

Abstract
VEGF secretion by the human retinal pigment epithelium (hRPE) plays an important role in retinal and choroidal neovascularization. In this study, transforming growth factor-β2 (TGF-β2)-induced vascular endothelial growth factor (VEGF) gene expression was investigated in hRPE cells. Treatment of hRPE cells with TGF-β2 for 24 and 48 hr as compared to 8 hr resulted in markedly increased VEGF secretion by 5-fold and 9-fold, respectively. Induced VEGF mRNA peaked within 3 hr of stimulation and remained above the basal at 36 hr. Stimulation of VEGF expression by TGF-β2 was blocked by cycloheximide, suggesting that de novo protein synthesis is required. Induced VEGF production was strongly inhibited by anti-inflammatory agents, dexamethasone and cyclosporin A. Despite of the weak stimulation of VEGF expression by TNF-α or bFGF alone, co-administration of either of these two cytokines synergized the effect of TGF-β2 on VEGF mRNA expression and protein production. Quantitative RT-PCR revealed that the synergy was predominantly at the level of VEGF transcription. Moreover, TGF-β2-induced RPE VEGF secretion was significantly reduced by inhibitors of mitogen-activated protein (MAP) kinase (MEK) (U0126), p38 (SB202190), c-Jun NH2-terminal kinase (JNK), Sp600125, protein tyrosine kinase (PTK) (Genistein), and phosphatidylinositol 3-kinase (PI3K) (Ly294002). Induced VEGF expression was completely abrogated by inhibitors of protein kinase C (PKC) (Ro318220), nuclear factor-κB (NF-κB) [caffeic acid phenethyl ester (CAPE)], and reactive oxygen species (ROS) [N-acetyl-cysteine (Nac) and diphenyleneiodonium (DPI)]. These results suggest that MEK, p38, JNK, PI3K, and NF-κB as well as multiple essential signaling intermediates, including PKC, PTK and ROS, are involved in hRPE VEGF upregulation by TGF-β2.
Keywords: human retinal pigment epithelium, TGF-β2, VEGF, bFGF, TNF-α, synergy
1. Introduction
Human retinal pigment epithelium (hRPE) cells are strategically interposed between the neurosensory retina and the choroid, and play a major role in retinal and choroidal neovascularization. A number of angiogenic factors, prominent among which is vascular endothelial growth factor (VEGF), are involved in the initiation and development of choroidal and retinal neovascularization. VEGF protein predominantly exists as a homodimer of four alternative spice variants VEGF121, VEGF165, VEGF189, and VEGF206 of a single gene (Keck et al., 1989). VEGF121 and VEGF165 are soluble proteins, whereas VEGF189 and VEGF206 are bound to heparin-containing proteoglycans on cell surfaces or in basement membranes (Houck et al., 1992). VEGF is an endothelial cell-specific mitogen, promoting vascular permeability (Keck et al., 1989). A clear temporal and spatial relationship has been found between VEGF and ocular neovascularization (Miller et al., 1994). VEGF stimulates neovascularization in corneal micropocket assays (Connolly et al., 1989) and in chicken chorioallantoic membranes (Wilting et al., 1992). It has been known that the intraocular VEGF levels are elevated in humans with proliferative diabetic retinopathy (PDR) (Adamis et al., 1994). Significantly increased VEGF immunoreactivity has been reported in retinal vascular endothelium, blood vessel walls, choriocapillaris endothelium, choroidal neovascular endothelium, and migrating human RPE cells in diabetic subjects (Lutty et al., 1996). In addition, the VEGF levels detected in vitreous from patients with active PDR have also been shown to be 15 to 30 times higher than that of patients with nonproliferative or quiescent diabetic retinopathy or nondiabetic control (Aiello et al., 1994).
An important function of hRPE cells, like that of endothelial cells and Müller cells, is to secrete VEGF (Adamis et al., 1993). Expression of VEGF has been known to be induced by various stimuli, including hypoxia (Aiello et al., 1995), mechanical stress, advanced glycation endproducts (AGE), vasopressor hormones such as angiotensin II and vasopressin, and cytokines such as interleukine-1 (IL-1), transforming growth factor-beta (TGF-β), basic fibroblast growth factor (bFGF) and platelet-derived growth factor (PDGF) (Chiarelli et al., 2000a).
