

Abstract
Direct in vivo evidence is still lacking for α4-integrin–mediated T cell interaction with VCAM-1 on blood-brain barrier–endothelium in experimental autoimmune encephalomyelitis (EAE). To investigate a possible α4-integrin–mediated interaction of encephalitogenic T cell blasts with VCAM-1 on the blood-brain barrier white matter endothelium in vivo, we have developed a novel spinal cord window preparation that enabled us to directly visualize CNS white matter microcirculation by intravital fluorescence videomicroscopy. Our study provides the first in vivo evidence that encephalitogenic T cell blasts interact with the spinal cord white matter microvasculature without rolling and that α4-integrin mediates the G protein–independent capture and subsequently the G protein–dependent adhesion strengthening of T cell blasts to microvascular VCAM-1.
Introduction
The CNS is considered an immunoprivileged site where the endothelial blood-brain barrier (BBB) tightly controls lymphocyte entry into the CNS. Under physiological conditions lymphocyte traffic into the CNS is low, whereas during inflammatory diseases of the CNS, such as multiple sclerosis (MS) or in the animal model experimental autoimmune encephalomyelitis (EAE), a large number of circulating lymphocytes readily gain access to the CNS. EAE is a CD4+ T cell–mediated autoimmune disease of the CNS, which is initiated by autoaggressive T cells activated outside the CNS. These encephalitogenic T cell blasts enter the CNS parenchyma across the healthy BBB and start the molecular events leading to inflammation, edema, and demyelination. Thus, the interaction of encephalitogenic T cells with the healthy BBB endothelium is a critical initial step in the pathogenesis of EAE.
In general, lymphocyte recruitment across the vascular wall is regulated by the sequential interaction of different adhesion or signaling molecules on lymphocytes and endothelial cells lining the vessel wall (1). An initial transient contact of the circulating leukocyte with the vascular endothelium, generally mediated by adhesion molecules of the selectin family and their respective carbohydrate ligands, slows down the leukocyte in the bloodstream. Subsequently, the leukocyte rolls along the vascular wall with greatly reduced velocity. The rolling leukocyte can receive endothelial signals resulting in its firm adhesion to the endothelial surface. These signals are transduced by chemokines via G protein–coupled receptors on the leukocyte surface. Binding of a chemokine to its receptor results in a pertussis toxin–sensitive activation of integrins on the leukocyte surface. Only activated integrins mediate the firm adhesion of the leukocytes to the vascular endothelium by binding to their endothelial ligands, which belong to the Ig superfamily. This ultimately leads to the extravasation of the leukocyte. Successful recruitment of circulating leukocytes into the tissue depends on the productive leukocyte/endothelial interaction during each of these sequential steps.
The adhesion molecules mediating the initial contact of T lymphocytes with the BBB endothelium are not yet defined. Selectins are not involved in T cell interaction with the BBB. Encephalitogenic T cell blasts lack expression of L-selectin (2), and E- and P-selectin are neither expressed at the BBB (3) nor functionally involved in the recruitment of autoaggressive T lymphocytes across the BBB during EAE (4). In contrast, constitutive expression of VCAM-1 in the CNS microvasculature of healthy SJL/N mice has been observed (5), and upregulation of ICAM-1 and VCAM-1 was reported on cerebral vessels during EAE preceding the perivascular infiltration by lymphocytes and the onset of disease (6).
It has become commonly accepted that the interaction of α4β1 and VCAM-1 contributes to T cell recruitment across the BBB. This assumption was based on findings that blocking Ab’s directed against α4-integrin or VCAM-1 inhibit or delay the development of EAE (7) and that surface expression of α4-integrin by antigen-specific T cell clones can enhance the pathogenicity of those clones in EAE models (8, 9). In support of this notion encephalitogenic T lymphoblasts adhere via α4-integrin to VCAM-1 on brain endothelium in vitro (5), and lymphocytes bind via α4/VCAM-1 to inflamed cerebral vessels in a modified Stamper-Woodruff frozen section adhesion assay (6). Nevertheless, alternative functions of α4-integrin in the pathogenesis of EAE have also been suggested. Evidence for an involvement of α4-integrin in antigen-specific T cell proliferation (2) or maintaining the residency of inflammatory cells within the CNS parenchyma during EAE (10) has been provided.
Therefore, definite in vivo evidence for α4-integrin–mediated T cell interaction with VCAM-1 on BBB-endothelium in EAE is still lacking. Observation of the CNS microcirculation by intravital fluorescence microscopy is hampered by the protected localization of the brain and spinal cord within the skull and the spinal column, respectively. To gain intravital microscopic access to the CNS microcirculation, acute and chronic cranial window techniques have been developed for rodents, which allow observation of the pial and cortical (i.e., CNS grey matter) microcirculation, respectively (11, 12). In EAE, however, inflammation is located in the CNS white matter with preference of the spinal cord. Thus, in order to investigate the physiologically relevant interaction of autoaggressive T lymphocytes with the BBB endothelium it is mandatory to gain intravital microscopic access to the CNS white matter microcirculation.
Here we introduce a novel spinal cord window technique, which allows direct visualization of white matter microcirculation by intravital fluorescence videomicroscopy. Using this model we were able to assess the interaction of encephalitogenic T cell blasts with the white matter BBB-endothelium in SJL/N mice. To our knowledge, this study provides the first in vivo evidence that α4-integrin mediates the G protein–independent capture and subsequently the G protein–dependent adhesion strengthening of encephalitogenic T cell blasts to microvascular VCAM-1 in spinal cord white matter microvasculature.
Methods
Mice.
Female SJL/N mice were obtained from Bomholdgård Breeding (Ry, Denmark) and used for experiments at the age of 17 to 19 weeks.
Pertussis toxins and mAb’s.
