

Abstract
Tp0655 of Treponema pallidum, the causative agent of syphilis, is predicted to be a 40-kDa membrane lipoprotein. Previous sequence analysis of Tp0655 noted its homology to polyamine-binding proteins of the bacterial PotD family, which serve as periplasmic ligand-binding proteins of ATP-binding-cassette (ABC) transport systems. In the current study, the 1.8-Å crystal structure of Tp0655 demonstrated structural homology to E. coli PotD and PotF. The latter two proteins preferentially bind spermidine and putrescine, respectively. All of these proteins contain two domains that sandwich the ligand between them. The ligand-binding site of Tp0655 can be occupied by 2-(N-morpholino)ethanesulfanoic acid, a component of the crystallization medium. To discern the polyamine binding preferences of Tp0655, the protein was subjected to isothermal titration calorimetric experiments. The titrations established that Tp0655 binds polyamines avidly, with a marked preference for putrescine (Kd = 10 nM) over spermidine (Kd = 430 nM), but the related compounds cadaverine and spermine did not bind. Structural comparisons and structure-based sequence analyses provide insights into how polyamine-binding proteins recognize their ligands. In particular, these comparisons allow the derivation of rules that may be used to predict the function of other members of the PotD family. The sequential, structural, and functional homology of Tp0655 to PotD and PotF prompt the conclusion that the former likely is the polyamine-binding component of an ABC-type polyamine transport system in T. pallidum. We thus rename Tp0655 as TpPotD. The ramifications of TpPotD as a polyamine-binding protein to the parasitic strategy of T. pallidum are discussed.
Keywords: ABC transporter, isothermal titration calorimetry, lipoprotein, polyamine receptor, Treponema pallidum
Introduction
Syphilis, caused by the spirochetal bacterium Treponema pallidum, is a sexually transmitted infection (STI) of major global importance 1,2. Despite its recognition for centuries, syphilis remains among the most poorly understood of all STIs. The dearth of knowledge concerning syphilis pathogenesis stems primarily from the fact that T. pallidum, despite decades of intensive efforts, still cannot be cultivated continuously in vitro 3. This obstacle has severely impeded progress in understanding many features of this enigmatic pathogen, particularly the structures and functions of key treponemal membrane proteins that likely serve at the host-pathogen interface to facilitate virulence 4-6. Along these lines, the initial discovery of membrane lipoproteins in T. pallidum 7,8 prompted new avenues of investigation into treponemal membrane biology. In other bacteria, membrane lipoproteins have importance as virulence factors, modular components of ATP-binding cassette (ABC) transporters, enzymes, receptors, protective immune targets, and proinflammatory agonists that contribute to the innate immune response 9-16. Genome analysis now predicts that T. pallidum devotes as much as 4.3% of its genetic coding capacity to lipoproteins 17. The absence of in vitro cultivation and genetic manipulation of T. pallidum, however, has greatly hindered the ability to characterize the functional aspects of T. pallidum’s membrane lipoproteins.
As a strategy for investigating the unusual membrane biology of T. pallidum, we have been pursuing a structure-to-function approach, with the objective of formulating new testable hypotheses regarding the potential function(s) of a number of the treponemal lipoproteins. This approach has led to the structural and functional characterization of at least four other T. pallidum lipoproteins 18-21. In the current study, we have focused our efforts on Tp0655, predicted to be a lipoprotein. The genome of T. pallidum contains four contiguous genes, annotated as potABCD, that encode a putative spermidine/putrescine ABC transporter 22. Putrescine, spermidine, and spermine are polyamines that play important roles in cellular processes such as cell growth, differentiation, and cell death 23-25. Tp0655 has sequence homology to PotD of E. coli; in E. coli, PotD is the spermidine-preferring periplasmic ligand-binding protein (pLBP) component of the ABC transporter system 26,27. E. coli encodes another ABC-type polyamine transport system (potFGHI), wherein the binding protein, PotF, specifically binds putrescine 28.
Little is known about polyamine transport in mammals, but many bacterial uptake systems have been identified. Some species have a single carrier for the three major polyamines, but others have at least two transport systems that have different specificities. E. coli, for example, harbors both spermidine- and putrescine-specific transporters in addition to the enzymes required for de novo polyamine synthesis 29,30. By contrast, T. pallidum appears to have only one polyamine uptake system 22,29. Also, unlike E. coli, T. pallidum lacks the enzymes necessary to synthesize polyamines, hence its reliance on an uptake system to maintain optimum levels for metabolism.
To investigate whether the PotD homolog (Tp0655) is likely to fulfill the polyamine requirement for T. pallidum, we determined the crystal structure of the binding component of Tp0655 and elucidated its substrate specificity. The results demonstrated that Tp0655, which is likely a pLBP, preferentially binds putrescine, but also binds spermidine. The importance of Tp0655 as an ABC-type receptor that is tethered to the cytoplasmic membrane (via its lipid anchors) and the potential relationship of Tp0655′s affinity for polyamines to treponemal survival within the human host are discussed. These findings add to our knowledge of key physiological functions for the treponemal membrane lipoproteins and their potential relationships to T. pallidum membrane biology and virulence expression.
Results & Discussion
Structure Determination and Description
The crystal structure of rTp0655, the non-lipidated version of Tp0655 (residues 2-324, lacking the N-terminal cystein), was determined by sulfur single-wavelength anomalous dispersion (sulfur SAD) using Cu-Kα X-rays from a rotating anode; diffraction data to a resolution of 1.78 Å were used (Table 1). The asymmetric unit contains one molecule of rTp0655 with dimensions of about 55 × 42 × 30 Å. The first 25 residues belonging to the cloning linker and the following four native residues of the crystallized protein could not be located in the electron density.
Table 1.
Data collection and refinement statistics
| sulfur SADa | native | |
|---|---|---|
| Data Collection | ||
| Space group | P212121 | P212121 |
| Unit cell dimensionsa | 51.7 | 51.6 |
| b | 86.4 | 86.3 |
| c | 89.5 | 89.5 |
| Resolution (Å) | 20.00 - 2.30 (2.32-2.30) | 31.04-1.78 (1.80-1.78) |
| Completeness | 99.5 (97.6) | 99.7 (97.2) |
| Unique reflections | 34,327 (849) | 39,206 (911) |
| Multiplicity | 11.9 (6.2) | 6.7 (4.4) |
| Rmerge (%) | 1.4 (3.5) | 4.8 (82.8) |
| I/σ(I) | 166.4 (63.2) | 39.4 (2.0) |
| Wilson B factor (Å2) | 29.8 | 28.9 |
| Phase Determination | ||
| anomalous scatterer | sulfur (11 sites) | |
| figure of merit | 0.2 to 2.3Å (0.7 after density modification) | |
| Refinement | ||
| Resolution (Å) | 30.0 - 1.78(1.82-1.78) | |
| No. of reflections Rwork/Rfree | 35,504/1,893 | |
| Rwork/Rfree (%) | 23.4/26.0(29.0/31.1) | |
| Average B-factor (Å2) | 32.7 | |
| Atoms non-H protein | 2,654 | |
| water molecules | 137 | |
| others | 27 | |
| R.m.s. deviations | ||
| Bond lengths (Å) | 0.011 | |
| Angles (°) | 1.29 | |
| Correlation coefficient Fo-Fc | 0.949 | |
| Ramachandran Plot (%) (favored/add./dis.)b | 98.1/2.9/0.0 | |
Data for the outermost shell are given in parentheses.
Bijvoet-pairs were kept separate for data processing
as defined by the validation suite MolProbity 56
The structure of rTp0655 is typical for group I pLBP’s 31. It consists of two globular domains, each containing a central five-stranded mixed β-sheet. The sheets are flanked on both sides by six and eight α-helices in the N-and C-terminal domains, respectively (Figure 1A). The N-terminal domain (N-domain) contains residues 7-109 and 241-291, and the C-terminal domain (C-domain) contains residues 114-236 and 292-325. A region (residues 110-113 and 237-240) that links the N- and C-domains is composed of parts of a two-stranded central β-sheet. Also, this linker region contains a loop around Pro292. Our crystal structure of rTp0655 contains two bound molecules of 2-(N-morpholino)ethanesulfonic acid (MES), a buffer component included in the crystallization medium (one of which is shown in Figure 1B).
Figure 1.