TGF-β is a multifunctional regulator mediating cell proliferation, differentiation, apoptotic death and angiogenesis. However, the molecular mechanism of TGF-β–mediated angiogenesis remains poorly understood. TGF-β is mitogenic in some circumstances, but anti-mitogenic in others (Sporn and Roberts, 1992). Recent studies have demonstrated that TGF-β1, 2 and 3 all induce VEGF expression in several cell types including hRPE cells (Nagineni et al., 2003). Despite of the fact that TGF-β alone is able to induce VEGF expression, little has been known whether TGF-β may work in concert with other angiogenic cytokines such as PDGF, tumor necrosis factor-alpha (TNF-α) and bFGF in VEGF gene expression (Tolentino and Adamis, 1998). In addition, most of previous reports have focused on TGF-β1, not TGF-β2 or TGF-β3. In contrast to TGF-β3 which is functionally similar to TGF-β1, TGF-β2 appears distinct (Merwin et al., 1991a, b). TGF-β2 is the predominant form of TGF-β in ocular tissues (Pfeffer et al., 1994). Depending on the cell types and experimental conditions (Merwin et al., 1991b), the cellular responses to TGF-β2 differ from that to TGF-β1 in proliferation, migration and in vitro angiogenesis. In this study we investigated the effects of TGF-β2 alone and in combination with TNF-α or bFGF on hRPE VEGF mRNA expression and protein secretion, and examined the signaling pathways involved in VEGF expression in hRPE cells.
2. Materials and Methods
2.1. Materials
Recombinant human IL-1β, TNF-α, bFGF, PDGF, TGF-β2 was purchased from R&D System (Minneapolis, MN). U0126 was obtained from Promega (Madison, WI), SB202190, Sp600125, Ly294002, Genistein, Ro318220, and AG490 from Calbiochem (San Diego, CA), CAPE (caffeic acid phenethyl ester) from BIOMOL (Plymouth Meeting, PA), Nac (N-acetyl-cysteine) and DPI (diphenyleneiodonium chloride) from Sigma-Aldrich Company (St. Louis, MO). Antibodies against actin, phosphorylated p38, and protein kinase C (PKC) were purchased from Cell Signaling Technology (Beverly, MA). Affinity purified polyclonal antibodies against phosphorylated-PKCβ1 was obtained from the Sigma-Aldrich Company. The polyclonal anti-TGF-β2 antibody was purchased from R&D System, which has been selected for its ability to neutralize the biological activity of TGF- β2, but not TGF-β1, TGF-β3, or TGF-β5. All other reagents were obtained from Sigma-Aldrich Company. The signal transduction inhibitors used in this study are summarized in Table 1.
Table 1.
Inhibitors used to study pathways for hRPE VEGF expression.
| Inhibitor | Protein Target |
|---|---|
| U0126 | MEK |
| SB202190 | P38 |
| Sp600125 | JNK |
| AG490 | Jak2 |
| Genistein | PTK |
| Ro318220 | PKC |
| CAPE | NF-κB |
| Ly294002 | PI3K |
| Nac | ROS |
| DPI | ROS |
* MEK, mitogen-activated protein kinase; JNK, c-Jun NH2-terminal kinase; jak2, Janus kinase 2; PTK, protein tyrosine kinase; PKC, protein kinase C; NF-κB, nuclear factor-κB; PI3K, phosphatidylinositol 3-kinase; Nac, N-acetyl-cysteine; DPI, diphenyleneiodonium chloride; ROS, reactive oxygen species.