The rat anti-mouse mAb’s used in this study were PS/2 (anti–α4-integrin rat IgG2b), 6C7.1 (anti–VCAM-1 rat IgG1), and MJ7/18 (anti-endoglin IgG2a), and they have been described in great detail previously (2, 5). The mAb’s were purified from serum-free hybridoma supernatants, and endotoxin levels were determined by Fresenius (Taunusstein, Germany) were below detection levels (< 0.6 EU/ml). Pertussis toxin (PTX) was obtained from Sigma-Aldrich Chemie GmbH (Deisenhofen, Germany). Mutant PTX (MTX; PT9K/129G) lacking the enzymatic activity of PTX was used as a control. MTX was kindly provided by R. Rappuoli (Chiron SpA., Siena, Italy) (13).
T lymphocyte lines and induction of EAE.
Establishment and culture of the CD4+ MHC class II–restricted protein lipid protein-specific (PLP-specific) T cell lines derived from SJL/N mice and induction and treatment of EAE have all been described in great detail previously (2, 4).
Labeling of T lymphoblasts.
T cell blasts were labeled with 20 nM Cell Tracker Orange (Molecular Probes, Eugene, Oregan, USA) in RPMI-1640 with 10% FCS at 5 × 106 cells/ml and 37°C for 45 minutes, according to the protocol by Hamann and Jonas (14). Cell Tracker Orange labeling did not impair encephalitogenicity of PLP-specific T cell blasts, because transfer of 3 × 106 PLP-specific Cell Tracker Orange–labeled T cell blasts into syngeneic animals induced EAE with the same time, kinetic, and clinical severity as the transfer of unlabeled PLP-specific T cell blasts when compared in six mice per group in two different experiments.
Flow cytometry.
Flow cytometry using Ig chimeras was performed exactly as described previously (4). Human recombinant VCAM-1–IgG (kindly provided by Dirk Seiffge, Aventis Pharma, Frankfurt, Germany) or murine HT7-IgG (4) used as negative control IgG-fusion protein was used at 2.5 μg/ml.
Preparation of the spinal cord window.
The surgical preparations and experiments were performed in accordance with the German legislation on the protection of animals and the Guide for the Care and Use of Laboratory Animals. Animals were anesthetized by subcutaneous injection of ketamine/xylazine. For systemic administration of fluorescent markers and injection of cells, a polyethylene catheter (PE-10) was inserted into the right common carotid artery of a thermocontrolled mouse placed on a heating pad. Next, the animal was turned to the prone position, and the head was fixed in a stereotactic rodent head holder. After a midline skin incision of 3–4 cm, the paravertebral musculature was detached from the cervical spinous processes and retracted laterally, exposing the vertebral laminae. Using microsurgical techniques, a laminectomy of C1 to C7 was performed, and the dura was opened over the dorsal spinal cord avoiding trauma to the parenchyma and the spinal microvasculature. To prevent dehydration and the influence of the ambient oxygen, the site was covered with an impermeable transparent membrane. Mice that were traumatized during surgery or revealed signs of acute inflammation (distorted vessels, hyperemia, stagnant blood flow) were excluded from the experiments.
Intravital fluorescence videomicroscopy.
For intravital fluorescence videomicroscopy the animals were transferred to the microscope stage, remaining within the stereotactic head holder. Intravital fluorescence videomicroscopy was performed by epi-illumination techniques using a modified Axiotech Vario microscope with a 100-W mercury lamp emitting an adjustable light intensity (Attoarc; Carl Zeiss Jena GmbH, Jena, Germany), which was attached to a combined blue (450–490 nm) and green (520–570 nm) filter block (Carl Zeiss Jena GmbH). Observations were made using ×3.2 long-distance, ×10 long-distance, and ×20 water immersion working objectives (all from Carl Zeiss Jena GmbH), resulting in magnifications of ×50, ×200, and ×400, respectively. Microscopic images were recorded by means of a low-light level charged coupled device video camera with an optional image intensifier for weak fluorescence (Kappa Opto-Electronics GmbH, Gleichen, Germany) and were transferred to a S-VHS videosystem (Panasonic, Hamburg, Germany) for off-line evaluation.
The spinal cord microvasculature was visualized by contrast enhancement with 2% FITC-conjugated dextran (0.1 ml FITC-dextran150, intravenously; MW = 150,000; Sigma Chemical Co., St. Louis, Missouri, USA) and use of the blue-light epi-illumination. Simultaneously, injected Cell Tracker Orange–labeled T cell blasts were visualized within the spinal cord microcirculation using the green-light epi-illumination. This combination of two fluorescent markers with distinct excitation wavelengths allowed localization of Cell Tracker Orange–labeled T cell blasts with respect to FITC-stained vessel lumina.
Experimental protocol.
Following visualization of the spinal cord microvasculature by FITC-dextran150, 3 × 106 Cell Tracker Orange–labeled PLP-specific T cell blasts were injected in aliquots of 100 μl, each containing 106 T cells, and their interaction with the endothelium was assessed over an observation period of 60 seconds within the spinal capillary bed and postcapillary venules (20–60 μm) of three different microvascular regions of interest. In contrast, due to the angioarchitecture of the spinal microvasculature, T cell blast/endothelium interaction within precapillary arterioles, which are located at the depth of the spinal cord parenchyma, could not be visualized by intravital fluorescence videomicroscopy. To assess permanent T cell blast adhesion, the spinal cord microvasculature was scanned at 10 minutes, 1 hour, and 2 hours after cell injection.
To study the role of α4-integrin and VCAM-1 for T cell blast/endothelial interaction, 3 × 106 T cell blasts were preincubated with 90 μg PS/2 in 300 μl PBS for 20 minutes (n = 3 animals) or mice were injected with 90 μg 6C7.1 in 150 μl PBS 20 minutes before infusion of T lymphoblasts (n = 3), respectively. Animals treated with PBS (n = 5) or the control mAb MJ7/18 (n = 2) served as controls. MJ7/18 was chosen since it binds to the vascular wall and has been shown previously to have no influence to the development of EAE (2). In separate experiments T lymphoblasts were incubated with PTX (n = 3) or MTX (n = 2) (each 100 ng/ml) for 2 hours at 37°C in order to study the relevance of G protein–mediated activation of α4-integrin (n = 3). Unbound PTX or MTX was removed by washing the T cells before injection into the mice.