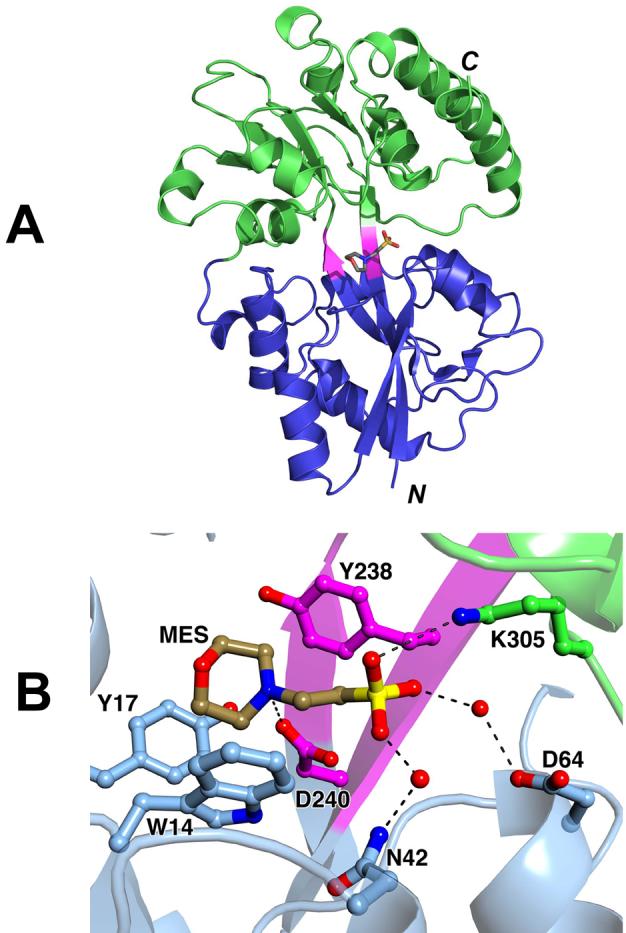
The structure of rTp0655. (A) Ribbons-style representation, with the N-domain in blue, the C-domain in green, and the connection region in purple. A stick model of one of the bound molecules of MES is also shown; oxygen atoms are red, carbon atoms are gray, the nitrogen atom is blue, and the sulfur atom is yellow. (B) A close-up of the bound MES. The color scheme for atoms is the same as in part (A), except carbon atoms belonging to the N-domain are colored light blue, carbon atoms from the C-domain are green, and carbon atoms from the connector region are purple. Hydrogen bonds or ionic interactions are shown as dashed black lines.
rTp0655 is structurally very similar to the ligand-bound, closed forms of PotD, the binding protein for the spermidine-preferential uptake system 27, and PotF, the binding protein for the putrescine-specific uptake system in E. coli 32. The superposition of rTp0655 with PotD complexed with spermidine (PDB entry 1POY) shows that 185 of rTp0655′s Cα atoms are equivalent to Cα atoms in PotD, with an r.m.s.d. between these atoms of 1.33 Å. The superposition of rTp0655 with PotF complexed with putrescine (PDB entry 1A99) results in an r.m.s.d. of 1.36 Å for 183 equivalent Cα atoms (Figure 2). PotD and PotF bind their respective ligands in a deep cleft at the interface between the N- and C-domains 27,32. In contrast to PotF, rTp0655 does not contain a disulfide bond. One of the MES-binding sites in rTp0655 coincides with the polyamine-binding sites in PotD and PotF. Ligand binding to PotD and PotF leads these proteins to adopt their closed structures in a manner typical for most periplasmic binding proteins. In comparison, the rTp0655 structure does not appear to be as fully closed.
Figure 2.
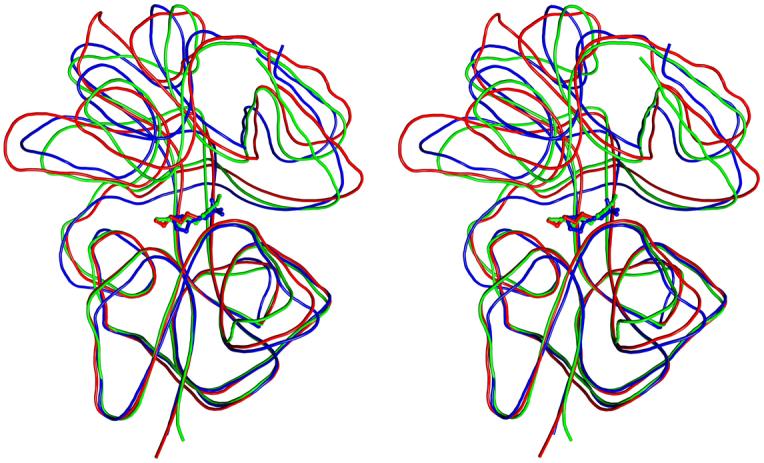
Stereo view of the superposition of rTp0655 (blue), PotD (green), and PotF (red). The lines are smoothed traces through the Cα backbone of the respective proteins. The bound ligand found in each structure is shown as a stick model.
The second MES-binding site is located on the “back” surface of the molecule as oriented in Figure 1A (not shown). The site, which is about 13 Å distant from the putative polyamine-binding site, is likely adventitious.
Binding Studies
Based on the sequential and structural similarities between rTp0655, PotD and PotF, we hypothesized that Tp0655 is a polyamine-binding protein. In order to study the binding of polyamine molecules to Tp0655, it was desirable to prepare ligand-free protein. Previously, we found that overexpressed pLBPs sometimes contain tightly bound ligands procured from the cytoplasm of the expression organism 19-21. Therefore, to remove bound ligands that could interfere with binding assays, Tp0655 was subjected to an unfolding/washing/refolding protocol as outlined in Methods. MES was excluded from all buffers used to prepare the protein because of that compound’s potential for competing with the polyamines for binding to rTp0655. The refolded protein is hereafter termed “apo-rTp0655.” Apo-rTp0655 was a well-behaved monomer in solution; in this preparation, ∼95% of the material exhibited a sedimentation coefficient of 3.2 S and a frictional ratio of about 1.2. The estimated mass from the AUC experiment (∼34 kD) is consistent with the monomeric molecular mass of apo-rTp0655 (∼39 kD). These data allow the conclusion that apo-rTp0655 is a well-folded monomer in solution.
Isothermal titration calorimetry (ITC) was used to examine the ability of apo-rTp0655 to bind to five polyamines: 1,3-diaminopropane (DAP), putrescine, cadaverine, spermidine, and spermine (Figure 3). All of the ITC data exhibiting significant heats were fitted using a single-site binding model (Figure 4, Table 2). Putrescine exhibited the strongest binding to apo-rTp0655 (dissociation constant, Kd = 10 nM), while spermidine (Kd = 430 nM) and DAP (Kd = 5 μM) bound significantly more weakly. No evidence of cadaverine or spermine binding was observed in the titrations. All of the interpretable titrations demonstrated that one polyamine molecule bound to one site on apo-rTp0655; that is, the stoichiometry (n) of binding was 1. The seemingly low value of n for spermidine (0.65) almost certainly does not imply half-site binding; rather, it probably indicates that the concentration for the batch of spermidine used in these experiments was higher than assumed, or that a portion of the refolded protein was incompetent to bind polyamine. Supporting this contention is the fact that, in other experiments under similar conditions, the n-value of spermidine binding to apo-rTp0655 was 1 (data not shown). We conclude that apo-rTp0655 has a single binding site for one polyamine molecule, and that this protein has a pronounced preference for putrescine over all other polyamines examined.
Figure 3.

Structures and atom numbering convention for the polyamines studied in this report. The structures are aligned such that they demonstrate their respective homologies to spermidine.
Figure 4.

Assessment of polyamine binding to apo-rTp0655 using ITC. The upper panel of each part is a time course of the power supplied to the reaction cell during the course of the experiment to keep the cell at a constant temperature. In the bottom parts, the circles represent the integrated heats from each injection. The solid line in these parts is the fitted binding isotherm assuming a 1:1 interaction between polyamines and apo-rTp0655. In the three titrations shown, the compounds injected into solutions of apo-rTp0655 were putrescine (A), spermidine (B), and 1,3-DAP (C).
Table 2.
