP40, 10 mM iodoacetamide, 1 mM EDTA, 1 μg/ml leupeptin, 1 μg/ml pepstatin, and 0.4 mM PMSF (14). IL-1α, IL-1β, TNF-α, and IL-6 levels in the collected samples were determined by use of ELISA kits (R&D Systems Inc., Minneapolis, Minnesota, USA, and Amersham Pharmacia Biotech, Piscataway, New Jersey, USA).
Cell adhesion and MAPK phosphorylation.
To analyze MAPK phosphorylation in response to integrin ligation in primary human keratinocytes, the cells were harvested with trypsin/EDTA and either suspended in serum-free medium supplemented with 10 ng/ml EGF for 30 minutes or plated at 37°C for various times onto bacteriological dishes coated with different ECM proteins or antibodies (25 μg/ml fibronectin from Becton Dickinson; 50 μg/ml collagens type I and type IV from Sigma Chemical Co.; or 100 μg/ml mAb13) and blocked with 1% heat denatured BSA in PBS for 1 hour at 37°C. Control dishes were coated with 20 μg/ml poly-L-lysine for 2 hours at 37°C and not blocked. Coating with laminin 5 was performed as described previously (4).
To assess the ability of IL-1α, IL-1β, IL-6, and EGF to activate ERK1/2 and p38 MAPK in adherent primary human keratinocytes, dishes of preconfluent keratinocytes were starved overnight in serum-free medium after removing the feeders. Cells were then stimulated with FAD supplemented with cytokine or FAD alone for 15 minutes before harvesting. Recombinant human IL-1 and IL-6 were purchased from R&D Systems Inc. and EGF from Peprotech (Rocky Hill, New Jersey, USA).
To analyze modulation of MAPK phosphorylation by the MAPKK1 and MANA mutants, preconfluent keratinocytes were starved overnight after removing the feeders and then treated with FAD ± FCS/HICE for 10 minutes. To analyze MAPK phosphorylation in suspension, cells were held in FAD + FCS + HICE supplemented with methylcellulose (3) for 24 hours and then harvested.
The ability of suprabasal integrins to activate MAPK was determined using mouse keratinocytes expressing the human β1 integrin subunit under the control of the involucrin promoter (8). The cells were held in suspension in FAD + FCS + HICE with methylcellulose for 16 hours, harvested, resuspended in FAD supplemented with 10 ng/ml EGF, and either held in suspension for 30 minutes or plated onto dishes coated with the human β1 integrin–specific antibodies mAb13 or P5D2 (4) or with 25 μg/ml fibronectin.
Lysates of cultured keratinocytes were prepared and immunoblotted as described previously (4). In addition, lysates were prepared from a shaved biopsy of a psoriatic lesion and a control biopsy of normal human skin. Before extraction, the skin samples were frozen in liquid nitrogen and ground with a mortar and pestle. Thereafter, skin samples were processed in the same way as cultured cells. Activated MAPK was detected with antibodies specific for phosphorylated ERK1/2 or p38MAPK (NEB). Blots were reprobed with antibodies to ERK2 or p38MAPK (Santa Cruz Biotechnology Inc.). Protein bands were visualized with horseradish peroxidase–coupled secondary antibodies using enhanced chemiluminescence (ECL; Amersham, Buckinghamshire, United Kingdom).
Analysis of suspension-induced terminal differentiation.
Suspension culture of human keratinocytes was carried out as described previously (3), except that the FCS used was depleted of fibronectin using Gelatin Sepharose 4B (Amersham Pharmacia Biotech, St. Albans, United Kingdom). Cells harvested from suspension culture were stained for flow cytometry with antibodies to involucrin (12), transglutaminase 1 (B.C1; gift of R. Rice, University of California, Davis, California, USA; ref. 15) and α cornifin (SQ37C; gift of A. Jetten, National Institute of Environmental Health Sciences, Research Triangle Park, North Carolina, USA; ref. 16) using essentially the method described for involucrin (12).
Results
MAPK is activated in hyperproliferative keratinocytes.
Nuclear translocation of the MAP kinases ERK1 and ERK2 is strictly dependent on enzyme activation (17). To confirm that nuclear localization was a reporter of MAPK activation in keratinocytes, we starved normal human epidermal keratinocytes overnight in FAD medium without serum or growth factors (HICE cocktail) and then treated the cultures for 15 minutes with FAD alone or FAD supplemented with 10% FCS and HICE before fixation. In serum-starved keratinocytes, there was no staining above background with an antibody specific for phosphorylated MAPK (ERK1,2) (Figure 1a). However, in cells treated with serum and growth factors, there was strong nuclear staining, and cytoplasmic staining was also increased (Figure 1b). A similar, though slightly less pronounced, translocation of MAPK to the cell nucleus was observed with an anti-ERK2 antibody that detects both phosphorylated and unphosphorylated ERK2 (p42mapk) (data not shown).
Figure 1.

Distribution and activation of p42mapk in cultured keratinocytes and in normal and hyperproliferative human epidermis. Confocal immunofluorescence microscopy of growth factor–starved cultured keratinocytes (a) or keratinocytes treated with FCS + HICE for 15 minutes (b); psoriatic (c, f, and h–j) or normal human epidermis (e and g). Green staining in a–c, e–h, and j is for MAPK with antibody specific for activated (a–c and e) or total MAPK (f–h and j); red staining in h is nuclear dye, Topro III. The field shown in h is part of the field shown in f. (i) Staining for β1 integrins (green) and involucrin (red). Scale bars: 50 μm (a, b, and f–h), 100 μm (c, e, i, and j). (d) Western blot of shaved biopsies of normal human skin (N) or psoriatic lesion (Ps) probed with phosphorylation-specific antibody to ERK1/2 (P-ERK) or total ERK2.