Intravital microscopic image analysis.
Quantitative analysis of the spinal cord microcirculation included the diameter of postcapillary venules (d) and the velocity of nonadherent T lymphoblasts. The highest cell velocity per venule, Vmax, was used to calculate the mean blood flow velocity, as described previously (15): Vmean = Vmax /(2 – ε2) (μm/s) where ε is the ratio of the T lymphoblast diameter to vessel diameter (d). From Vmean and d, wall shear rate (γ) was estimated as γ = 8 × Vmean /d (s–1) (15), and the shear stress (τ) was approximated by τ = γ × 0.025 (dyn/cm2) (16).
In postcapillary venules permanently adherent T lymphoblasts were identified as cells that stuck to the vessel wall without moving or detaching from the endothelium within an observation period of at least 20 seconds. Nonpermanently adherent T lymphoblasts were further categorized using a velocity criterion derived from the assumption of a parabolic velocity profile in the microvessel (15). Therefore, Vcrit, the velocity of an idealized noninteracting cell traveling at the vessel wall, was calculated as Vcrit = Vmean × ε × (2 – ε). Consequently, any cell traveling below Vcrit was regarded as a cell interacting with the vessel wall; any cell traveling above Vcrit was regarded as a noninteracting cell (15, 17). Within the capillary network, plugging T lymphoblasts were defined as cells that did not move and obviously blocked the capillary lumen, inducing blood flow stasis in the corresponding vascular segment. Permanent T lymphoblast adhesion at 10 minutes, 1 hour, and 2 hours after cell injection is expressed as the number of both adherent and plugging T lymphoblasts per region of interest (mm–2).
Detection of Cell Tracker Orange–labeled PLP-specific T lymphoblasts by immunofluorescence.
Mice were perfused with 1% formaldehyde in PBS 2, 4, 6, and 8 hours after injection of the PLP-specific T cell blasts. Spinal cords were removed, embedded in Tissue-Tek (OCT; Miles Inc., Vogel, Giessen, Germany) and snap-frozen in a 2-methylbutane (Merck KGaA, Darmstadt, Germany) bath at –80°C. Serial longitudinal cryo-sections (6 μm) of the spinal cords were cut, air-dried overnight, and acetone fixed. Cell Tracker Orange–labeled T lymphoblasts were detected by immunofluorescence microscopy. Vessels on sections containing T lymphoblasts were visualized by staining with either the mAb MJ7/18, a rat anti–mouse endoglin, or with the mAb 9DB3, a rat anti–mouse VCAM-1, both at 10 μg/ml in PBS/0.1% BSA, followed by staining with a goat anti-rat–FITC IgG (10 μg/ml in PBS supplemented with 10% normal mouse serum; Jackson, Dianova GmbH, Hamburg, Germany). Alternatively, bound 6C7.1 or bound MJ7/18 Ab’s were detected by staining with a goat anti-rat–FITC (Jackson, Dianova GmbH). Sections were coverslipped with Mowiol 4-88 (Calbiochem-Novabiochem GmbH, Bad Soden, Germany) and immediately analyzed.
Statistics.
Quantitative data are given as mean values plus or minus SD. Mean values were calculated from the average values in each animal. For analysis of differences between the groups, ANOVA followed by unpaired Student t test and Bonferroni correction for repeated measurements were performed. Results with P values less than 0.05 were considered significant and are marked with an asterisk.
Results
Spinal window technique for intravital microscopic observation of white matter microcirculation.
We established a novel spinal window technique, which enabled us to directly visualize spinal cord white matter microcirculation by intravital fluorescence videomicroscopy. Following preparation of the spinal window, five animals had to be excluded from the experiments due to surgical trauma to the spinal cord (n = 2); BBB breakdown, as indicated by extravasation of fluorescent marker (n = 1); or activation of the endothelium, as visualized by accumulation of leukocytes at the vessel wall (n = 2). In contrast, a total of 18 animals could be entered into this study. The laminectomy of C1 to C7 allowed intravital microscopic access to the entire posterior aspect of the cervical spinal cord (Figure 1). The anatomy of the spinal cord white matter microvasculature was characterized by a large collecting venule in the midline of the dorsal medulla, which drained blood in a caudal direction (Figure 1). The function of this vessel is analogous to that of the superior sagittal sinus in the cerebral microcirculation. From both lateral aspects of the medulla, the majority of postcapillary venules drained blood flow from the capillaries directly into this large collecting venule (Figure 1). The capillary network of the spinal cord white matter microcirculation was characterized by multiple vessel loops that could be followed by intravital fluorescence microscopy down to a depth of approximately 100 μm (Figure 1). Precapillary arterioles, however, were not visible in these preparations. While the blood flow in the major collecting venule revealed a respiratory-dependent flow motion, blood in the capillary network and postcapillary venules was even and characterized by physiologic microhemodynamic parameters (Table 1).
Figure 1.

Spinal window preparation for direct intravital microscopic assessment of white matter microcirculation. Stereomicroscopic (a) and intravital fluorescence microscopic (b) view of spinal window preparation exposing the dorsal spinal cord. Bar, 500 μm. High-magnification intravital fluorescence microscopy of spinal microvasculature depicting white matter capillary network (arrows) (c) and white matter postcapillary venules draining into the central dorsal vein (d). Bar, 100 μm.
Table 1.
Microhemodynamic parameters in spinal postcapillary venules

T lymphoblasts are captured without rolling in the spinal cord white matter microvasculature.