Thermodynamic values for polyamine binding to apo-rTp0655
| Compound | Kd (nM) | ΔG (kcal/mol) | ΔH (kcal/mol) | ΔS (cal/K·mol) | N |
|---|---|---|---|---|---|
| DAP | 5,000 ± 1,000 | -7.1 ± 0.1 | -2.4 ± 0.2 | 16 ± 1 | .99 ± .01 |
| Putrescine | 10.1 ± 0.1 | -10.79 ± 0.1 | -7.34 ± 0.07 | 11.7 ± 0.3 | .93 ± .01 |
| Cadaverine | N/Aa | - | NHb | - | - |
| Spermidine | 430 ± 10 | -8.54 ± 0.02 | 3.22 ± 0.03 | 40.1 ± 0.1 | 0.65 ± .01 |
| Spermine | N/A | - | NH | - | N/A |
Not applicable
No heat evolved upon titration
The ITC data also permitted the calculation of the apparent contributions that enthalpy (ΔHapp) and entropy (ΔSapp) make to the overall binding of polyamines to apo-rTp0655. Apparent values were derived because the observed ΔHapp values include both the intrinsic heat of binding and also any heat derived from the release or uptake of protons by the buffer (Bicine) during the binding event. For putrescine binding to apo-rTp0655, most of the free energy of association (ΔG = -10.8 kcal/mol) was contributed by the enthalpic term (ΔHapp =-7.3 kcal/mol), but there was also a significant contribution from the entropic term (-TΔSapp = -3.5 kcal/mol). For spermidine, the situation was reversed; the enthalpy (ΔHapp = +3.2 kcal/mol) did not favor binding, but a large entropic term (-TΔSapp = -11.7 kcal/mol) compensated and resulted in a favorable ΔG (-8.5 kcal/mol). The thermodynamic parameters of the association of DAP and apo-rTp0655 appeared to be intermediate between those of putrescine and spermidine. Although the enthalpy was favorable (ΔHapp = -2.4 kcal/mol), the majority of ΔG was contributed by the entropic component (-TΔSapp = -4.7 kcal/mol). These observations can be explained by a model in which the polyamine-binding site of apo-rTp0655 is occupied by water molecules that are displaced upon ligand binding, resulting in a positive entropic contribution to the free energy of the interaction. In addition, the desolvation of the ligand, which may be a prerequisite of polyamine binding, could also increase ΔSapp. Upon putrescine binding to apo-rTp0655, waters are liberated by desolvation and ligand displacement. The larger entropies observed upon the binding of spermidine or DAP suggest that more waters are liberated by ligand binding and/or desolvation when either of these two polyamines binds to Tp0655.
The existence of ligand-induced oligomerization or conformational change was studied using AUC experiments. These studies demonstrated that, regardless of the presence or absence of putrescine or spermidine, the s20,w value of the protein remained constant at 3.2 S. The sedimentation velocity methodology employed in this report should be able to detect changes in sedimentation coefficient as small as about 0.1 S. Therefore, any conformational changes that might occur upon ligand binding are probably small. These data agree with the finding during the purification of rTp0655 that the protein was a well-behaved monomer in solution (not shown).
Based on its sequential homology to PotD, Tp0655 has tentatively been annotated as a “spermidine/putrescine transport system substrate-binding protein” in the genomic database. An examination of the T. pallidum genome revealed that two putative ABC-type permeases (Tp0653, Tp0654) and an ABC-type ATP-binding protein (Tp0652) are encoded directly adjacent to Tp0655. The current results demonstrate the structural similarity of rTp0655 to PotD and PotF. Also, we have demonstrated that apo-rTp0655 binds to polyamines. Given all of these data, there is a strong likelihood that Tp0655 is the polyamine-binding component of an ABC-type polyamine-transport system in T. pallidum.
Polyamine Binding Site in rTp0655
Based on the presented binding studies and the three-dimensional structure, it is likely that the polyamine-binding sites of rTp0655, PotD, and PotF are structurally similar. The polyamine-binding site is located in the deep cleft in the center of the molecules. Using Figure 1A as a reference, the “bottom” and “top” of the cleft are lined by residues from the N- and C-domains, respectively. Residues from the linker region form the back of the binding site. In the rTp0655 crystal structure, the polyamine-binding site is occupied by a MES molecule (Figures 1B & 2). It is situated deeply in the binding cleft, reminiscent of the way spermidine is bound to PotD, with the sulfonate group being at a position equivalent to the N10 nitrogen of spermidine. The MES molecule establishes an interaction between its sulfonate group and the side chain of Lys305, a direct hydrogen bond between the N4 nitrogen atom of MES and Asp240, two water-mediated hydrogen bonds bridging sulfonate oxygen atoms and Asn42 and Asp64, as well as hydrophobic interactions with Trp14, Tyr17, and Tyr238, which cradle the morpholino moiety of the MES molecule.
Structural Determinants of Polyamine Binding
A specific problem presents itself with establishing specificity in proteins that bind very similar ligands made up of smaller subunits, such as oligosaccharides or polyamines. A protein that specifically binds a larger oligomer must ensure that a smaller oligomer does not become a significant competitor. Likewise, because proteins that bind similar ligands often have significant structural similarities and are often even derived from a common ancestor, mechanisms must exist that prevent binding of larger ligands when specificity for a smaller one is required. The presence of multiple interaction sites for the subunits of an oligomeric ligand also poses the problem of overlapping binding modes for smaller oligomers or monomers. Finally, for those proteins that must bind both smaller and larger variants of oligomers, specific sets of interactions must exist in the binding site that assure high-affinity binding for differently sized ligands. These aspects have been studied in great detail in some systems, such as for carbohydrates binding to oligosaccharide-converting enzymes 33,34. By contrast, despite the fact that structural analysis of PotD and PotF combined with mutagenesis studies revealed important insights into their ligand-binding properties, the mechanisms for achieving specificity in polyamine-binding proteins were not established.
As a first step in understanding the specificity of polyamine binding proteins, we note that many of the residues lining the clefts in PotD, PotF and rTp0655 are strictly or functionally conserved (Figure 5), yet the ligand specificities of these proteins vary. Although no rigorous binding studies have been carried out for PotD and PotF, it seems that PotD prefers spermidine, but also binds putrescine 35, whereas PotF binds only putrescine 36. As described above, Tp0655 can bind both spermidine and putrescine, but clearly prefers putrescine. A superposition of PotD and PotF shows that putrescine (1,4-diaminobutane) in PotF binds into the polyamine-binding cleft at the same location as the N1-N6 portion of spermidine in PotD (Figure 2). The extent of the binding sites is about the same in all three proteins. No large side chains protrude into the cleft in PotF that would prevent access of spermidine. It is thus likely that the specific interactions with ligands determine the specificities of polyamine-binding proteins.
Figure 5.
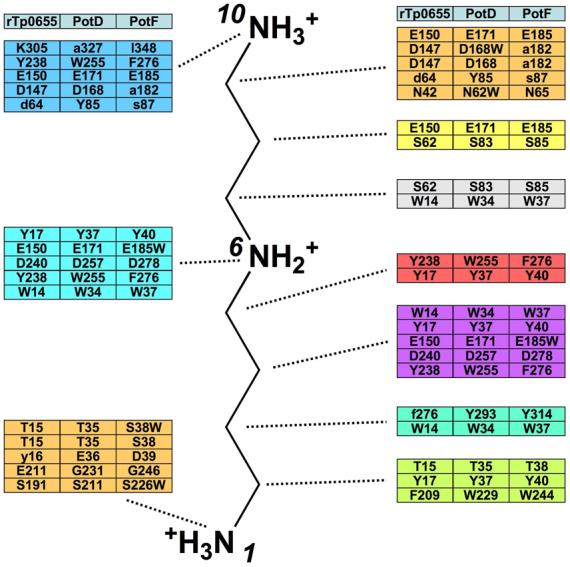
Schematic of the polyamine-binding site in Tp0655, PotD, PotF with spermidine as a reference. Listed are all residues that interact with ligand in PotD and PotF as well as structurally equivalent (based on Cα positions) residues in the other proteins. Residues in lower-case letters are structurally equivalent but their side chains do not establish interactions with the ligand.
One can define three main a priori principles that are expected to govern the binding specificity for putrescine or spermidine in architecturally similar proteins. The first principle specifies that these proteins must have some means of excluding chemically similar competitor molecules. Polyamine binding appears to be dominated by specific ionic and hydrogen-bond interactions that anchor the presumably positively charged amino groups and place them at strategic locations in the binding cleft. We here define three “anchor regions,” each corresponding to the position of a nitrogen of spermidine when bound to PotD. Structurally analogous regions of other PotD-like proteins are referenced in the same way. Hence, the “N1 anchor region” refers to the residues surrounding N1 of bound spermidine; likewise for the N6 and N10 anchor regions. Additional hydrophobic interactions are established with the aliphatic portions of bound polyamines. The anchor regions must be fairly rigid in their spacing, because the asymmetric spermidine binds directionally to PotD, i.e., with only one orientation, although the lengths of the two portions of spermidine differ only by a single methylene group. The strict ruler-like spacing is not compatible with cadaverine (1,5-diaminopentane) binding, likely explaining why this molecule does not associate with rTp0655. The spacings between the two amino moieties of 1,3-DAP and between N6 and N10 of spermidine are identical, which suggests that apo-rTp0655 uses the same residues to bind DAP and the aminopropane moiety of spermidine. This hypothesis is analogous to the supposition 27 that PotD uses the same residues to bind both putrescine and the putrescine moiety of spermidine.