2.2. Cell isolation and culture
The hRPE cells were isolated within 24 hr of death from the donor eyes obtained from the Midwest Eye Bank as previously described (Elner et al., 1992). In brief, the sensory retina tissue was separated gently from the hRPE monolayer, and the hRPE cells were removed from Bruch's membrane with papain (5 U/ml). The hRPE cells were cultured in Dulbecco's modified essential medium (DMEM), containing 15% fetal bovine serum, penicillin G (100 U/ml), streptomycin sulfate (100 μg/ml), and amphotericin B (0.25 μg/ml) in Falcon Primaria culture plates to inhibit fibroblast growth. The hRPE monolayers exhibited uniform immunohistochemical staining for cytokeratin 8/18, fibronectin, laminin, and type IV collagen in the chicken-wire distribution characteristic for these epithelial cells. Cells were subcultured, grown to reach confluency, and used for experiments. The cells were in culture up to four to six passages,
2.3. VEGF ELISA
The levels of antigenic VEGF in the serial dilutions of hRPE supernatants were quantitated by modification of a double ligand ELISA method as previously described (Bian et al, 2004). Standards included 0.5 log dilutions of recombinant VEGF (R&D Systems) from 5pg to 100 ng/well.
2.4. Quantitative real-time PCR (qRT-PCR) for VEGF
Total cellular RNA was isolated from nearly confluent cultures of hRPE cells using TRIzol reagent according to the manufacturer's procedure (Invitrogen, Carlsbad, CA). Then total RNA was treated with RQ1 RNase-Free DNase (Promega, Madison, WI) to remove genomic DNA contamination. First-strand cDNA was synthesized by reverse transcriptase from 5 μg total RNA with oligo-d (T) primers. Real-time PCR was performed by using iCycler IQ real-time PCR detection system (Bio-Rad, Hercules, CA) to measure the fluorescence produced by SYBR Green I dye (Molecular Probes, Eugene, OR) that intercalates into PCR product (Yu et al., 2003). The PCR reactions were performed in triplicate on each cDNA template along with triplicate reactions of a housekeeping gene, β-actin. Negative control was obtained by performing PCR without cDNA. The synthetic oligonucleotide primers were 5’-TTGCCTTGCTGCTCTACCTC-3’ (sense) and 5’-AAATGCTTTCTCCGCTCTGA-3’ (antisense) for human VEGF (Kanuga et al., 2002), and 5’-GTGGGGCGCCCCAGGCACCA-3’ (sense) and 5’-GCTCGGCCGTGGTGGTGAAGC-3’ (antisense) for human β-actin. The thermal cycling conditions were: 3 min at 95°C, followed by 45 cycles at 95°C for 30 sec, 57°C for 30 sec, and 72°C for 30 sec. All PCR reaction products were verified by melting curve analysis and agarose gel electrophoresis. The VEGF mRNA expression levels were quantified by calculating the average value of triplicate reactions, normalized by the average value of triplicate reactions for the housekeeping β-actin gene (Yu et al., 2003).
2.5. Western Blot Analysis of human RPE cells
For preparation of whole-cell extracts, the hRPE cells were lysed in lysis buffer, containing 50 mM HEPES (pH 7.4), 1% Triton X-100, 0.15 M sodium chloride, 10% glycerol, 1.5 mM magnesium chloride, 1 mM EDTA, and protease inhibitors by sonication and centrifugation. The protein concentrations were determined with a commercial kit (Sigma-Aldrich Company). Western blot analysis of the cellular extracts from hRPE cells was carried out according to the manufacturer's procedure. Briefly, the samples containing 20 to 50 μg of proteins were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and then were electrotransferred to nitrocellulose membranes. For signal protein detection, samples were blocked with a solution of Tris-buffered saline (PH 7.4) containing 5% dry milk and 0.1% Tween-20 (TBST) at room temperature for 1 hr and then incubated at 4°C overnight with appropriate rabbit polyclonal antibodies. After the washes in TBST, the membranes were incubated with horseradish peroxidase-conjugated polyclonal anti-rabbit secondary antibodies for 1 hr at room temperature and washed three additional times with TBST. The membranes were then visualized using an enhanced chemiluminescent (ECL) technique, according to the manufacturer's instructions.