Freshly stimulated encephalitogenic T lymphoblasts were labeled with Cell Tracker Orange and infused via the right carotid artery into healthy SJL/N mice (3 × 106/mouse). A high number of circulating T lymphoblasts (∼100–200) could be observed in the individual regions of interest within the spinal cord white matter microvasculature. While most of the T lymphoblasts passed through the spinal cord capillaries and postcapillary venules without interacting with the vessel wall, 5–10% of the T lymphoblasts passing through the region of intravital microscopic observation were abruptly arrested within the spinal cord microvasculature. The mechanism of this T lymphoblast arrest was twofold: 50% of the T lymphoblasts plugged capillaries either permanently or temporarily; in the latter case T lymphoblasts finally squeezed through the capillary segment and reentered the blood stream within 4 to 7 seconds. The other 50% of the T lymphoblasts were promptly captured at the endothelium of postcapillary venules without influencing blood flow in this vascular segment (Figure 2). Similar to the capillaries, capture of T lymphoblasts to the endothelium resulted either in permanent adhesion or remained temporary, with the cell reentering the blood circulation within 1 to 7 seconds. It should be noted that classical rolling or tumbling of T lymphoblasts along the venular endothelium prior to their capture was never visible. Objective assessment of the interaction of nonpermanently adherent T lymphoblasts with the endothelium revealed that 5% of T lymphoblasts were captured (Figure 3). Plotting the normalized velocities of T lymphoblasts confirmed the complete lack of rolling because none of the nonpermanently adherent T lymphoblasts traveled below Vcrit, i.e., the velocity of an idealized noninteracting cell traveling at the wall of a vessel with a parabolic flow profile, whereas 95% of T lymphoblasts traveled above Vcrit, i.e., traveled free in the bloodstream (15, 17).
Figure 2.

T lymphoblast/endothelium interaction within spinal white matter postcapillary venules during cell infusion. (a) Postcapillary venular segment before cell infusion after contrast enhancement of spinal microvasculature using FITC-dextran150. Arrows indicate direction of microvascular blood flow. (b–e) Intravital microscopic sequence of two Cell Tracker Orange–labeled T lymphocytes (1 and 2) over 0.24 seconds within the identical postcapillary segments indicated in a. Cells either lacked interaction with endothelium or were captured to endothelium without prior rolling. Bar, 100 μm.
Figure 3.

Normalized velocity of T cell blasts. Objective assessment of T lymphoblast/endothelial interaction was obtained by comparing the velocity distribution of T cells observed in vessels of comparable size. For the control group, 198 cells in eight venules of two mice, for the anti–α4-group, 440 cells in 22 venules of four mice, and for the anti–VCAM-1 group 236 cells in 15 venules of three mice were analyzed. Vcrit, the velocity of an idealized noninteracting T cell blast, was calculated as described in Methods. Five percent of circulating T cells were transiently captured at the vascular wall (a, c). Blocking α4-integrin (a) or VCAM-1 (c) resulted in a significantly reduced number of captured T cells (a, c). Lack of T lymphoblast rolling is demonstrated by the lack of T cells traveling at velocities below Vcrit (b, d).
VCAM-1 and α4-integrin mediate T lymphoblast interaction with the spinal cord white matter microvasculature.
To investigate to what extent α4-integrin and VCAM-1 are involved in the interaction of T lymphoblasts with the spinal cord microvasculature, blocking Ab’s directed against α4-integrin or against VCAM-1 were applied. Without affecting microhemodynamic parameters (Table 1), pretreatment of T lymphoblasts with an Ab directed against α4-integrin, as well as pretreatment of mice with an Ab directed against VCAM-1, almost completely abolished initial T lymphoblast capture (Figure 3). In contrast, injection of control Ab’s into mice before T lymphoblast infusion did not result in any reduction of T cell capture within spinal cord microvessels when compared with untreated mice.
To determine whether inhibition of the initial α4-integrin/VCAM-1 mediated T lymphoblast capture to the endothelium also translated into a reduced permanent adhesion of T lymphoblasts within the spinal cord microvasculature, we determined the number of T lymphoblasts permanently adhering within the microvasculature at 10 minutes, 1 hour, and 2 hours after T lymphoblast infusion (Figure 4). In accordance with the extent of initial T lymphoblast capture and capillary plugging, permanently adherent T lymphoblasts were observed in both capillaries (51% ± 6%) and postcapillary venules (49% ± 6%) of the spinal cord white matter microvasculature (Figure 4a). Blocking α4-integrin reduced the number of permanently adherent T lymphoblasts by 70% ± 14% (76% ± 6% residing in capillaries and 24% ± 6% in postcapillary venules) and blocking VCAM-1 by 78% ± 9% (65% ± 19% residing in capillaries and 34% ± 19% in postcapillary venules) when compared with controls (Figure 4, b and c). Given the magnitude of inhibition of permanent T lymphoblast adhesion, blocking α4-integrin or VCAM-1 reduced the number of both T lymphoblasts permanently adherent in postcapillary venules and permanently plugging capillaries (Figure 4c).
Figure 4.

Involvement of α4-integrin and VCAM-1 in permanent T lymphoblast adherence within spinal cord white matter microvasculature. (a–d) Permanent T lymphoblast adherence in control mice 10 minutes after cell injection. T cell blasts were permanently adherent either within the capillary network (a and b) or within postcapillary venules (c and d). Intravital fluorescence videomicroscopy using epi-illumination techniques. Contrast enhancement of spinal microvasculature using FITC-dextran150 (a and c; arrows mark localization of T cells). Cell Tracker Orange–labeled T lymphocytes (b and d). Bar, 100 μm. (e–j) Permanent T lymphoblast adherence 1 hour after cell injection. Control (e and f), anti–α4-integrin Ab (g and h), anti–VCAM-1 (i and j). Intravital fluorescence videomicroscopy using epi-illumination techniques. Contrast enhancement of spinal microvasculature using FITC-dextran150 (e, g, and i; arrows mark localization of T cells). Cell Tracker Orange–labeled T lymphocytes (f, h, and j). Bar, 100 μm. (k) Quantitative analysis of permanent T lymphoblast adherence. T lymphoblasts permanently adhering within spinal cord white matter microvasculature were counted 10 minutes, 1 hour, and 2 hours after infusion of 3 × 106 PLP-specific T cell blasts by intravital fluorescence videomicroscopy using epi-illumination techniques as described in Methods. Number of mice included in this analysis per group: control, n = 5 mice; antibody-control, n = 2 mice; anti–α4-integrin group, n = 3 mice; and anti–VCAM-1 group, n = 3 mice. Asterisks indicate significant differences. p.i., postinjection.