The second principle specifies that, to prefer putrescine to spermidine, a protein should emphasize binding the N1 and/or N6 amino groups of the polyamine, because a large part of the binding energy must come from these moieties. Likewise, to prefer spermidine to putrescine, a protein should de-emphasize binding the N1 and/or N6 amino groups of the polyamine. This principle manifests itself in the more intimate interactions between PotF and the N1 anchor region of putrescine compared to spermidine binding to PotD. Although the locations of the anchor regions are preserved in PotD, PotF and rTp0655, their exact architectures vary. In PotD, the N1 amino group of spermidine forms an ionic interaction with Glu36 and a hydrogen bond with the side chain of Thr35, both of which belong to the N-domain. By contrast, in PotF, the N1 amino group of putrescine engages in a larger number of interactions, a fact that has been deemed important for putrescine to bind with high affinity 32. Specifically, the putrescine N1 amino group establishes an ionic interaction to Asp247 from the C-domain and two hydrogen bonds to the main chain carbonyls of Ser38 and Asp39, respectively. In addition, two water molecules are hydrogen-bonded to the N1 amino group as well as to each other. One of these water molecules establishes a hydrogen bond with the side chain of Ser38. The second water molecule establishes hydrogen bonds with the main chain of Ser226 and with the side chain of Asp247. PotF has an acidic residue (Asp39) in the N-domain that is structurally equivalent (in terms of Cα position) to the critical Glu36 in PotD. Replacing this residue with asparagine did not affect putrescine binding 32. Indeed, the crystal structure of PotF shows that the Asp39 side chain is pointing away from the ligand-binding site and does not establish interactions with putrescine. Instead, the side chain of Asp247 provides the stabilizing negative charge. Replacing this residue with an alanine completely abolishes putrescine binding 32. Asp39 is apparently not able to substitute. The water molecules involved in the N1 anchor region appear to be important for binding as well. Substitution by alanine of Ser38, whose side chain hydrogen-bonds with one of the stabilizing water molecules, led to an ∼80% reduction in putrescine binding. The equivalent residue in PotD is Thr38, but its side chain is not involved in a water-mediated hydrogen bond to the N1 amino group of spermidine. The absence of stabilizing water molecules is probably due to the lack of the acidic residue in the C-domain that is equivalent to Asp247 in PotF (the structurally equivalent residue is Ser232).
By contrast, the N6 anchor region does not appear to be a major specificity determinant. This anchor region consists primarily of an ionic interaction with an aspartate (Asp257 in PotD and Asp278 in PotF, respectively). The electrostatic interaction with a negatively charged residue is crucial for stabilizing the N6 amino group. Substitution of Asp278 with asparagine in PotF abolished putrescine binding 32. PotF has an additional, water-mediated hydrogen bond to Glu185. However, this residue is sequentially and spatially conserved in PotD (Glu 171), making it unlikely that this residue could provide for putrescine specificity.
The third principle specifies that the binding site of a putrescine-preferring protein should provide an architecture that is unfavorable to binding of the aminopropane moiety of spermidine. Conversely, the binding site of a spermidine-preferring protein should provide an architecture that is favorable to binding of the aminopropane moiety. The veracity of this principle can be assessed by examining the N10 anchor region in PotF and the analogous area in PotD. In PotD, the N10 amino group forms an ionic interaction with Glu171 (distance of about 2.6 Å) and a presumably weaker interaction with the side chain of Asp168 (distance of about 3.8 Å). In addition, it engages in hydrogen bonds with the amide oxygen of Gln327 and the hydroxyl group of Tyr85. Although PotF contains Glu185, which is structurally equivalent to Glu171 in PotD, it does not have residues that are structurally equivalent to the other residues in PotD that interact with the spermidine N10 amino group. In all cases, the substituting residues in PotF are smaller, and in two cases (Asp168/Ala182 and Gln327/I348), the equivalent PotF residue is hydrophobic. The loss of hydrogen-bonding potential and the increase in hydrophobicity in the binding pocket may act to disfavor the binding of the polar aminopropyl moiety of spermidine to PotF. As is the case for the N1 and N6 anchor regions, a negatively charged residue is crucial for binding of the N10 amino group. Interestingly, Glu185 in PotF is structurally equivalent to the crucial Glu171 in PotD. As mentioned above, in PotF, this residue forms a water-mediated hydrogen bond to the N6 of putrescine, but it is in principle poised to also bind spermidine through an electrostatic interaction. Overall, the N10 anchor region in PotF appears less well suited to bind the aminopropyl portion of spermidine. This region in PotF should in fact disfavor binding of an N10 amino group; otherwise spermidine would become a significant competitor. The architecture of the N10 anchor region in PotF does not appear to sterically exclude spermidine. Therefore, any potential favorable interactions in this region must be over-compensated by unfavorable effects upon spermidine binding, for example, a large loss of entropy or the necessity to displace tightly bound water molecules from the binding cleft.
Given that the three a priori principles hold true for PotD and PotF, we investigated whether they can be extended to rTp0655, which can bind both spermidine and putrescine, but prefers putrescine (Figure 4; Table 2). rTp0655 is only the third receptor component of polyamine transporter systems for which a crystal structure is available. Based on its polyamine-binding characteristics, one can hypothesize that rTp0655 shares structural features of the PotF N1 and N6 anchor regions as well as structural features of the PotD N10 anchor region. An examination of the rTp0655 crystal structure shows that the N1 anchor region indeed bears the hallmarks of the PotF N1 anchor region. Specifically, the negatively charged residue is provided by the C domain and not by the N-terminal region. This residue is Glu211 in rTp0655, which is equivalent to Asp247 in PotF. Tp0655 does not have an N-terminal acidic residue equivalent to Glu36 in PotD.
The anchor region at the N6 position in rTp0655 is very well conserved and comprises equivalent residues that can establish interactions with a bound polyamine as observed in PotD and PotF. These similarities support the hypothesis that the N6 anchor region does not appear to provide any polyamine-specificity determinants.
The N10 anchor region in rTp0655 contains the negatively charged residue (Glu150) that is conserved in PotD and PotF. In addition, it also has the second acidic residue (Asp147) that is found in PotD (Asp168) but not in PotF. Likewise, in Lys305, rTp0655 has a residue whose side chain is capable of establishing a hydrogen bond with the N10 of spermidine, like Gln327 in PotD. PotF does not have an equivalent residue that could engage in such a stabilizing interaction. It should be noted that, in the rTp0655-spermidine interaction, either the lysine or N10 must not be charged in order to avoid electrostatic repulsion. Taken together, the N10 anchor region in rTp0655 shares some aspects of that in PotD and is less similar to that in PotF. The rTp0655 protein therefore fits well into the principles laid out above - it has rigid anchoring regions that exclude the binding of cadaverine, it is similar to PotF in its N1 anchor region, and its N10 anchor region does not provide the same favorable interactions that are present there for PotD. In fact, Tp0655 provides at the N10 anchor region a positively charged residue, which could discourage the binding of spermidine that is positively charged at N10. The large entropy gain and smaller enthalpy upon spermidine binding to apo-rTp0655 suggest that the requirement for the displacement of bound water deleteriously affects spermidine binding relative to that of putrescine.
With three crystal structures available for polyamine-binding proteins with binding profiles, one can attempt to establish rules for predicting the specificity of polyamine binding in other, homologous proteins.
First, in the N1 anchor region, the presence of an acidic residue equivalent to Asp247 in PotF or Glu211 in rTp0655 indicates a preference for putrescine. Likewise, the absence of such an acidic residue and the presence of an acidic residue equivalent to Glu36 in PotD indicate a preference for spermidine.
Second, putrescine-binding proteins should also have a residue equivalent to Ser38 in PotF that is able to participate in the water network in the N1 region.
Third, in the N10 anchor region, the presence of a second negatively charged residue equivalent to Asp168 in PotD or Asp147 in rTp0655 and/or a potential hydrogen bond partner equivalent to Gln327 in PotD or Lys305 in rTp0655 indicate a preference for spermidine.
It should be noted that there might be polyamine-binding proteins with specific mechanisms of achieving specificity that do not obey the above rules. For example, the N10 anchor region could be occupied by a bulky side chain, preventing the longer spermidine from entering the cleft.
Sequence conservation in the PotD family
Proteins with sequence homologies to PotD have been placed in a Cluster of Orthologous Groups family called COG0687. This family contains many PotF homologues as well. Based on the homologous structures of rTp0655, PotD, and PotF, a careful structural alignment was performed. The other 110 sequences from COG0687 were aligned to the structure-based alignment. The result is shown in Figure 6 (the rTp0655 numbering will be used for this discussion). The mean similarity among proteins in this family is about 60%. Residues whose side chains putatively make direct polar contacts to bound ligand at the N1 or N6 anchor positions are very well conserved (83-84% similarity). However, residues whose side chains make such contacts at the N10 anchor position are not as well conserved (45-58%). This discrepancy is to be expected because the family consists of both putative spermidine- and putrescine-binding proteins, and the latter are not expected to have residues conducive to spermidine binding at the N10 anchor position. Notably, the hydrophobic residues whose side chains putatively form van der Waals contacts with bound polyamines are well conserved (similarities range from 77% to 93%). It should be noted that, in this alignment, two positions are strictly conserved: 133 and 257. These positions always have hydrophobic residues. However, they are not located at the spermidine/putrescine binding sites. Instead, they are parts of the hydrophobic cores of the proteins. Their strict conservation indicates that these positions are especially sensitive to substitution, implying that they may be important for protein folding.