2.6. Statistic Analysis
The mean VEGF concentration ± SEM was determined for each assay condition. Various assay conditions were compared using ANOVA and t-test in Statview, and p<0.05 was considered to be statistically significant.
3. Results
3.1. TGF-β2 Induces hRPE VEGF mRNA Expression and Protein Production
To examine stimulated VEGF secretion, hRPE cells were challenged by 10 ng/ml of TGF-β2. The growth medium was harvested at the time points ranging from 0 to 48 hr after stimulation. As shown in Fig. 1A, TGF-β2 induced time-dependent, statistically-significant increases in VEGF secretion. The induced VEGF expression appeared at 8 hr and increased substantially at 24 and 48 hr after the stimulation. When compared to 8 hr, the VEGF levels at 24 and 48 hr after stimulation were further increased by 5-fold and 9-fold, respectively. We next induced VEGF secretion by various concentrations of TGF-β2 (2, 10, 20 ng/ml) for 24 hr. As compared to the low basal level of VEGF in unstimulated RPE cells, TGF-β2 clearly induced dose-dependent, statistically-significant increases in VEGF protein, although the additional increase at concentrations from 10 to 20 ng/ml of TGF-β2 was small (Fig. 1B). Induction of VEGF expression by TGF-β2 was completely abrogated by cycloheximide (Fig. 2), indicating that TGF-β2 induced de novo synthesis of VEGF in hRPE cells. Moreover, the stimulated VEGF secretion was markedly neutralized with highly specific anti-TGF-β2 antibody, suggesting that the increased VEGF expression was TGF-β2–specific (Fig. 2).
Figure 1.
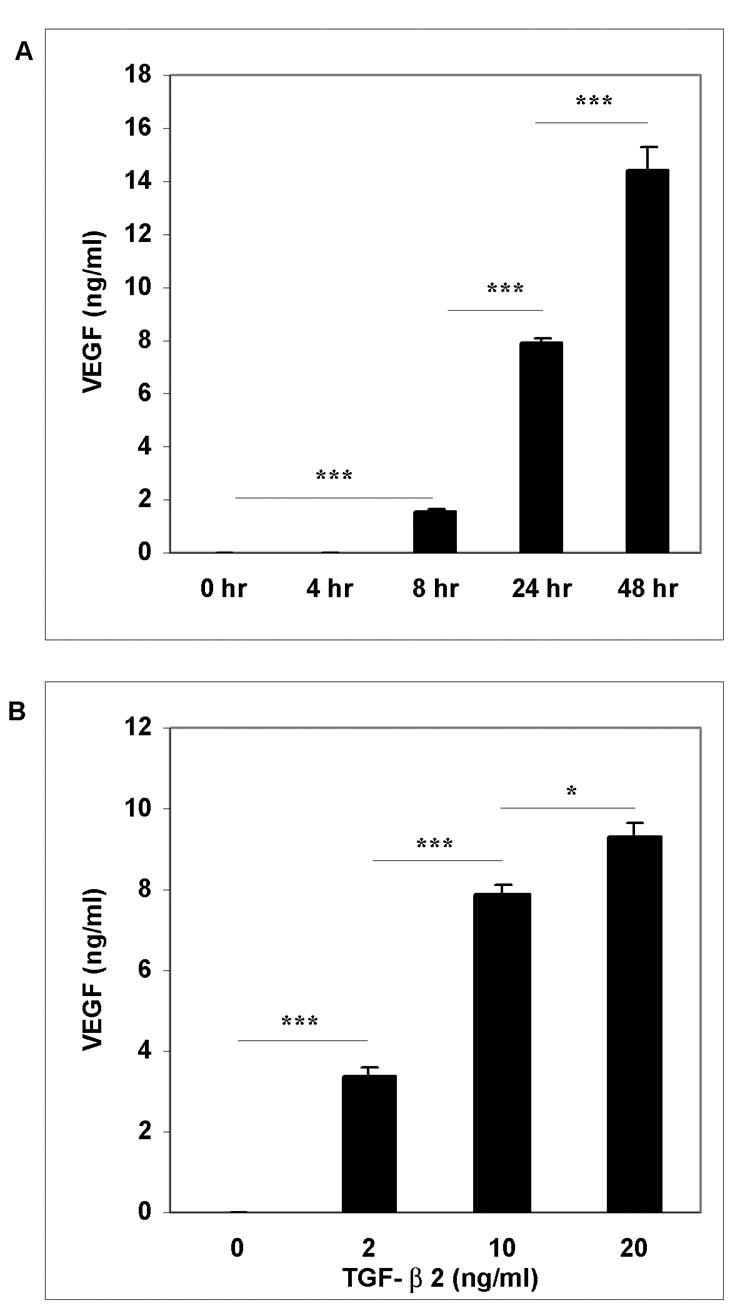

Time-and dose-dependent effects of TGF-β2 on hRPE VEGF mRNA synthesis and protein secretion. A, hRPE cells were incubated with TGF-β2 (10 ng/ ml) for 0, 4, 8, 24, 48 hr. B, hRPE cells were stimulated in the media containing various concentrations of TGF-β2 (0 to 20 ng/ml) for 24 hr. The conditioned media were assayed for VEGF using ELISA. The values represent means ± SEM (n=3). C, qRT-PCR analysis of time-dependent VEGF mRNA expression that was induced by TGF-β2 in hRPE cells. The hRPE cells were incubated with TGF-β2 (10 ng/ ml) for 0, 1.5, 3, 7, 24, 36 hr. The total RNA was isolated, reverse transcribed, and subjected to RT PCR. Relative expression units of samples were calculated by their threshold cycle differences from control level at 0 hr, which was arbitrarily assigned as value of 100. These representative data are from two independent experiments.