G proteins are not involved in T cell capture within the spinal cord white matter microvasculature.
Encephalitogenic T lymphoblasts were shown to have high-affinity α4-integrin on their surface by binding soluble recombinant VCAM-1 (Figure 5a). We therefore asked whether T lymphoblast/endothelium interaction required G protein–mediated increase of α4-integrin avidity in situ. T lymphoblasts were pretreated with PTX before infusion. Compared with untreated T lymphoblasts, PTX treatment had no effect on the number of T lymphoblasts captured in postcapillary venules (Figure 5b). However, pretreatment of T lymphoblasts with PTX but not MTX dramatically reduced the number of permanently adherent T cells at 10 minutes, 1 hour, or 2 hours after injection by 78% ± 9% (69% ± 18% residing in capillaries and 31% ± 18% in postcapillary venules) (Figure 5c). Given the total inhibition of permanent T cell adhesion and the ratio of permanently adhering T cells in capillaries (46% ± 1%) and postcapillary venules (54% ± 1%) in the control group, PTX treatment reduced both permanent T cell adhesion in capillaries and postcapillary venules. Thus, permanent adhesion of T lymphoblasts within the spinal cord white matter microvasculature but not initial capture of T lymphoblasts to the endothelium was dependent on G protein–mediated signals in situ. In line with our intravital microscopic results, pretreatment of encephalitogenic T lymphoblasts with PTX resulted in a highly significant delayed onset of passively transferred EAE (mean day of onset of clinical disease = 9.75 ± 0.5; n = 4 mice) when compared with controls (mean day of onset of clinical disease = 6.5 ± 0.5; n = 4 mice). Pertussis toxin did not influence the surface expression of α4-integrins on the encephalitogenic T cells as determined by FACS analysis (data not shown).
Figure 5.

Requirement for G proteins in T lymphoblast interaction with the spinal cord white matter microvasculature. (a) Presence of high-affinity α4-integrin on encephalitogenic T cell blasts but not on resting T cells is demonstrated by binding of VCAM-1–Ig (thick line) as determined by FACS-analysis. Binding of HT7-Ig was used as control (thin line). (b and c) Capture events (b) and normalized velocity (c) of pertussis toxin–pretreated T cell blasts. Objective assessment of PTX-pretreated T lymphoblast/endothelial interaction was obtained by comparing the velocity distribution of T cells observed in vessels of comparable size. For the control group 160 cells in seven postcapillary venules of two mice and for the pertussis toxin group 190 cells in 15 postcapillary venules of three mice were analyzed. Vcrit, the velocity of an idealized noninteracting T cell blast, was calculated as described in Methods. Ten percent of circulating T cells were transiently captured at the vascular wall (b). PTX did not influence the number of captured T cells (b). Lack of T lymphoblast rolling is demonstrated by the lack of T cells traveling at velocities below Vcrit (c). (d) Quantitative analysis of permanent T lymphoblast adherence within spinal cord white matter microvasculature. T lymphoblasts permanently adhering within spinal cord white matter microvasculature were counted 10 minutes, 1 hour, and 2 hours after infusion of 3 × 106 PLP-specific T cell blasts by intravital fluorescence videomicroscopy using epi-illumination techniques as described in Methods. Number of mice included in this analysis per group: PBS-control, n = 2; MTX:, n = 2; PTX, n = 3. Asterisks indicate significant differences.
G protein–dependent α4-integrin–mediated adhesion of T lymphoblasts to microvascular VCAM-1 is essential for their emigration into the spinal cord parenchyme.
To determine whether T lymphoblasts, which were permanently arrested within spinal cord white matter microvessels, also emigrate into the spinal cord parenchyma, mice were perfused with formaldehyde 2, 4, 6, and 8 hours after infusion of T lymphoblasts, and the spinal cords were removed for histological examinations. In longitudinal sections a significant number (23 ± 9 cells per section, n = 63 counted sections) of Cell Tracker Orange–positive T lymphoblasts could be detected within the spinal cord parenchyma derived from control animals 6 hours after infusion of T lymphoblasts. Immunofluorescence counterstaining for endothelial endoglin localized 32% of T lymphoblasts still within and 68% of T lymphoblasts outside spinal cord microvessels (Figure 6). In contrast, within spinal cords derived from mice pretreated with either anti–VCAM-1 Ab or infused with T lymphoblasts pretreated with anti–α4-integrin Ab, only a few fluorescent T lymphoblasts could be detected exclusively within the lumen of spinal cord microvessels (data not shown). These observations show that G protein–dependent α4-integrin–mediated firm adhesion of T lymphoblasts to VCAM-1 on the endothelial surface is essential for their successful emigration into the spinal cord parenchyme.
Figure 6.