Figure 6.
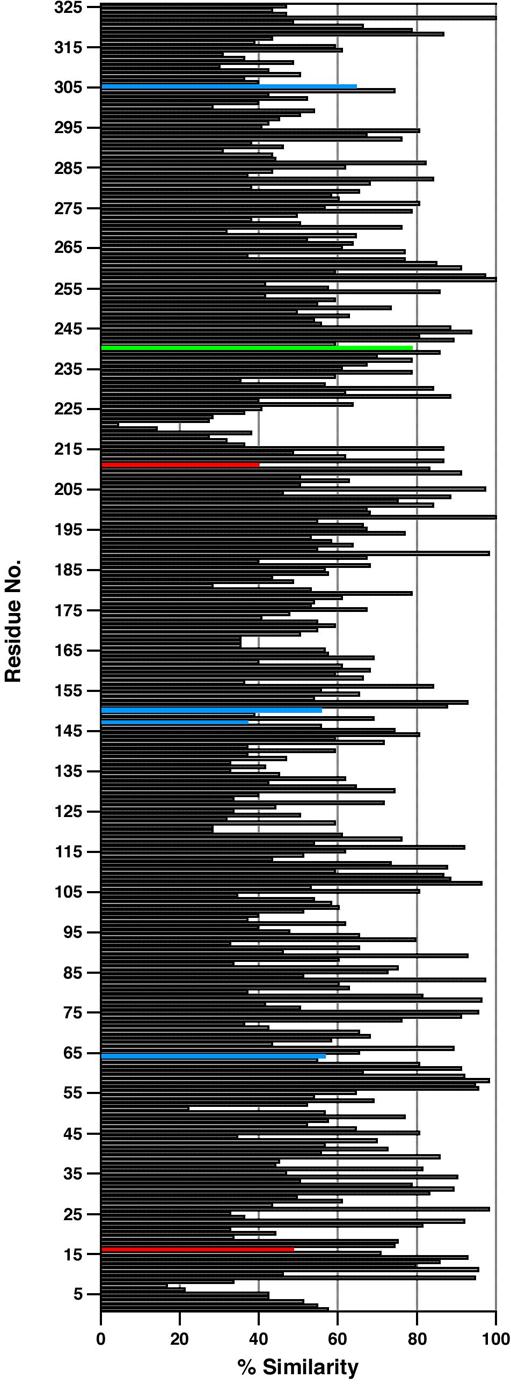
Sequence similarity among COG0687 PotD-like proteins. The y-axis is the sequence number in Tp0655 numbering. The horizontal bars represent the percentage of COG0687 members that have an amino acid similar to the predominant type at that position. Four types of amino acids were defined: hydrophobic, polar, acidic, and basic. The colored bars denote positions in the alignment that putatively contact bound polyamine at anchor positions N1 (red), N6 (green), or N10 (blue).
The three rules for polyamine binding delineated above can be applied to the structure-based sequence alignment. Proteins obeying rules 1 and 2 should bind to putrescine; 36 of the 110 proteins in COG0687 fall into this category. Those proteins conforming to both provisions of rule 3 should bind spermidine; 23 of the proteins fit this criterion. Eight proteins of the COG obey all of the rules. Tp0655, of course, is one of them. The seven other proteins may therefore be polyamine-binding proteins that prefer putrescine to spermidine. These proteins are AF1605m (Archaeoglobus fulgidus), APE0945 (Aeropyrum pernix), PAE0953 (Pyrobaculum aerophilum), VC0704 (Vibrio cholerae), SMc00991 (Sinorhizobium meliloti), DR1305 (Deinococcus radiodurans), and UU110 (Ureaplasma urealyticum). Only two of these organisms (S. meliloti and V. cholerae) are known to have more than one polyamine-binding LBP. It is therefore possible that these proteins serve as the sole means of binding extracellular polyamines for transport into the cytoplasms of the remainder of the above-named organisms. Of the 110 proteins that make up COG0687, 25 do not obey any of the rules. The outlying proteins could use alternative, as yet undefined mechanisms. In addition, some of these proteins simply may have been wrongly annotated as PotD-like.
Spermidine/putrescine-binding proteins are a prime example for how specificity is obtained in proteins that bind ligands with very similar structures. The available structural and sequence data have allowed us to identify general strategies for determining specificity in this class of proteins. Based on this analysis, we surmise that most of the proteins in the COG0687 family bind polyamines using means similar to those described in this report.
Summary & Implications
Polyamines, such as putrescine, spermidine, and spermine, are small, positively charged molecules present in both prokaryotic and eukaryotic cells 30,37. In bacteria, it is clear that polyamines play important roles in cell growth, as well as in transcription and translation 25,30,38. Although de novo biosynthesis typically contributes to the intracellular levels of polyamines, bacteria often also posses a polyamine uptake system that, when needed, can substitute for de novo synthesis 29,37. Putrescine can be synthesized from L-ornithine by ornithine decarboxylase (ODC; EC 4.1.1.17), which is the target for the Trypanosoma brucei (agent of sleeping sickness) drug difluoromethylornithine 39. Also, putrescine may be synthesized indirectly from L-arginine by arginine decarboxylase (ADC; EC 4.1.1.19) 23,30,37. Both the ODC and ADC pathways are common in virtually all living cells, and often they are found together in the same organism. Putrescine can be converted to spermidine and spermine by spermidine synthase (EC 2.5.1.16) and spermine synthase (EC 2.5.1.22), respectively.
Most of our knowledge of polyamine physiology and biosynthesis in prokaryotes is derived from studies with E. coli. Apart from its biosynthetic capability, E. coli also depends on three polyamine uptake systems to maintain optimal intracellular polyamine levels 29,40. The first is an ABC-type system that is preferential for spermidine and that consists of the PotA, PotB, PotC, and PotD proteins. The second system, which is also of the ABC-type but with a preference for putrescine, is comprised of the PotF, PotG, PotH, and PotI proteins. A third system is involved in the excretion of putrescine; it relies on a single protein, PotE, which is a putrescine-ornithine antiporter. Both T. pallidum and Borrelia burgdorferi (the agent of Lyme disease) are predicted to lack ODC- and ADC-mediated biosynthetic pathways 22,41, and thus likely are polyamine auxotrophs. However, they are postulated to encode ABC-type operons, denoted as potABCD, whose putative functions are to import polyamines from their extracellular environments. In T. pallidum, the four contiguous genes Tp0652, Tp0653, Tp0654, and Tp0655 are orthologs of E. coli PotA (ATP-binding protein), PotB (permease component), PotC (permease component), and PotD (spermidine/putrescine-binding protein), respectively 22,29 although Tp0655 also has structural similarity to E. coli PotF (current study). Relative to the function of Tp0655 investigated herein, it is noteworthy that T. pallidum has a limited genome, and thus limited biosynthetic capability 41, which likely is a reflection of its restricted (human) host range. In contrast, the closely related saprophytic treponeme T. denticola encodes an ortholog of ODC (TDE1109), and thus T. denticola seems to rely on a biosynthetic pathway that is ODC-based to satisfy its polyamine requirement(s) 42.
The pathogenesis of syphilis is complex, occurring in stages and characterized by protracted asymptomatic periods (e.g., latency) 43. Many details of the pathogenesis of syphilis remain unknown. One such information gap concerns where viable spirochetes reside within the human body during latency. Based on decades of histopathological studies, there is no compelling evidence that T. pallidum resides inside cells during human infection, and hence it is widely accepted that T. pallidum is an extracellular pathogen 43. On the other hand, the paucibacillary nature of syphilis makes demonstrating viable treponemes in any tissue or body fluid a formidable challenge. An intracellular residence by T. pallidum, however, could partly explain its characterization as a “stealth pathogen” 5: that is, a bacterium that escapes the host’s vigorous immune responses, leading to a state of bacterial chronicity 43.
If T. pallidum is an extracellular pathogen, as conventionally thought, our data raise an interesting paradox. Polyamines are present in very low concentrations in vaginal secretions, interstitial fluids, and serum 44-48, all presumed to be relevant to treponemal survival during one of more phases of transmission, infection, and dissemination. Despite these low levels, the sub-micromolar binding constants we determined for polyamine binding to Tp0655 must still be sufficient to supply T. pallidum with these essential nutrients. On the other hand, intracellular concentrations of polyamines in eukaryotic cells are high (millimolar) 30,38. It is therefore tempting to revisit the notion that T. pallidum may invade host cells at some point during disease progression, wherein the uptake of host-derived polyamines via the PotABCD transporter would have an important role in parasitic growth. Regardless, our ITC-based ligand-binding analyses demonstrated that Tp0655 has a 40-fold preference for putrescine (over spermidine), implying that its function is more akin to the putrescine-binding protein PotF. However, T. pallidum is predicted to lack spermidine synthase for the conversion of putrescine to spermidine. It is thus plausible that Tp0655 mediates the uptake of both putrescine and spermidine to fulfill the metabolic requirements of T. pallidum. Given the available data, we conclude that Tp0655 is the polyamine-binding component of an ABC-type polyamine transport system (potABCD) in T. pallidum. Consequently, we propose that Tp0655 be denoted as TpPotD.