We next used immunofluorescence staining to examine MAPK activation in normal and hyperproliferative human epidermis. Frozen sections were stained with an mAb against total ERK2 or antibodies specific for phosphorylated MAPK. In healthy skin (from five individuals), the antibody to total MAPK predominantly stained the membrane region of epidermal keratinocytes with little cytoplasmic and no nuclear staining (Figure 1g), whereas anti–phospho-MAPK gave no staining above background (Figure 1e). In psoriatic epidermis (from eight individuals), there was both membranous and cytoplasmic staining with the antibody to total MAPK; in addition, the majority of cells in the basal and lower suprabasal layers showed a positive nuclear signal for MAPK (Figure 1, f, h, and j). Staining with the antibody to phosphorylated MAPK was predominantly nuclear in psoriatic lesions (Figure 1c). The suprabasal layers that had nuclear MAPK also showed strong expression of β1 integrins (compare Figure 1i, green, with Figure 1j, which is a serial section) and were further characterized by the absence of involucrin expression (Figure 1i, red). The enhanced activation of MAPK in psoriatic skin compared with normal skin was confirmed by Western blotting of shaved biopsies (Figure 1d).
Normal and hyperproliferative epidermis from transgenic mice that expressed β1 integrins suprabasally under the control of the involucrin promoter (7) was stained with an antibody to ERK2 or anti–phospho-MAPK (Figure 2). In phenotypically normal epidermis from transgenic mice and transgene-negative littermate controls, there was weak cytoplasmic and plasma membrane staining and no nuclear staining with the antibody to total MAPK (Figure 2a and data not shown). The antibody to phospho-MAPK gave no staining above background (Figure 2c). In contrast, spontaneous hyperproliferative lesions from transgenic mice had pronounced nuclear phosphorylated MAPK (Figure 2d).
Figure 2.

Distribution and activation of p42mapk in phenotypically normal and hyperproliferative transgenic mouse epidermis. (a and b) Antibody to total MAPK. (c–e) Antibody to activated MAPK. Hyperproliferative β1 transgenic mouse epidermis 7 days after wounding (b and e) or unwounded transgenic mouse skin (a). Phenotypically normal epidermis (c) and psoriatic lesion (d) of α6β1 transgenic mouse. Red staining in a and b is nuclear dye, Topro III. Scale bar: 50 μm.
Epidermal hyperproliferation can be induced by wounding. We stained sections of hyperproliferative epidermis from β1 transgenic mice and littermate controls 6 or 7 days after creating full thickness wounds. Immunostaining showed localization of ERK2 in the nuclei of many basal and suprabasal keratinocytes in the hyperproliferative area covering the wound (Figure 2b), but not in the adjacent nonaffected skin (data not shown). The nuclear localization of MAPK was confirmed by double staining with the DNA dye, Topro III (Figure 2b). The antibody to phosphorylated MAPK also gave strong nuclear staining of wounded, hyperproliferative epidermis (Figure 2e). Activation of MAPK was observed in wounds of wild-type and transgenic epidermis (Figure 2, b and e, and data not shown).
We conclude that MAPK was activated in hyperproliferative mouse and human epidermis, nuclear staining being observed in suprabasal integrin-positive cells. However, suprabasal integrin expression did not result in constitutive activation of MAPK, as no nuclear staining was observed in phenotypically normal epidermis from the transgenic mice.
Suprabasal integrins can activate MAPK.
The MAPK cascade is a primary target of signaling pathways originating both from growth factor receptors and integrins, with growth factor–stimulated MAPK activity being dependent on integrin engagement (18, 19). In suspended primary human keratinocytes or keratinocytes plated onto the nonspecific adhesion substrate, poly-L-lysine MAPK activity was low or undetectable, even in the presence of 10 ng/ml EGF (Figure 3a). MAPK activation was observed upon engagement of β1 integrins by collagen, fibronectin, and the β1 integrin–specific antibody mAb13 and also upon attachment to laminin 5, which is mediated by the α6β4 and α3β1 integrins. MAPK activation was observed in cells plated on laminin 5 under conditions in which binding via the α3β1 laminin receptor was inhibited with the β1-specific antibody P5D2, thereby confirming that attachment to laminin via α6β4 was sufficient to trigger MAPK activation (Figure 3a). MAPK activation by cell attachment was transient, phosphorylation of the enzyme being highest after 30 minutes and then gradually decreasing to reach background levels after 4 hours (ref. 4; data not shown). These results indicate that activation of MAPK in human keratinocytes is integrin dependent, even in the presence of a high concentration of EGF.
Figure 3.

Integrin-mediated MAPK phosphorylation. Western blot analysis of MAPK phosphorylation: upper panels show threonine/tyrosine phosphorylation of ERK1/2 visualized with a phosphorylation-specific antibody; lower panels show the same blots incubated with an antibody against total ERK2 as a loading control. (a) Human keratinocytes in FAD medium containing 10 ng/ml EGF were held in suspension or plated for 30 minutes onto dishes coated with poly-L-lysine or the ECM proteins and antibodies shown. (b) Invβ1 mouse keratinocytes were cultured in suspension for 12 hours to induce terminal differentiation and transgene expression and then held in suspension or plated for 30 minutes in the presence of 10 ng/ml EGF onto fibronectin or the human β1–specific antibodies mAb13 and P5D2.