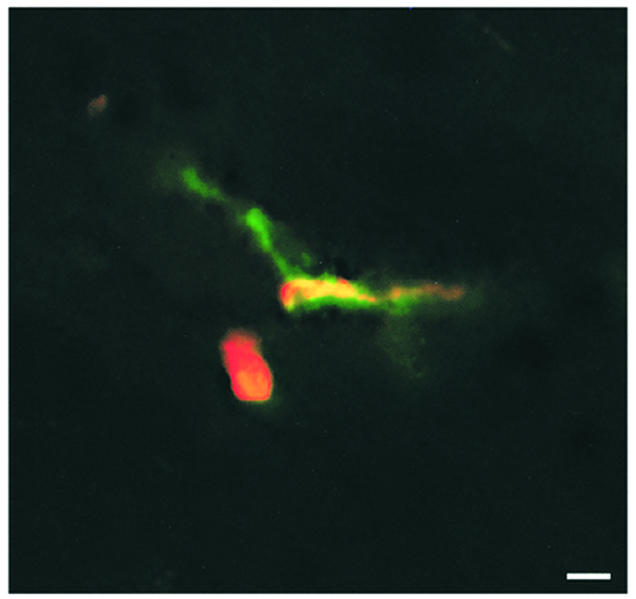
Localization of T lymphoblasts within the spinal cord parenchyma in control animals. Six hours after infusion of T lymphoblasts, the Cell Tracker Orange–labeled T cell blasts (red fluorescence) could be localized outside the spinal cord microvasculature (green fluorescence) within the spinal cord parenchyma of control animals. Superimposed fluorescence is shown, which demonstrates one T cell blast attached within the venule (yellow fluorescence) and one T cell blast within the spinal cord parenchyma (red fluorescence). Bar, 10 μm.
Discussion
To gain direct in vivo evidence for an involvement of α4/VCAM-1–mediated T cell interaction with the BBB endothelium it is mandatory to perform intravital microscopy on the CNS microvasculature. Because in EAE inflammation is located in the CNS white matter, preferentially in the spinal cord, we have developed a novel spinal cord window preparation that allows observation of the interaction of fluorescently labeled encephalitogenic T cell blasts with the spinal cord white matter microvasculature in healthy SJL/N mice. T lymphoblasts did not roll along the endothelium, but rather were promptly captured within capillaries and at the vascular wall of postcapillary venules. α4/VCAM-1–mediated T cell capture to the white matter BBB endothelium preceded G protein–mediated increase in integrin avidity on the T cell surface in situ, which was, however, necessary for adhesion strengthening.
It has been demonstrated that α4β1-integrin/VCAM-1 interaction might mediate several steps of the recruitment cascade, namely tethering, rolling, and adhesion (18–21). The molecular basis of the versatility of adhesion functions mediated by a single receptor/ligand pair (i.e., α4-integrin /VCAM-1) may reside in the multiple avidity states α4β1-integrin can adopt on the cell surface (21). Due to the lack of requirement of previous integrin activation by external stimuli it was suggested that α4β1-integrin tethering to VCAM-1 does not require integrin activation (20, 21), indicating that α4β1/VCAM-1 interaction precedes activation events required for integrin-mediated stable arrest on VCAM-1. On the other hand, under physiological shear in vitro–purified CD4+ T memory cells were shown to exhibit a significantly greater adhesiveness to VCAM-1 than naive T cells, which could be correlated to the increased presence of activation/ligand-induced epitopes on β1-integrin on memory T cells as compared with naive T cells (22). These findings suggest that on activated T cells α4β1 is intrinsically activated as compared with resting T cells. Finally, Lim et al. have demonstrated that α4β1-activation is necessary for efficient T cell interaction with VCAM-1 under flow in vitro (23). Interestingly, two different behavioral patterns of α4-integrin–mediated T cell interaction with VCAM-1 under physiological shear could be observed in vitro (20). After α4-mediated tethering, some of the T lymphocytes engaged in α4-integrin–mediated rolling, whereas others promptly arrested without prior rolling even in the absence of external activating stimuli. High-affinity α4β1 was mandatory for latter behavior, where the tether bond was sufficiently strong to immediately arrest the tethering T cell. These observations provoked the conclusion that at least in vitro interaction of α4 with VCAM-1 alone can support all consecutive adhesive steps required to lead to permanent arrest of lymphocytes on VCAM-1 in the absence of exogenous stimuli such as chemokines (20). Other in vitro studies investigating lymphocyte interaction with VCAM-1 under shear have, however, demonstrated a requirement for chemokine activation of lymphocytes to trigger α4-integrin–mediated arrest on VCAM-1 (24).
Due to the lack of Ab’s recognizing ligand- or activation-dependent epitopes on the α4-integrin subunit in the mouse, the affinity state of α4β1 on encephalitogenic T lymphoblasts cannot be tested directly. Presence of high-affinity α4-integrin on the surface of freshly activated T cell blasts is, however, suggested by binding of soluble VCAM-1–Ig. Because only freshly activated but not resting autoaggressive T cells can pass the BBB and transfer EAE (25, 26), it is tempting to speculate that T cell activation induces the presence of high-avidity α4-integrin on their surface, which allows their capture on constitutively expressed VCAM-1 in the CNS white matter microvasculature under physiological shear.
To test whether α4-integrin–mediated capture within the spinal cord white matter postcapillary venules requires G protein–mediated increase in α4-integrin avidity at the endothelial contact zone, in situ G protein–mediated signaling of T lymphoblast was inhibited by PTX. Whereas PTX did not interfere with the initial T cell capture to the vascular wall, as demonstrated by the unchanged velocity profile of PTX pretreated T lymphoblasts, it significantly reduced the number of permanently adhering T lymphoblasts. Necessity of G protein–mediated increase in integrin avidity for T lymphoblast entry into the CNS is underlined by our observation that development of EAE is delayed upon injection of pertussis toxin pretreated T lymphoblasts when compared with controls. On the other hand, it has been demonstrated recently that chemokines can augment α4-mediated tethering within less than 0.1 second of contact through G protein–mediated signals (27), suggesting that chemokines could increase α4-integrin–mediated capture on VCAM-1. For T lymphoblast interaction with the spinal cord microvasculature this mechanism seems unlikely because we did not observe any influence of PTX on the velocity profile of the circulating T lymphocytes. Also, PTX-mediated inhibition of adhesion strengthening of T lymphoblasts without influencing T cell capture argues against a short circuit of the multistep paradigm at the BBB, where α4/VCAM-1–mediated capture would be followed by adhesion strengthening without additional chemokine–mediated increase in integrin avidity. Preliminary observations in our laboratory have demonstrated that blocking LFA-1 on encephalitogenic T lymphoblasts neither interfered with T cell capture nor their adhesion strengthening to the spinal cord microvasculature (data not shown). Thus, it is most likely that α4-integrin–mediated capture of T lymphoblasts to VCAM-1 allows T lymphocytes to bind an as yet unknown chemokine on the BBB surface, which then leads to secondary G protein–dependent α4/VCAM-1–mediated adhesion strengthening of T lymphoblasts, allowing their diapedesis into the spinal cord white matter. Our findings are in contrast to the observations made by recently Carrithers et al. (28), who were not able to demonstrate an involvement of α4-integrin in lymphocyte recruitment across the BBB when performing Ab-inhibition studies followed by immunohistology to detect fluorescently labeled cells in the CNS. The different results might be easily explained because these authors used an anti–α4-integrin Ab (R1.2), which, in contrast to the mAb used in the present study (PS/2), has been proven to be a poor inhibitor of lymphocyte migration in vivo (2, 29).