Experimental Procedures
Construction of His6-tagged rTp0655
To produce a non-lipidated, recombinant derivative of Tp0655 (rTp0655) in E. coli, a DNA fragment encoding amino acid residues 2-324 (cloned without the post-translationally modified N-terminal Cys residue) was amplified by PCR from treponemal genomic DNA using primer pairs encoding the predicted 5′-and 3′-termini. The PCR primers were 5′-GCGGCCGCCTGCAGACACGACAGGATGTCCTG-3′ and 5′-GGATCCTATTACCTAAATCGCACCGCGTTCCAG-3′. In the preceding sequences, the region of each primer complementary to the tp0655 sequence is underlined. The forward primer contained a NotI site (bold); the reverse primer contained a BamHI site (bold). The PCR was performed using TaKaRa Ex Taq DNA polymerase (TaKaRa Bio Inc.). The resulting PCR product was cloned into the pGEM-T Easy vector (Promega) and transformed into E. coli XL1-Blue cells (Stratagene) and the plasmid DNA isolated from colonies that tested positive by restriction digest were verified using standard DNA sequencing techniques. The insert fragment was excised by digestion with NotI and BamHI and then ligated into the corresponding restriction sites of the expression vector pIVEX2.4d vector (Roche). The resultant construct encoded a fusion protein with a His6-tag at its N-terminus; this His6-tag region is cleavable by Factor Xa. Ligation mixtures were transformed into E. coli XL1-Blue cells and plasmid DNA isolated from colonies were verified by DNA sequencing. The sequenced verified plasmid was then transformed into E. coli BL21 AI (Invitrogen) cells for protein expression.
Expression and Purification of rTp0655
E. coli BL21 AI cells were grown at 37 °C in LB medium containing 0.1% (w/v) glucose and 100 μg/mL of ampicillin until the cell density reached an A600 of 0.5. The culture was then induced for 3 h with 0.2% (w/v) L-arabinose. Cells derived from one liter of culture were harvested by centrifugation and lysed at room temperature with gentle rocking for 30 min using 50 mL of B-PER II (Pierce). The resulting suspension was centrifuged at 25,000 × g for 15 min to remove cell debris. rTp0655 was isolated from the supernatant by affinity chromatography using Ni-NTA Agarose (Qiagen).
The protein was then subjected to size exclusion chromatography using a HiLoad 16/60 Superdex 75 prep grade column (GE Healthcare) equilibrated with Buffer A (20 mM Hepes, 0.1 M NaCl, pH 7.0, 2 mM n-octyl β-D-glucopyranoside (βOG)). Subsequent to elution, peak fractions were analyzed by SDS-PAGE. At this stage, the protein was pure to apparent homogeneity (i.e., >95%). Fractions containing purified rTp0655 were pooled and concentrated to 20 mg/mL in buffer A for protein crystallization. Protein concentration was determined spectrophotometrically using an extinction coefficient of 54,780 M-1cm-1 at 280 nm, which was calculated using the ProtParam utility of ExPASy (www.expasy.org).
Preparation of Apo-rTp0655
Apo-rTp0655 used for ITC analyses was prepared by a reversible denaturation/renaturation method 20 in ITC buffer (20 mM Bicine, 0.1 M KCl, pH 7.6, 2 mM βOG).
Crystallization, data collection, and structure determination
Crystals were grown at 20 °C using the vapor diffusion method in hanging drop mode by mixing 1 μl protein (20 mg/mL) in Buffer A with 1 μl reservoir solution (10-12% (w/v) PEG 20K, 0.1 M MES, pH 6.5) and equilibrating against 1 mL of reservoir solution. Crystals appeared within several hours and grew to a final size of 0.2 × 0.2 × 0.8 mm in about 2 days. The crystals were cryo-protected in reservoir solution supplemented with 20% (v/v) ethylene glycol, and then flash-cooled in liquid nitrogen. The crystals exhibit the symmetry of space group P212121 with cell dimensions of a = 51, b = 86 Å, c = 89 Å, and contain one molecule per asymmetric unit and 52% solvent. Crystals diffracted to a minimum Bragg spacing (dmin) of about 1.7 Å. The diffraction data exhibited significant anisotropy.
Phases for rTp0655 were obtained from a sulfur single-wavelength anomalous dispersion (SAD) experiment using Cu-Ka X-rays from an FR-E Ultra-Bright X-ray generator (Rigaku). Oscillation images were collected on an R-Axis IV++ imaging-plate detector (Rigaku). A highly redundant dataset (2,123 frames; overall multiplicity = 17) was collected over a period of 48 hours and processed to a minimum Bragg spacing (dmin) of 2.3 Å. Data reduction with the program HKL2000 49 included experimental procedures for radiation-damage correction (to be presented elsewhere). Eleven heavy atom sites were identified with the programs ShelXC, D, and E 50 using data to a resolution of 2.6 Å, refined with the program MLPhaRe of the CCP4 package 51 using data to 2.3 Å (resulting figure of merit = 0.2), and subjected to density modification with the program DM 52 (resulting figure of merit = 0.7). The resultant electron density map was of sufficient quality to automatically construct an initial model using the program ARP/warp 53. Manual rebuilding was carried out with the program Coot 54. Refinement was performed using the program REFMAC5 55.
A higher resolution native dataset was subsequently collected at beamline 19BM (SBC-CAT) at the Advanced Photon Source (Argonne National Laboratory, Argonne, Illinois, USA) to a dmin of about 1.78 Å. The model derived from the sulfur SAD experiment served as the starting model for the refinement of the higher resolution dataset. After the protein model was completed, water molecules were added where chemically reasonable. There was clear electron density for two bound molecules of morpholino-ethanesulfonic acid (MES).
The final model has an Rwork of 23.4% and Rfree of 26.0%. These R values are rather high, given the resolution of the data, probably due to the anisotropy of the diffraction data. Five N-terminal residues could not be located in the electron density. Data collection and refinement statistics are shown in Table 1.
Isothermal titration calorimetry
Before the binding experiment, solutions of apo-rTp0655 were exhaustively exchanged into ITC Buffer. Solid polyamine compounds were dissolved in ITC Buffer to a concentration of 10 mM. These stock solutions were further diluted in ITC Buffer as needed. Thirty-one 8-μL injections of the polyamine (230-520 μM) were made into a 1.4-mL stirred reaction cell that contained 19-23 μM apo-rTp0655. The temperature of the reaction cell was held constant at 20 °C. The heats that resulted from polyamine binding to apo-rTp0655 were measured using a VP-ITC microcalorimeter (MicroCal, Inc.). The resultant thermograms were integrated to generate binding isotherms for the polyamines. A one-site binding model was fitted to the isotherms using the program SEDPHAT 57, which yielded fitted values for ΔH and Kd. Usually, heats of dilution of the ligand are subtracted from the thermogram. However, obtaining heats of dilution for spermidine proved to be difficult. Further, the heats obtained from the spermidine titration were very small, and subtracting the noisy heats of dilution introduced an unacceptable noise level into the isotherm. Therefore, we utilized a feature of SEDPHAT that estimates a “baseline” heat of dilution from the experimental data, eliminating the need to establish the heat of dilution of ligand alone. For consistency, this methodology was followed for the putrescine and DAP titrations, also. All titrations were performed in triplicate, and all quoted values for Kd and ΔH are the means of values obtained from the three titrations ± their standard deviations. The quantity ΔS was calculated from the relationships ΔG = ΔH - TΔS and ΔG = -RTlnKAwhere KA is the equilibrium association constant (KA = 1/Kd), R is the gas constant in units of calories per Kelvin-mole, and T is the temperature in Kelvin. Specifically, ΔG was substituted in the above equations and they were rearranged to yield the relationship
which was used to calculate the values given in Table 2.