To test whether integrins expressed by suprabasal keratinocytes were able to signal to MAPK, we used a line of keratinocytes isolated from the skin of an invβ1 transgenic mouse (8). When held in suspension, these cells, like normal human keratinocytes, are induced to undergo terminal differentiation: expression of the endogenous integrins is downregulated while expression of involucrin and the β1 transgene is induced (8). The transgenic integrin is human and can therefore be distinguished from the endogenous mouse integrins using species-specific antibodies (8). The cells were held in suspension for 16 hours and then plated onto fibronectin or the human β1–specific antibodies mAb13 and P5D2 (Figure 3b). As observed in primary human keratinocytes (compare Figure 3a with 3b) MAPK activity was low in transgenic keratinocytes in suspension but increased when the transgenic integrin was ligated by ECM or anti–β1 antibody (Figure 3b). Thus β1 integrins expressed on the surface of terminally differentiating keratinocytes can signal through MAPK.
Increased production of IL-1α by keratinocytes expressing suprabasal integrins.
Although MAPK activation in vivo correlated with suprabasal integrin expression in hyperproliferative mouse and human epidermis (Figures 1 and 2) and suprabasal integrins were able to activate MAPK (Figure 3b), constitutive activation of MAPK was not observed in phenotypically normal transgenic mice expressing suprabasal integrins (Figure 2, a and c). We therefore sought to identify an additional signal that might be required for MAPK activation. Given that elevated expression of growth factors and proinflammatory cytokines is a feature of psoriasis (20), we examined whether suprabasal integrin expression correlated with altered cytokine production. We examined four proinflammatory cytokines known to be synthesized by keratinocytes (21): IL-1α, IL-1β, TNF-α, and IL-6.
We compared keratinocytes isolated from a wild-type mouse with keratinocytes isolated from a transgenic mouse expressing the α2 and β1 integrin subunits under the control of the involucrin promoter (Figure 4). We measured cytokine production by preconfluent keratinocytes cocultured overnight with a 3T3 feeder layer, keratinocytes held in suspension for 8 or 24 hours to stimulate terminal differentiation, and keratinocytes induced to differentiate in suspension for 8 hours and then replated on type IV collagen to ligate endogenous and transgenic α2β1. In all experimental situations, keratinocytes expressing suprabasal α2β1 secreted three- to fourfold the IL-1α into the medium (Figure 4a) and had larger intracellular stores of IL-1α (Figure 4b) than did wild-type keratinocytes. Suspension-induced differentiation increased the amount of IL-1α released by transgenic keratinocytes, and this increase was independent of ligation of α2β1 (Figure 4a; compare 24-hour suspension with 8-hour suspension + 16-hour attachment to collagen). Intracellular stores of IL-1α decreased with time in suspension and were restored by replating on collagen (Figure 4b).
Figure 4.

Proinflammatory cytokine production and cytokine-induced MAPK phosphorylation. Secretion of IL-1α (a), IL-1β (c), and IL-6 (d) by nontransgenic (open bars) and α2β1 transgene-expressing (filled bars) mouse keratinocytes. Intracellular IL-1α levels are shown in b. In the case of overnight keratinocyte/feeder cocultures (o/n coculture), cytokine secreted represents the total produced by both cell types, whereas intracellular levels were measured in keratinocytes alone. (e) Western blot analysis of ERK1/2 and p38 MAPK phosphorylation (P-ERK1/2 and P-p38 MAPK, respectively) and total ERK2 and p38 MAPK. Preconfluent human keratinocytes had feeders removed and were starved overnight before a 15-minute stimulation with medium alone (FAD) or medium supplemented with IL-1α/β, IL-6, or EGF alone or in combinations. IL-1 α/β and IL-6 were added to a final concentration of 10 ng/ml while EGF was added to 10 ng/ml when the only cytokine present (10 EGF) or 0.1 ng/ml when used in combination with other cytokines (0.1 EGF).
The difference in cytokine production between transgenic and control keratinocytes was specific for IL-1α. IL-1β release was induced equally in suspended transgenic and control keratinocytes (Figure 4c), and the greater amount released by transgenic compared with control keratinocytes after replating on collagen was not statistically significant (Figure 4c). The amount of intracellular IL-1β was below the detection limit of the assay. There was no difference in IL-6 release between transgenic and control cultures (Figure 4d), and intracellular IL-6 levels mirrored the levels secreted (data not shown). The amount of IL-6 released in overnight cocultures was an order of magnitude greater than the amount released by keratinocytes in suspension or suspended and replated, and this probably reflects IL-6 release by the feeder cells (22). TNF-α production (secreted and intracellular) in transgenic and wild-type keratinocytes was below 25 ng/ml, the limit of sensitivity of the assay (data not shown).
We next tested whether IL-1α activated MAPK in adherent human keratinocytes, using 10 ng/ml EGF as a positive control (Figure 4e). IL-1α and β activated ERK, although to a lesser extent than 10 ng/ml EGF did. IL-6 also had a stimulatory effect, but it was lower than the effect of IL-1. No further activation was observed with higher concentrations of the cytokines. There was no synergy in ERK activation between IL-1 and IL-6, nor between IL-1 and EGF (0.1 or 1 ng/ml EGF; Figure 4e and data not shown). As reported previously (23), IL-1α and β, but not IL-6 or EGF, activated p38MAPK (Figure 4e).
We conclude that suprabasal integrin expression by keratinocytes is correlated with increased IL-1α release and that IL-1 can activate ERK in keratinocytes.
Effect of MAPK activation on keratinocyte proliferation.