Besides T cell interaction with the wall of postcapillary venules, 50% of infused T cell blasts were observed to plug capillaries either transiently or permanently. While transient plugging of capillaries by activated leukocytes might be a physiological process, permanent capillary plugging by leukocytes has been regarded as a passive rather than a receptor-mediated process attributed to the leukocyte size exceeding the capillary diameter of 4–5 μm. In support of this interpretation was the observation that activated leukocytes are characterized by an increasing rigidity compared with nonstimulated leukocytes (30). In contrast to this notion the results of the present study suggest that G protein–dependent α4-integrin–mediated binding to VCAM-1 is involved in the firm adhesion of encephalitogenic T cell blasts within CNS white matter capillaries, since the number of T lymphoblasts permanently plugging capillaries was significantly reduced after blocking α4-integrin, VCAM-1, or G-proteins. It should be noted that presence of VCAM-1 in spinal cord capillaries can be observed during EAE (ref. 6 and unpublished observations). Nevertheless, as extravasation of leukocytes across CNS capillaries was never observed by us, capillaries seem to lack other promigratory signals that are required for emigration of leukocytes across the vascular wall. Alternatively, blocking α4/VCAM-1–mediated interactions or G protein–mediated signals could influence the actin-cytoskeleton of T cells such that an increased deformability of T lymphocytes would lead to a better ability to squeeze through capillaries, resulting in a lower number of T cells permanently plugging capillaries.
Taken together, to our knowledge this study provides the first direct in vivo evidence that in the spinal cord white matter microvasculature constitutively expressed VCAM-1 mediates the G protein–independent capture of circulating encephalitogenic T cell blasts via α4-integrin. Transient capture is followed by G protein–dependent α4-integrin/VCAM-1–mediated adhesion strengthening and their subsequent entry into the spinal cord white matter. Absence of rolling and the predominant if not exclusive involvement of α4-integrin and VCAM-1 make this T lymphoblast interaction with the BBB unique and suggests that VCAM-1 serves as a CNS-specific addressin, allowing effector T lymphoblasts with high-affinity α4-integrin as a CNS homing receptor to travel to this immunoprivileged site in absence of inflammation.
Acknowledgments
We owe great thanks to Monika Bruckner for her outstanding and enthusiastic technical support. The excellent technical assistance of Veronika Schmidt and Irene Küchenmeister are also gratefully acknowledged. Thanks go to Urban Deutsch and Friedemann Kiefer for their critical discussion of the manuscript. Part of this work has been funded by the DFG-grants En214/3-4 and VA151/4-1.
Footnotes
See the related Commentary beginning on page 517.
Peter Vajkoczy and Melanie Laschinger contributed equally to this work.
References
- 1.Butcher EC, Williams M, Youngman K, Rott L, Briskin M. Lymphocyte trafficking and regional immunity. Adv Immunol. 1999;72:209–253. doi: 10.1016/s0065-2776(08)60022-x. [DOI] [PubMed] [Google Scholar]
- 2.Engelhardt B, et al. The development of experimental autoimmune encephalomyelitis in the mouse requires alpha4-integrin but not alpha4beta7-integrin. J Clin Invest. 1998;102:2096–2105. doi: 10.1172/JCI4271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barkalow FJ, Goodman MJ, Gerritsen ME, Mayadas TN. Brain endothelium lack one of two pathways of P-selectin-mediated neutrophil adhesion. Blood. 1996;88:4585–4593. [PubMed] [Google Scholar]
- 4.Engelhardt B, Vestweber D, Hallmann R, Schulz M. E- and P-selectin are not involved in the recruitment of inflammatory cells across the blood-brain barrier in experimental autoimmune encephalomyelitis. Blood. 1997;90:4459–4472. [PubMed] [Google Scholar]
- 5.Laschinger M, Engelhardt B. Interaction of alpha4-integrin with VCAM-1 is involved in adhesion of encephalitogenic T cell blasts to brain endothelium but not in their transendothelial migration in vitro. J Neuroimmunol. 2000;102:32–43. doi: 10.1016/s0165-5728(99)00156-3. [DOI] [PubMed] [Google Scholar]
- 6.Steffen BJ, Butcher EC, Engelhardt B. Evidence for involvement of ICAM-1 and VCAM-1 in lymphocyte interaction with endothelium in experimental autoimmune encephalomyelitis in the central nervous system in the SJL/J mouse. Am J Pathol. 1994;145:189–201. [PMC free article] [PubMed] [Google Scholar]
- 7.Yednock TA, et al. Prevention of experimental autoimmune encephalomyelitis by antibodies against alpha 4 beta 1 integrin. Nature. 1992;356:63–66. doi: 10.1038/356063a0. [DOI] [PubMed] [Google Scholar]
- 8.Baron JL, Madri JA, Ruddle NH, Hashim G, Janeway CA., Jr Surface expression of alpha 4 integrin by CD4 T cells is required for their entry into brain parenchyma. J Exp Med. 1993;177:57–68. doi: 10.1084/jem.177.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kuchroo VK, et al. Cytokines and adhesion molecules contribute to the ability of myelin proteolipid protein-specific T cell clones to mediate experimental allergic encephalomyelitis. J Immunol. 1993;151:4371–4382. [PubMed] [Google Scholar]
- 10.Graesser D, Mahooti S, Madri JA. Distinct roles for matrix metalloproteinase-2 and α4-integrin in autoimmune T cell extravasation and residency in brain parenchyma during experimental autoimmune encephalomyelitis. J Neuroimmunol. 2000;109:121–131. doi: 10.1016/s0165-5728(00)00275-7. [DOI] [PubMed] [Google Scholar]
- 11.Uhl E, Pickelmann S, Rohrich F, Baethmann A, Schurer L. Influence of platelet-activating factor on cerebral microcirculation in rats: part 2. Local application. Stroke. 1999;30:880–886. doi: 10.1161/01.str.30.4.880. [DOI] [PubMed] [Google Scholar]
- 12.Vajkoczy P, Ullrich A, Menger MD. Intravital fluorescence videomicroscopy to study tumor angiogenesis and microcirculation. Neoplasia. 2000;2:53–61. doi: 10.1038/sj.neo.7900062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pizza M, et al. Mutants of pertussis toxin suitable for vaccine development. Science. 1989;246:497–500. doi: 10.1126/science.2683073. [DOI] [PubMed] [Google Scholar]
- 14.Hamann, A., and Jonas, P. 1997. Lymphocyte migration in vivo: the mouse model. In Immunology methods manual. I. Lefkovits, editor. Academic Press Ltd. London, United Kingdom. 1333–1341.