Analytical Ultracentrifugation
For characterization of hydrodynamic properties via sedimentation velocity (SV) analytical ultracentrifugation (AUC), apo-rTp0655 was diluted with a solution of AUC Buffer (20 mM Tris-Cl, 100 mM NaCl, pH 7.5). The concentration of apo-rT0655 used was 9.5 μM; where polyamines were included, their concentrations were 100 μM. Solutions of apo-rTp0655 (390 μL) were centrifuged in a Beckman XL-I ultracentrifuge at a speed of 50,000 rpm in twin-sector epon-filled centerpieces that were housed in an An60-Ti rotor. The sedimentation of the macromolecule toward the bottom of the cells was monitored using both absorbance spectrophotometry and laser interferometry. The SV data were analyzed using SEDFIT 58, which yields a continuous distribution of sedimentation coefficients that best fits the data (the distribution is termed c(s)). The sedimentation coefficients of Tp0655 were taken as the weight-averaged position under the single peaks in the c(s) distributions. All sedimentation coefficients were corrected to standard conditions (s20,w). Partial specific volume, buffer density, and buffer viscosity were calculated using SEDNTERP 59.
Protein Data Bank accession codes
Coordinates and structure factors for the MES-bound TpPotD are deposited in the RCSB Protein Data Bank with the PDB ID 2v84.
Acknowledgments
We thank Sandra Hill for technical assistance and the staff at the Advanced Photon Source for synchrotron data collection support. Results shown in this report are derived from work performed at Argonne National Laboratory, Structural Biology Center at the Advanced Photon Source. Argonne is operated by UChicago Argonne, LLC, for the U.S. Department of Energy, Office of Biological and Environmental Research under contract DE-AC02-06CH11357. We are grateful to NIH (grant AI-56305) and the Welch Foundation (grant I-0940) for financial support.
The abbreviations used are
- ABC
ATP-binding cassette
- ADC
arginine decarboxylase
- AUC
analytical ultracentrifugation
- βOG
n-octyl β-D-glucopyranoside
- DAP
1,3-diaminopropane
- ITC
isothermal titration calorimetry
- MES
2-(N-morpholino)ethanesulfonic acid
- ODC
ornithine decarboxylase
- pLBP
periplasmic ligand-binding protein
- r.m.s.
root-mean-square
- SAD
single-wavelength anomalous dispersion
- SV
sedimentation velocity
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chao JR, Khurana RN, Fawzi AA, Reddy HS, Rao NA. Syphilis: reemergence of an old adversary. Ophthamol. 2006;113:2074–2079. doi: 10.1016/j.ophtha.2006.05.048. [DOI] [PubMed] [Google Scholar]
- 2.Kerani RP, Handsfield HH, Stenger MS, Shafii T, Zick E, et al. Rising rates of syphilis in the era of syphilis elimination. Sex. Transm. Dis. 2007;34:154–161. doi: 10.1097/01.olq.0000233709.93891.e5. [DOI] [PubMed] [Google Scholar]
- 3.Norris SJ. Polypeptides of Treponema pallidum: progress toward understanding their structural, functional, and immunologic roles. Microbiol. Rev. 1993;57:750–779. doi: 10.1128/mr.57.3.750-779.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cox DL, Chang P, McDowall A, Radolf JD. The outer membrane, not a coat of host proteins, limits the antigenicity of virulent Treponema pallidum. Infect. Immun. 1992;60:1076–1083. doi: 10.1128/iai.60.3.1076-1083.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Radolf JD. Role of outer membrane architecture in immune evasion by Treponema pallidum and Borrelia burgdorferi. Trends Microbiol. 1994;2:307–311. doi: 10.1016/0966-842x(94)90446-4. [DOI] [PubMed] [Google Scholar]
- 6.Radolf JD. Treponema pallidum and the quest for outer membrane proteins. Mol. Microbiol. 1995;16:1067–1073. doi: 10.1111/j.1365-2958.1995.tb02332.x. [DOI] [PubMed] [Google Scholar]
- 7.Chamberlain NR, Brandt ME, Erwin AL, Radolf JD, Norgard MV. Major integral membrane protein immunogens of Treponema pallidum are proteolipids. Infect. Immun. 1989;57:2872–2877. doi: 10.1128/iai.57.9.2872-2877.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schouls LM, Mout R, Dekker J, Van Embden JDA. Characterization of lipid-modified immunogenic proteins of Treponema pallidum expressed in Escherichia coli. Microbial Pathogen. 1989;7:175–188. doi: 10.1016/0882-4010(89)90053-3. [DOI] [PubMed] [Google Scholar]
- 9.Hayashi S, Wu HC. Lipoproteins in bacteria. J. Bioenerget. Biomem. 1990;22:451–471. doi: 10.1007/BF00763177. [DOI] [PubMed] [Google Scholar]
- 10.Sutcliff IC, Russell RRB. Lipoproteins of gram-positive bacteria. J. Bacteriol. 1995;177:1123–1128. doi: 10.1128/jb.177.5.1123-1128.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Becker PS, Akins DR, Radolf JD, Norgard MV. Similarity between the 38-kilodalton lipoprotein of Treponema pallidum and the glucose/galactose-binding (MglB) protein of Escherichia coli. Infect. Immun. 1994;62:1381–1391. doi: 10.1128/iai.62.4.1381-1391.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haake DA. Spirochetal lipoproteins and pathogenesis. Microbiology. 2000;146:1491–1504. doi: 10.1099/00221287-146-7-1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Steere AC, Sikand VK, Meurice F, Parenti DL, Fikrig E, et al. Vaccination against Lyme disease with recombinant Borrelia burgdorferi outer-surface lipoprotein A with adjuvant. N. Engl. J. Med. 1998;339:209–215. doi: 10.1056/NEJM199807233390401. [DOI] [PubMed] [Google Scholar]
- 14.Ochsner UA, Vasil AI, Johnson Z, Vasil ML. Pseudomonas aeruginosa fur overlaps with a gene encoding a novel outer membrane lipoprotein, OmlA. J. Bacteriol. 1999;181:1099–1109. doi: 10.1128/jb.181.4.1099-1109.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Radolf JD, Arndt LL, Akins DR, Curetty LL, Levi ME, et al. Treponema pallidum and Borrelia burgdorferi lipoproteins and synthetic lipopeptides activate monocytes/macrophages. J. Immunol. 1995;154:2866–2877. [PubMed] [Google Scholar]
- 16.Norgard MV, Arndt LL, Akins DR, Curetty LL, Harrich DA, et al. Activation of human monocytic cells by Treponema pallidum and Borrelia burgdorferi lipoproteins and synthetic lipopeptides proceeds via a pathway distinct from that of lipopolysaccharide but involves the transcriptional activator NF-kB. Infect. Immun. 1996;64:3845–3852. doi: 10.1128/iai.64.9.3845-3852.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Setubal JC, Reis M, Matsunaga J, Haake DA. Lipoprotein computational prediction in spirochaetal genomes. Microbiology. 2006;152:113–121. doi: 10.1099/mic.0.28317-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deka RK, Machius M, Norgard MV, Tomchick DR. Crystal structure of the 47-kilodalton lipoprotein of Treponema pallidum reveals a novel penicillin-binding protein. J. Biol. Chem. 2002;277:41857–41864. doi: 10.1074/jbc.M207402200. [DOI] [PubMed] [Google Scholar]
- 19.Deka RK, Neil L, Hagman KE, Machius M, Tomchick DR, et al. Structural evidence that the 32-kilodalton lipoprotein (Tp32) of Treponema pallidum is an L-methionine-binding protein. J. Biol. Chem. 2004;279:55644–55650. doi: 10.1074/jbc.M409263200. [DOI] [PubMed] [Google Scholar]
- 20.Deka RK, Brautigam CA, Yang XF, Blevins JS, Machius M, et al. The PnrA (Tp0319; TmpC) lipoprotein represents a new family of bacterial purine nucleoside receptor encoded within an ATP-binding cassette (ABC)-like operon in Treponema pallidum. J. Biol. Chem. 2006;281:8072–8081. doi: 10.1074/jbc.M511405200. [DOI] [PubMed] [Google Scholar]
- 21.Deka RK, Brautigam CA, Tomson FL, Lumpkins SB, Tomchick DR, et al. Crystal structure of the Tp34 (TP0971) lipoprotein of Treponema pallidum: implications of its metal-bound state and affinity for human lactoferrin. J. Biol. Chem. 2007;282:5944–5958. doi: 10.1074/jbc.M610215200. [DOI] [PubMed] [Google Scholar]
- 22.Fraser CM, Casjens S, Huang WM, Sutton GG, Clayton R, et al. Genomic sequence of a Lyme disease spirochaete, Borrelia burgdorferi. Nature. 1997;390:580–586. doi: 10.1038/37551. [DOI] [PubMed] [Google Scholar]