To modulate MAPK activity independently of integrins and growth factors, we introduced mutants of MAPK kinase 1 into primary human keratinocytes by means of retroviral infection, as described previously (4). Expression of a MAPK kinase 1 construct containing two activating point mutations (MAPKK1; ref. 11) led to constitutive activation of the MAPK cascade, as indicated by an increased level of phosphorylated MAPK in both adherent (Figure 5a) and suspended (Figure 5b) keratinocytes. The elevated level of MAPK phosphorylation achieved by expression of MAPKK1 in the absence of growth factors was lower than that in control (transduced with empty vector, puro) adherent cells stimulated for 10 minutes with medium containing FCS/HICE (Figure 5a), but was stable for at least 24 hours in adherent and suspended keratinocytes (Figure 5b and data not shown). Thus, introduction of MAPKK1 led to a sustained, submaximal activation of MAPK in adherent and suspended keratinocytes. Expression of a dominant negative mutant of MAPK kinase 1, MANA, caused reduced MAPK activation after stimulation of starved cells with medium containing FCS/HICE (Figure 5a), similar to the effect of MANA on attachment-induced MAPK phosphorylation (4).
Figure 5.

Effect of MAPK activity on keratinocyte proliferation. (a and b) Western blot detection of MAPK phosphorylation (upper panels) and total MAPK (lower panels) as described in the legend to Figure 3. (a) Adherent keratinocytes infected with empty vector puro, MAPKK1, or MANA were starved overnight and lysed 10 minutes after addition of fresh medium with or without FCS/HICE. (b) Keratinocytes infected with puro or MAPKK1 were trypsinized and held in suspension for 15 minutes (start) or 24 hours (susp). (c) Growth curves: 500 (puro, MAPKK1) or 1,000 (MANA) keratinocytes were plated per 35-mm dish (to achieve equal numbers of adherent cells; ref. 4) and cultured for 21 days. Triplicate dishes were harvested on the days shown. Error bars = SD.
We next used cells infected with constitutively active (MAPKK1) or dominant negative (MANA) mutants of MAPK kinase 1 to investigate the role of this signaling pathway in proliferation of primary human keratinocytes. Expression of MAPKK1 led to an increased growth rate compared with the empty vector control, puro, whereas introduction of MANA caused a reduction of cell proliferation (Figure 5c).
Although constitutive MAPK activity is sufficient to transform mammalian cell lines (11, 24), primary human keratinocytes expressing MAPPK1 still underwent growth arrest in suspension. The proportion of S-phase cells fell from 21.5 ± 3.5% in adherent cultures of cells expressing puro to 1.8 ± 0.9% after 24 hours in suspension. In cells expressing MAPKK1, it fell from 27.7 ± 1.2% to 1.0 ± 0.14%. Data are mean values from triplicate samples ± SD.
Effect of MAPK activation on suspension-induced terminal differentiation.
In keratinocytes, loss of integrin engagement is a strong trigger for the onset of terminal differentiation (3) and simultaneously results in disabled signal transduction via MAPK (Figure 3a). To examine the effects of MAPK activation on terminal differentiation, human keratinocytes infected with puro or MAPKK1 were placed in suspension for different lengths of time (Figure 6). In cells infected with puro, expression of involucrin, cornifin, and transglutaminase 1 was induced, with the proportion of cells expressing each protein increasing from 8 to 24 hours in suspension; this is revealed as an increase in the size of the cell peak with high fluorescence and a corresponding decrease in the cell peak with low fluorescence. Keratinocytes infected with MAPKK1 also showed an increase in expression of the differentiation markers; however, the number of cells expressing each protein (cell peak with high fluorescence) was always much lower than in puro-infected cells (Figure 6). Thus forced activation of MAPK signaling delayed the onset of suspension-induced keratinocyte differentiation.
Figure 6.

Effect of constitutive MAPK activation on suspension-induced terminal differentiation. Flow cytometry profiles of keratinocytes fixed immediately after trypsinization (0 hours) or after 8 or 24 hours in suspension. Cells were labeled with antibodies to the differentiation markers indicated. Black lines: Cells expressing empty vector puro; red lines: cells expressing MAPKK1.
Effect of MAPK activation on proliferation and differentiation of reconstituted epidermis.
When keratinocytes are seeded on dead, de-epidermized dermis and cultured at the air-liquid interface, they are able to reconstitute an epidermis with many of the characteristics of the normal tissue (ref. 13 and references cited therein). We compared epidermis reconstituted by keratinocytes infected with puro, MAPKK1, and MANA. At day 11 of culture, keratinocytes infected with puro had formed a stratified epithelium with a single basal cell layer and several suprabasal layers, the uppermost of which had started to form the dead, cornified layers characteristic of the final stage of keratinocyte terminal differentiation (Figure 7a). By day 14, the epidermis formed by puro-infected cells had slightly increased in thickness, and a largely anucleate cornified layer had developed with an underlying granular layer, thus bearing a close resemblance to normal epidermis (Figure 7d).
Figure 7.

Effects of constitutive activation or inhibition of MAPK on reconstituted epidermis. Human keratinocytes infected with puro (a and d), MAPKK1 (b and e), or MANA (c and f) were cultured for 11 (a–c) or 14 (d–f) days on dead, de-epidermized dermis. Asterisks in d indicate cells in granular layer. Scale bar: 100 μm.
Keratinocytes expressing MAPKK1 formed an epidermis that was already slightly thicker than the puro controls at day 11 and by day 14 showed pronounced thickening and elongated dermal projections (Figure 7, b and e). Immunostaining with an antibody to type IV collagen demonstrated that the basement membrane was intact (data not shown) and so the downgrowths of keratinocytes were not invasive fronts, but probably reflected the topology of the dermal substrate. In sharp contrast to cells expressing puro, there was no granular layer and nuclei were retained in the thick cornified layer that had formed by day 14. Keratinocytes expressing MANA gave rise to a thinner epidermis than the puro controls in which the basal cell layer was not clearly distinguishable and which by day 14 had only a thin, anucleate cornified layer (Figure 7, c and f).