- 15.Ley K, Gaehtgens P. Endothelial, not hemodynamic, differences are responsible for preferential leukocyte rolling in rat mesenteric venules. Circ Res. 1991;69:1034–1041. doi: 10.1161/01.res.69.4.1034. [DOI] [PubMed] [Google Scholar]
- 16.Von Andrian UH, et al. L-selectin function is required for beta 2-integrin-mediated neutrophil adhesion at physiological shear rates in vivo. Am J Physiol. 1992;263:H1034–H1044. doi: 10.1152/ajpheart.1992.263.4.H1034. [DOI] [PubMed] [Google Scholar]
- 17.Robert C, et al. Interaction of dendritic cells with skin endothelium: a new perspective on immunosurveillance. J Exp Med. 1999;189:627–636. doi: 10.1084/jem.189.4.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Berlin C, et al. Alpha 4 beta 7 integrin mediates lymphocyte binding to the mucosal vascular addressin MAdCAM-1. Cell. 1993;74:185–195. doi: 10.1016/0092-8674(93)90305-a. [DOI] [PubMed] [Google Scholar]
- 19.Sriramarao P, von Andrian UH, Butcher EC, Bourdon MA, Broide DH. L-selectin and very late antigen-4 integrin promote eosinophil rolling at physiological shear rates in vivo. J Immunol. 1994;153:4238–4246. [PubMed] [Google Scholar]
- 20.Alon R, et al. The integrin VLA-4 supports tethering and rolling in flow on VCAM-1. J Cell Biol. 1995;128:1243–1253. doi: 10.1083/jcb.128.6.1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen C, et al. High affinity very late antigen-4 subsets expressed on T cells are mandatory for spontaneous adhesion strengthening but not for rolling on VCAM-1 in shear flow. J Immunol. 1999;162:1084–1095. [PubMed] [Google Scholar]
- 22.Lichtman AH, et al. CD45RA-RO+ (memory) but not CD45RA+RO- (naive) T cells roll efficiently on E- and P-selectin and vascular cell adhesion molecule-1 under flow. J Immunol. 1997;158:3640–3650. [PubMed] [Google Scholar]
- 23.Lim Y-C, et al. α4β1-integrin activation is necessary for high-efficiency T cell subset interactions with VCAM-1 under flow. Microcirculation. 2000;7:201–214. [PubMed] [Google Scholar]
- 24.Campbell JJ, Qin S, Bacon KB, Mackay CR, Butcher EC. Biology of chemokine and classical chemoattractant receptors: differential requirements for adhesion-triggering versus chemotactic responses in lymphoid cells. J Cell Biol. 1996;134:255–266. doi: 10.1083/jcb.134.1.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wekerle H, Linington C, Lassmann H, Meyermann R. Cellular immune reactivity within the CNS. Trends Neurosci. 1986;9:271–277. [Google Scholar]
- 26.Hickey WF, Hsu BL, Kimura H. T-lymphocyte entry into the central nervous system. J Neurosci Res. 1991;28:254–260. doi: 10.1002/jnr.490280213. [DOI] [PubMed] [Google Scholar]
- 27.Grabovsky V, et al. Subsecond induction of alpha 4 integrin clustering by immobilized chemokines stimulates leukocyte tethering and rolling on endothelial vascular cell adhesion molecule 1 under flow conditions. J Exp Med. 2000;192:495–505. doi: 10.1084/jem.192.4.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carrithers MD, Visintin I, Kang SJ, Janeway CA., Jr Differential adhesion molecule requirements for immune surveillance and inflammatory recruitment. Brain. 2000;123:1092–1101. doi: 10.1093/brain/123.6.1092. [DOI] [PubMed] [Google Scholar]
- 29.Hamann A, Andrew DP, Jablonski WD, Holzmann B, Butcher EC. Role of alpha 4-integrins in lymphocyte homing to mucosal tissues in vivo. J Immunol. 1994;152:3282–3293. [PubMed] [Google Scholar]
- 30.Rosengren S, Hernson PM, Worther GS. Migration associated volume changes in neutrophils facilitate the migratory process in vitro. Am J Physiol. 1994;267:C1623–C1632. doi: 10.1152/ajpcell.1994.267.6.C1623. [DOI] [PubMed] [Google Scholar]