- 23.Wallace HM, Fraser AV, Hughes A. A perspective of polyamine metabolism. Biochem. J. 2003;376:1–14. doi: 10.1042/BJ20031327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wallace HM. The physiological role of the polyamines. Eur. J. Clin. Invest. 2000;30:1–3. doi: 10.1046/j.1365-2362.2000.00585.x. [DOI] [PubMed] [Google Scholar]
- 25.Tabor CW, Tabor H. Polyamines. Annu. Rev. Biochem. 1984;53:749–790. doi: 10.1146/annurev.bi.53.070184.003533. [DOI] [PubMed] [Google Scholar]
- 26.Sugiyama S, Vassylyev DG, Matsushima M, Kashiwagi K, Igarashi K, et al. Crystal structure of PotD, the primary receptor of the polyamine transport system in Escherichia coli. J. Biol. Chem. 1996;271:9519–9525. doi: 10.1074/jbc.271.16.9519. [DOI] [PubMed] [Google Scholar]
- 27.Sugiyama S, Matsuo Y, Maenaka K, Vassylyev DG, Matsushima M, et al. The 1.8-Å X-ray structure of the Eschericia coli PotD protein complexed with spermidine and the mechanism of polyamine binding. Prot. Sci. 1996;5:1984–1990. doi: 10.1002/pro.5560051004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vassylyev DG, Kashiwagi T, Tomitori H, Kashiwagi K, Igarashi K, et al. Crystallization and preliminary X-ray analysis of the periplasmic receptor (PotF) of the putrescine transport system in Escherichia coli. Acta Crystallogr. D. Biol. Crystallogr. 1998;54:132–134. doi: 10.1107/s0907444997009049. [DOI] [PubMed] [Google Scholar]
- 29.Igarashi K, Kashiwagi K. Polyamine transport in bacteria and yeast. Biochem. J. 1999;344:633–642. [PMC free article] [PubMed] [Google Scholar]
- 30.Tabor CW, Tabor H. Polyamines in microorgansisms. Microbiol. Rev. 1985;49:81–99. doi: 10.1128/mr.49.1.81-99.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Quiocho FA, Ledvina PS. Atomic structure and specificity of bacterial periplasmic receptors for active transport and chemotaxis: variation of common themes. Mol. Microbiol. 1996;20:17–25. doi: 10.1111/j.1365-2958.1996.tb02484.x. [DOI] [PubMed] [Google Scholar]
- 32.Vassylyev DG, Tomitori H, Kashiwagi K, Morikawa K, Igarashi K. Crystal structure and mutational analysis of the Escherichia coli putrescine receptor. J. Biol. Chem. 1998;273:17604–17609. doi: 10.1074/jbc.273.28.17604. [DOI] [PubMed] [Google Scholar]
- 33.Machius M, Vertesy L, Huber R, Wiegand G. Carbohydrate and protein-based inhibitors of porcine pancreatic alpha-amylase: structure analysis and comparison of their binding characteristics. J. Mol. Biol. 1996;260:409–421. doi: 10.1006/jmbi.1996.0410. [DOI] [PubMed] [Google Scholar]
- 34.Kramhoft B, Bak-Jensen KS, Mori H, Juge N, Nohr J, et al. Involvement of individual subsites and secondary substrate binding sites in multiple attack on amylose by barely alpha-amylase. Biochemistry. 2005;44:1824–1832. doi: 10.1021/bi048100v. [DOI] [PubMed] [Google Scholar]
- 35.Kashiwagi K, Miyamoto S, Nukui E, Kobayashi H, Igarashi K. Functions of PotA and PotD proteins in spermidine-preferential uptake system in Escherichia coli. J. Biol. Chem. 1993;268:19358–19363. [PubMed] [Google Scholar]
- 36.Pistocchi R, Kashiwagi K, Miyamoto S, Nukui E, Sadakata Y, et al. Characteristics of the operon for a putrescine transport system that maps at 19 minutes on the Escherichia coli chromosome. J. Biol. Chem. 1993;268:146–152. [PubMed] [Google Scholar]
- 37.Yatin M. Polyamines in living organisms. J. Cell Molec. Biol. 2002;1:57–67. [Google Scholar]
- 38.Yoshida M, Kashiwagi K, Shigemasa A, Taniguchi S, Yamamoto K, et al. A unifying model for the role of polyamines in bacterial cell growth, the polyamine modulon. J. Biol. Chem. 2004;279:46008–46013. doi: 10.1074/jbc.M404393200. [DOI] [PubMed] [Google Scholar]
- 39.Fairlamb AH. Chemotherapy of human African trypanosomiasis: current and future prospects. Trends Parasitol. 2003;19:488–494. doi: 10.1016/j.pt.2003.09.002. [DOI] [PubMed] [Google Scholar]
- 40.Igarashi K, Ito K, Kashiwagi K. Polyamine uptake systems in Escherichia coli. Res. Microbiol. 2001;152:271–278. doi: 10.1016/s0923-2508(01)01198-6. [DOI] [PubMed] [Google Scholar]
- 41.Fraser CM, Norris SJ, Weinstock GM, White O, Sutton GG, et al. Complete genome sequence of Treponema pallidum, the syphilis spirochete. Science. 1998;281:375–388. doi: 10.1126/science.281.5375.375. [DOI] [PubMed] [Google Scholar]
- 42.Seshadri R, Myers GS, Tettelin H, Eisen JA, Heidelberg JF, et al. Comparison of the genome of the oral pathogen Treponema denticola with other spirochete genomes. Proc. Natl. Acad. Sci. U. S. A. 2004;101:5646–5651. doi: 10.1073/pnas.0307639101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Radolf JD, Hazlett KRO, Lukehart SA. Pathogenesis of syphilis. In: Radolf JD, Lukehart SA, editors. Pathogenic Treponema molecular and cellular biology. Caister Academic Press; Norfolk, England: 2006. [Google Scholar]
- 44.Wolrath H, Forsum U, Larsson PG, Boren H. Analysis of bacterial vaginosis-related amines in vaginal fluid by gas chromatography and mass spectrometry. J. Clin. Microbiol. 2001;39:4026–4031. doi: 10.1128/JCM.39.11.4026-4031.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen KCS, Forsyth PS, Buchanan TM, Holmes KK. Amine content of vaginal fluid from untreated and treated patients with nonspecific vaginitis. J. Clin. Invest. 1979;63:828–835. doi: 10.1172/JCI109382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marton LJ, Russell DH, Levy CC. Measurement of putrescine, spermidine, and spermine in physiolgical fluids by use of an amino acid analyzer. Clin. Chem. 1973;19:923–926. [PubMed] [Google Scholar]
- 47.El Baze P, Milano G, Verrando P, Renee N, Ortonne JP. Polyamine levels in normal human skin. Arch. Dermatol. Res. 1983;275:218–221. doi: 10.1007/BF00416663. [DOI] [PubMed] [Google Scholar]
- 48.Chen KC, Amsel R, Eschenbach DA, Holmes KK. Biochemical diagnosis of vaginitis: determination of diamines in vaginal fluid. J. Infect. Dis. 1982;145:337–345. doi: 10.1093/infdis/145.3.337. [DOI] [PubMed] [Google Scholar]
- 49.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Met. Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 50.Schneider TR, Sheldrick GM. Substructure solution with SHELXD. Acta Crystallogr. D. Biol. Crystallogr. 2002;58:1772–1779. doi: 10.1107/s0907444902011678. [DOI] [PubMed] [Google Scholar]
- 51.Collaborative Computational Project The CCP4 suite: programs for protein crystallography. Acta Cryst. 1994 Number 4;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 52.Cowtan K, Main P. Miscellaneous algorithms for density modification. Acta Crystallogr. D. Biol. Crystallogr. 1998;54:487–493. doi: 10.1107/s0907444997011980. [DOI] [PubMed] [Google Scholar]
- 53.Perrakis A, Morris RJ, Lamzin VS. Automated protein model building combined with iterative structure refinement. Nature Struc. Biol. 1999;6:458–463. doi: 10.1038/8263. [DOI] [PubMed] [Google Scholar]
- 54.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D. Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 55.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Cryst. 1997;D53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 56.Davis IW, Murray LW, Richardson JS, Richardson DC. MolProbity: structure validation and all-atom contact analysis for nucleic acids and their complexes. Nucleic Acids Res. 2004;32:W615–W619. doi: 10.1093/nar/gkh398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Houtman JCD, Brown PH, Bowden B, Yamaguchi H, Appella E, et al. Studying multisite binary and ternary protein interactions by global analysis of isothermal titration calorimetry data in SEDPHAT: application to adaptor protein complexes in cell signaling. Prot. Sci. 2007;16:30–42. doi: 10.1110/ps.062558507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schuck P, Braswell EH. Measuring protein-protein interactions by equilibrium sedimentation. In: Coligan JE, Kruisbeek AM, Margulies DH, Strober W, editors. Current protocols in immunology. John Wiley; New York: 2000. [DOI] [PubMed] [Google Scholar]
- 59.Laue TM, Shah BD, Ridgeway TM, Pelletier SL. Computer-aided interpretation of analytical sedimentation data for proteins. In: Harding SE, Rowe AJ, Horton JC, editors. Analytical ultracentrifugation in biochemistry and polymer science. The Royal Society of Chemistry; Cambridge: 1992. [Google Scholar]