To analyze proliferation in day 11 cultures, we used immunostaining with an antibody against the Ki 67 proliferation–associated antigen. The average number of Ki 67–positive cells per high-power field in 11-day cultures of keratinocytes expressing puro was 16.4 ± 1.6; in keratinocytes expressing MAPKK1, it was 29.6 ± 4.6; and in keratinocytes expressing MANA, it was 9.9 ± 2.4. The average number of basal cells per field was 115 for puro-, 134 for MAPKK1-, and 99 for MANA-expressing keratinocytes. Even allowing for differences in the cellularity of the basal layer, introduction of MAPKK1 led to an almost twofold increase in the number of Ki 67–positive cells, whereas MANA caused a reduction in proliferation. In the puro- and MANA-expressing cultures, the majority of Ki 67–positive cells were in the basal layer, whereas in the MAPKK1-expressing cultures, suprabasal Ki 67–positive cells were also present (data not shown).
MAPPK1 expression delayed the onset of involucrin expression in reconstituted epidermis (Figure 8). In cultures of keratinocytes expressing puro, basal cells were involucrin negative (red) and all suprabasal layers were involucrin positive (green or yellow in Figure 8a). In contrast, cultures infected with MAPKK1 contained several suprabasal involucrin–negative cell layers (Figure 8b, red) and involucrin expression was confined to a small compartment of cells directly beneath the cornified layer (Figure 8b, green or yellow). In epidermis reconstituted by MANA-expressing cells, the expression pattern of involucrin was similar to the puro control, in that all the suprabasal layers were positive; however, there were more involucrin-positive basal cells in the MANA cultures (Figure 8c).
Figure 8.
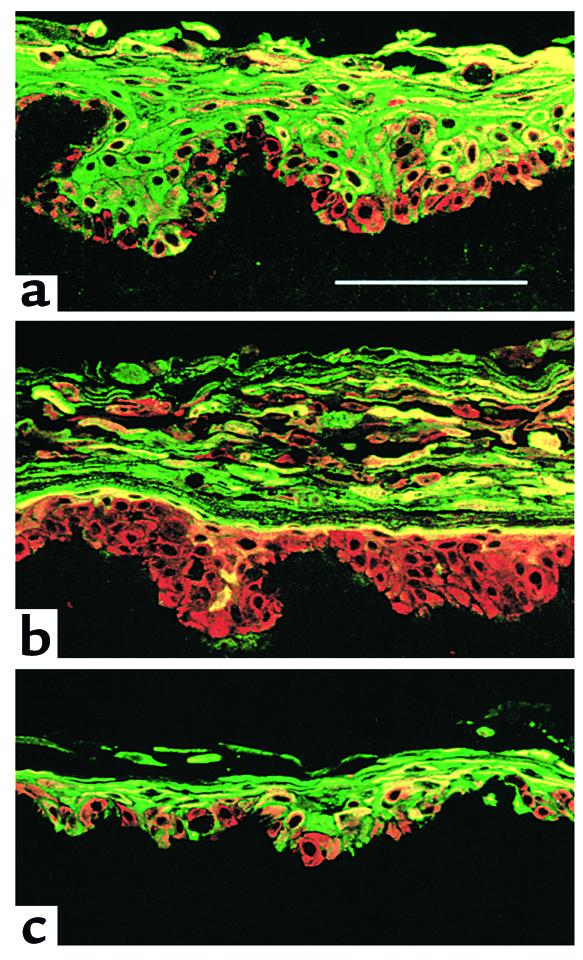
Effects of constitutive activation or inhibition of MAPK on involucrin expression in reconstituted epidermis. Keratinocytes infected with empty vector puro (a), MAPKK1 (b), or MANA (c) were cultured on dead, de-epidermized dermis for 11 days and stained with antibodies against keratin 14 (expressed by all cells; red) and involucrin (green). Scale bar: 100 μm.
Discussion
Using the nuclear localization and phosphorylation of MAPK as a readout for its activity (17, 25), we found that in hyperproliferative wounded or psoriatic epidermis, but not in normal epidermis, MAPK was activated in basal and suprabasal keratinocytes. The suprabasal cells with nuclear MAPK staining were cells that expressed β1 integrins. Ligation of suprabasal integrins activated MAPK, thus establishing that the receptors were capable of signal transduction. Although there is no evidence for suprabasal accumulation of the major basement membrane proteins in hyperproliferative epidermis (6), certain suprabasal integrins could potentially be ligated in vivo by minor collagens, such as type XIII collagen (26).
A second mechanism by which suprabasal integrins could activate MAPK is through stimulating keratinocytes to release IL-1α. The α2β1 transgenic keratinocytes in culture synthesized and released more IL-1α than control keratinocytes did, independent of integrin ligation, and IL-1α activated keratinocyte MAPK. In fibroblasts, IL-1α expression is increased by changes in cell shape, which can be induced by exposure to soluble or insoluble α5β1 integrin ligand (27), and in monocytes, ligation of αMβ2 or αXβ2 stimulates production of IL-1β (28). The concept that changes in cell shape could affect IL-1 release is consistent with the increase in IL-1α release when transgenic keratinocytes were placed in suspension (Figure 4a). In addition, mechanical deformation of human keratinocytes leads to rapid release of IL-1α, and this has been proposed to explain why psoriatic lesions are most commonly in locations where the skin is subject to repetitive stretch or trauma (29).
Even though suprabasal integrins were capable of activating MAPK directly or via stimulating release of IL-1α, examination of phenotypically normal transgenic epidermis established that suprabasal integrins do not activate MAPK constitutively. One explanation is that keratinocyte responsiveness to IL-1α does not correlate directly with the amount of IL-1α released. Keratinocytes express an IL-1 receptor antagonist and the nonsignaling type 2 IL-1 receptor, both of which decrease responsiveness to IL-1 (30). In addition, there are double paracrine pathways of growth regulation involving keratinocytes and dermal fibroblasts, such that keratinocyte release of IL-1 induces fibroblasts to produce KGF and IL-6, and fibroblasts reduce the level of keratinocyte IL-1 in the medium by a process of internalization (22, 31). IL-1α production by keratinocytes induces a dermal mononuclear infiltrate, leading to release of further cytokines and growth factors (32), and this could account for the inflammation seen in the lesions of transgenic mice expressing suprabasal integrins (7).
Although we ruled out any difference in expression of IL-1β, IL-6, and TNF-α between wild-type and transgenic keratinocytes in culture, it would obviously be premature to conclude that IL-1α is the only cytokine to be selectively upregulated when integrins are expressed suprabasally. The recent finding that the phorbol ester TPA, which can activate MAPK via protein kinase C (33), selectively induces suprabasal proliferation in transgenic epidermis (34) opens the way for an in vivo survey of all growth factors and cytokines that might contribute to the activation of MAPK in this model of psoriasis, one of the candidate cytokines being IL-12 (35). Equally, MAPK is certainly not the only signaling pathway that is altered in psoriasis; for example, IL-1 activates the NF-κB pathway, which is of pivotal importance in cutaneous inflammation (36, 37) and in normal epidermal homeostasis (38). Finally, the changes in epidermal gene expression observed in response to MAPK activation imply altered activity of a host of transcriptional regulators (38–40), and these remain to be examined in integrin transgenic mice.
MAPK activation contributed to epidermal hyperproliferation. Expression of MAPKK1 resulted in an increased growth rate of keratinocytes both under conventional culture conditions (Figure 5) and in reconstituted epidermis (Figure 7); conversely, inhibition of MAPK signaling by expression of MANA reduced cell proliferation. Previous work has shown that although MANA increases the proportion of transit-amplifying cells, MAPKK1 has no effect on the proportion of stem cells in culture (4). The stimulation of growth by MAPKK1 may therefore occur by delaying the onset of terminal differentiation, observed both in suspension and in reconstituted epidermis (Figures 6 and 8). An increase in the proliferative potential of the transit-amplifying compartment would have a dramatic effect on the number of keratinocytes: one additional round of cell division would double the number of cells in the epidermis. Interestingly, expansion of the transit-amplifying compartment is thought to occur in psoriasis, leading to elongation of the rete ridges (41), and, in epidermis reconstituted by MAPKK1 expressing cells, we observed more pronounced keratinocyte downgrowths than in controls (Figure 7e).
Whereas MAPK activity seems to stimulate proliferation in many cell types (9, 24) and is also implicated in senescence and apoptosis (42), its role in the regulation of differentiation is more variable. In MDCK cells, features of differentiation are suppressed (43), whereas in PC12 cells, MAPK stimulates neurite outgrowth (11). In skeletal muscle cells, MAPK activity inhibits early stages of differentiation but stimulates later phases (44). Activated MAPK delayed, but did not inhibit, the onset of keratinocyte differentiation; suspended keratinocytes transduced with MAPKK1 did not divide (data not shown), suggesting that they were committed to terminal differentiation (3). The histological changes in reconstituted epidermis after constitutive activation of MAPK mimicked the appearance of psoriatic lesions (45): the epidermis was thickened, with more proliferating cells, a reduction in the granular layer and a highly thickened, nucleated cornified layer. Whereas these changes reflect the consequences of pathological MAPK activation in vivo, a certain physiological level of MAPK activity is required in order to form a normal stratified epithelium, because epidermis reconstituted by MANA-expressing cells was hypocellular, with decreased proliferation and incomplete terminal differentiation.
In mammalian cell lines, constitutive activation of the MAPK pathway is sufficient for cell transformation (11, 24). Although activation of MAPK in keratinocytes stimulated proliferation and delayed differentiation cells expressing MAPKK 1 did not exhibit two key properties of transformed cells: they did not proliferate in suspension, and they showed no invasiveness in organotypic cultures. This is consistent with psoriatic lesions of the skin being fully reversible and psoriasis itself not increasing the risk of skin tumor development (45). It is likely that the pathological activation of the MAPK pathway that has been observed in various tumors (46) is due to mutational changes in upstream regulating molecules. Both in wound healing and in psoriasis, there seems to be a transient and reversible upregulation of MAPK activity that is mediated through upstream regulation by integrins and growth factors. This makes pharmacological interference with MAPK signaling a promising approach for the treatment of psoriasis.
Acknowledgments
We are very grateful to everyone who provided advice or reagents, particularly Marina Haitas-Haase and Karin Hartmann. This work was supported by the Imperial Cancer Research Fund and by a grant to I. Haase from the Deutsche Forschungsgemeinschaft (Ha 2623/1-1).
Footnotes
Ingo Haase’s present address is: Department of Dermatology, University of Cologne, Cologne, Germany.M. Rosario Romero’s present address is: Prolifix Ltd., Abingdon, United Kingdom.
Ingo Haase and Robin M. Hobbs contributed equally to this work.
References
- 1.Watt FM. Epidermal stem cells: markers, patterning and the control of stem cell fate. Philos Trans R Soc Lond B Biol Sci. 1998;353:1–7. doi: 10.1098/rstb.1998.0247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fuchs E. Epidermal differentiation. Curr Opin Cell Biol. 1990;2:1028–1035. doi: 10.1016/0955-0674(90)90152-5. [DOI] [PubMed] [Google Scholar]
- 3.Jones PH, Watt FM. Separation of human epidermal stem cells from transit amplifying cells on the basis of differences in integrin function and expression. Cell. 1993;73:713–724. doi: 10.1016/0092-8674(93)90251-k. [DOI] [PubMed] [Google Scholar]
- 4.Zhu AJ, Haase I, Watt FM. Signaling via β1 integrins and mitogen-activated protein kinase determines human epidermal stem cell fate in vitro. Proc Natl Acad Sci USA. 1999;96:6728–6733. doi: 10.1073/pnas.96.12.6728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Levy L, Broad S, Diekmann D, Evans RD, Watt FM. β1 integrins regulate keratinocyte adhesion and differentiation by distinct mechanisms. Mol Biol Cell. 2000;11:453–466. doi: 10.1091/mbc.11.2.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Watt, F.M., and Hertle, M.D. 1994. Keratinocyte integrins. In The keratinocyte handbook. I.M. Leigh, E.B. Lane, and F.M. Watt, editors. Cambridge University Press. Cambridge, United Kingdom. 153–164.
- 7.Carroll JM, Romero MR, Watt FM. Suprabasal integrin expression in the epidermis of transgenic mice results in developmental defects and a phenotype resembling psoriasis. Cell. 1995;83:957–968. doi: 10.1016/0092-8674(95)90211-2. [DOI] [PubMed] [Google Scholar]
- 8.Romero MR, Carroll JM, Watt FM. Analysis of cultured keratinocytes from a transgenic mouse model of psoriasis: effects of suprabasal integrin expression on keratinocyte adhesion, proliferation and terminal differentiation. Exp Dermatol. 1999;8:53–67. doi: 10.1111/j.1600-0625.1999.tb00348.x. [DOI] [PubMed] [Google Scholar]
- 9.Seger R, Krebs EG. The MAPK signaling cascade. FASEB J. 1995;9:726–735. [PubMed] [Google Scholar]
- 10.Alessi DR, et al. Identification of the sites in MAP kinase kinase-1 phosphorylated by p74raf-1. EMBO J. 1994;13:1610–1619. doi: 10.1002/j.1460-2075.1994.tb06424.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cowley S, Paterson H, Kemp P, Marshall CJ. Activation of MAP kinase kinase is necessary and sufficient for PC12 differentiation and for transformation of NIH 3T3 cells. Cell. 1994;77:841–852. doi: 10.1016/0092-8674(94)90133-3. [DOI] [PubMed] [Google Scholar]
- 12.Gandarillas A, Watt FM. c-Myc promotes differentiation of human epidermal stem cells. Genes Dev. 1997;11:2869–2882. doi: 10.1101/gad.11.21.2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rikimaru K, Molès J-P, Watt FM. Correlation between hyperproliferation and suprabasal integrin expression in human epidermis reconstituted in culture. Exp Dermatol. 1997;6:214–221. doi: 10.1111/j.1600-0625.1997.tb00165.x. [DOI] [PubMed] [Google Scholar]
- 14.Hogquist KA, et al. Generation of monoclonal antibodies to murine IL-1β and demonstration of IL-1 in vivo. J Immunol. 1991;146:1534–1540. [PubMed] [Google Scholar]
- 15.Thacher SM, Rice RH. Keratinocyte-specific transglutaminase of cultured human epidermal cells: relation to cross-linked envelope formation and terminal differentiation. Cell. 1985;40:685–695. doi: 10.1016/0092-8674(85)90217-x. [DOI] [PubMed] [Google Scholar]
- 16.Fujimoto W, Nakanishi G, Arata J, Jetten AM. Differential expression of human cornifin α and β in squamous differentiating epithelial tissues and several skin lesions. J Invest Dermatol. 1997;108:200–204. doi: 10.1111/1523-1747.ep12334240. [DOI] [PubMed] [Google Scholar]
- 17.Lenormand P, Brondello JM, Brunet A, Pouyssegur J. Growth factor-induced p42/p44 MAPK nuclear translocation and retention requires both MAPK activation and neosynthesis of nuclear anchoring proteins. J Cell Biol. 1998;142:625–633. doi: 10.1083/jcb.142.3.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Renshaw MW, Ren X-D, Schwartz MA. Growth factor activation of MAP kinase requires cell adhesion. Cell. 1997;16:5592–5599. doi: 10.1093/emboj/16.18.5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin TH, Chen Q, Howe A, Juliano RL. Cell anchorage permits efficient signal transduction between ras and its downstream kinases. J Biol Chem. 1997;272:8849–8852. [PubMed] [Google Scholar]
- 20.Krueger, J.G., and Gottlieb, A.B. 1994. Growth factors, cytokines and eicosanoids. In Psoriasis. L. Dubertret, editor. ISED Publishing Co. Brescan, Italy. 18–28.
- 21.Kondo S. The roles of keratinocyte-derived cytokines in the epidermis and their possible responses to UVA-irradiation. J Investig Dermatol Symp Proc. 1999;4:177–183. doi: 10.1038/sj.jidsp.5640205. [DOI] [PubMed] [Google Scholar]
- 22.Maas-Szabowski N, Stark H-J, Fusenig NE. Keratinocyte growth regulation in defined organotypic cultures through IL-1-induced keratinocyte growth factor expression in resting fibroblasts. J Invest Dermatol. 2000;114:1075–1084. doi: 10.1046/j.1523-1747.2000.00987.x. [DOI] [PubMed] [Google Scholar]
- 23.Ichijo H. From receptors to stress-activated MAP kinases. Oncogene. 1999;18:6087–6093. doi: 10.1038/sj.onc.1203129. [DOI] [PubMed] [Google Scholar]
- 24.Mansour SJ, et al. Transformation of mammalian cells by constitutively active MAP kinase kinase. Science. 1994;265:966–970. doi: 10.1126/science.8052857. [DOI] [PubMed] [Google Scholar]
- 25.Brunet A, et al. Nuclear translocation of p42/p44 mitogen-activated protein kinase is required for growth factor-induced gene expression and cell cycle entry. EMBO J. 1999;18:664–674. doi: 10.1093/emboj/18.3.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peltonen S, et al. A novel component of epidermal cell-matrix and cell-cell contacts: transmembrane protein type XIII collagen. J Invest Dermatol. 1999;113:635–642. doi: 10.1046/j.1523-1747.1999.00736.x. [DOI] [PubMed] [Google Scholar]
- 27.Kheradmand F, Werner E, Tremble P, Symons M, Werb Z. Role of Rac1 and oxygen radicals in collagenase-1 expression induced by cell shape change. Science. 1998;280:898–902. doi: 10.1126/science.280.5365.898. [DOI] [PubMed] [Google Scholar]
- 28.Rezzonico R, Chicheportiche R, Imbert V, Dayer J-M. Engagement of CD11b and CD11c β2 integrin by antibodies or soluble CD23 induces IL-1β production on primary human monocytes through mitogen-activated protein kinase-dependent pathways. Blood. 2000;95:3868–3877. [PubMed] [Google Scholar]
- 29.Lee RT, et al. Mechanical deformation promotes secretion of IL-1α and IL-1 receptor antagonist. J Immunol. 1997;159:5084–5088. [PubMed] [Google Scholar]
- 30.Kupper TS, Groves RW. The interleukin-1 axis and cutaneous inflammation. J Invest Dermatol. 1995;105:62S–66S. doi: 10.1111/1523-1747.ep12316087. [DOI] [PubMed] [Google Scholar]
- 31.Boxman ILA, Ruwhof C, Boerman OC, Löwick CWGM, Ponec M. Role of fibroblasts in the regulation of proinflammatory interleukin IL-1, IL-6 and IL-8 levels induced by keratinocyte-derived IL-1. Arch Dermatol Res. 1996;288:391–398. doi: 10.1007/BF02507108. [DOI] [PubMed] [Google Scholar]
- 32.Groves RW, Mizutani H, Kieffer JD, Kupper TS. Inflammatory skin disease in transgenic mice that express high levels of interleukin 1α in basal epidermis. Proc Natl Acad Sci USA. 1995;92:11874–11878. doi: 10.1073/pnas.92.25.11874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schonwasser DC, Marais RM, Marshall CJ, Parker PJ. Activation of the mitogen-activated protein kinase/extracellular signal-regulated kinase pathway by conventional, novel, and atypical protein kinase C isotypes. Mol Cell Biol. 1998;18:790–798. doi: 10.1128/mcb.18.2.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Owens DM, Watt FM. Influence of β1 integrins on epidermal squamous cell carcinoma formation in a transgenic mouse model: α3β1, but not α2β1, suppresses malignant conversion. Cancer Res. 2001;61:5248–5254. [PubMed] [Google Scholar]
- 35.Hong K, Chu A, Lúdvíksson BJ, Berg EL, Ehrhardt RO. Il-12, independently of IFN-γ, plays a crucial role in the pathogenesis of a murine psoriasis-like skin disorder. J Immunol. 1999;162:7480–7491. [PubMed] [Google Scholar]
- 36.Robert C, Kupper TS. Inflammatory skin diseases, T cells, and immune surveillance. N Engl J Med. 1999;341:1817–1828. doi: 10.1056/NEJM199912093412407. [DOI] [PubMed] [Google Scholar]
- 37.Murphy J-E, Robert C, Kupper TS. Interleukin-1 and cutaneous inflammation: a crucial link between innate and acquired immunity. J Invest Dermatol. 2000;114:602–608. doi: 10.1046/j.1523-1747.2000.00917.x. [DOI] [PubMed] [Google Scholar]
- 38.Kaufman CK, Fuchs E. It’s got you covered: NF-ΚB in the epidermis. J Cell Biol. 2000;149:999–1004. doi: 10.1083/jcb.149.5.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eckert RL, Crish JF, Banks EB, Welter JF. The epidermis: genes on - genes off. J Invest Dermatol. 1997;109:501–509. doi: 10.1111/1523-1747.ep12336477. [DOI] [PubMed] [Google Scholar]
- 40.Sinha S, Degenstein L, Copenhaver C, Fuchs E. Defining the regulatory factors required for epidermal gene expression. Mol Cell Biol. 2000;20:2543–2555. doi: 10.1128/mcb.20.7.2543-2555.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Iizuka H, Ishida-Yamamoto A, Honda H. Epidermal remodelling in psoriasis. Br J Dermatol. 1996;135:433–438. [PubMed] [Google Scholar]
- 42.Cobb MH. MAP kinase pathways. Prog Biophys Mol Biol. 1999;71:479–500. doi: 10.1016/s0079-6107(98)00056-x. [DOI] [PubMed] [Google Scholar]
- 43.Schramek H, Feifel E, Healy E, Pollack V. Constitutively active mutant of the mitogen-activated protein kinase kinase MEK1 induces epithelial dedifferentiation and growth inhibition in madin-darby canine kidney-C7 cells. J Biol Chem. 1997;272:11426–11433. doi: 10.1074/jbc.272.17.11426. [DOI] [PubMed] [Google Scholar]
- 44.Bennett AM, Tonks NK. Regulation of distinct stages of skeletal muscle differentiation by mitogen-activated protein kinases. Science. 1997;278:1288–1291. doi: 10.1126/science.278.5341.1288. [DOI] [PubMed] [Google Scholar]
- 45.Lever, W.F., and Schaumburg-Lever, G. 1983. Histopathology of the skin. J.B. Lippincott Co. Philadelphia, Pennsylvania, USA. 136–163.
- 46.Sebolt-Leopold JS, et al. Blockade of the MAP kinase pathway suppresses growth of colon tumors in vivo. Nat Med. 1999;5:810–816. doi: 10.1038/10533. [DOI] [PubMed] [Google Scholar]