Figure 2.

Effects of selected inhibitors on hRPE VEGF secretion that were induced by TGF-β2. The hRPE cells were incubated with TGF-β2 (10 ng/ml) for 24 hr in the presence or absence of cycloheximide (CHX) (5μg/ml), anti-TGF-β2 antibody (10μg/ml), cyclosporine A (CSA30, CSA at 30 ng/ml and CSA3, CSA at 3ng/ ml), and dexamethasone (Dex 1, 'Dex at 1μM, 'Dex 0.1, Dex at 0.1μM). The unstimulated hRPE cells were used as control (Con). The conditioned media were assayed for VEGF using ELISA. The values shown represent means ± S.E.M. (n=3). The significance of increases in VEGF secretion is presented by p values (*p<0.05, **p<0.01, ***p<0.001) as compared to cells stimulated by TGF-β2 (10 ng/ ml) alone.
To determine if the stimulation of VEGF secretion by TGF-β2 is at transcriptional and/or translational levels, steady-state VEGF mRNA was quantified by real-time PCR. As seen in Fig. 1C, TGF-β2-induced VEGF mRNA level was barely detectable up to 1.5 hr after the stimulation, and the induction reached peak levels at 3 hr after the stimulation with expression level about 6-fold higher than the basal. Following peak induction, the levels of induced VEGF mRNA dropped slightly, but remained significantly above the basal for at least 36 hr.
3.2. Suppression of TGF-β2-induced VEGF expression by anti-inflammatory agents
Dexamethasone is widely used as an immunosuppressive drug and cyclosporin A as an immunomodulating agent. Both suppress VEGF production and exhibit anti-angiogenic properties (Renner et al., 2002; Cho et al., 2002). Therefore, we hypothesized that these agents might also suppress TGF-β2-induced VEGF expression in hRPE cells. Consistent with previous observations that dexamethasone suppresses TGF-β-stimulated VEGF production in pituitary cells (Renner et al., 2002) and cyclosporin A in rheumatoid synovial fibroblasts (Cho et al., 2002), we observed that both dexamethasone (1 μM) and cyclosporin A (30 ng/ml) blocked TGF-β2 induced-hRPE VEGF secretion by 50% and 100%, respectively (Fig. 2).
3.3. Synergistic induction of VEGF expression by TGF-β2 in combination with TNF-α and bFGF
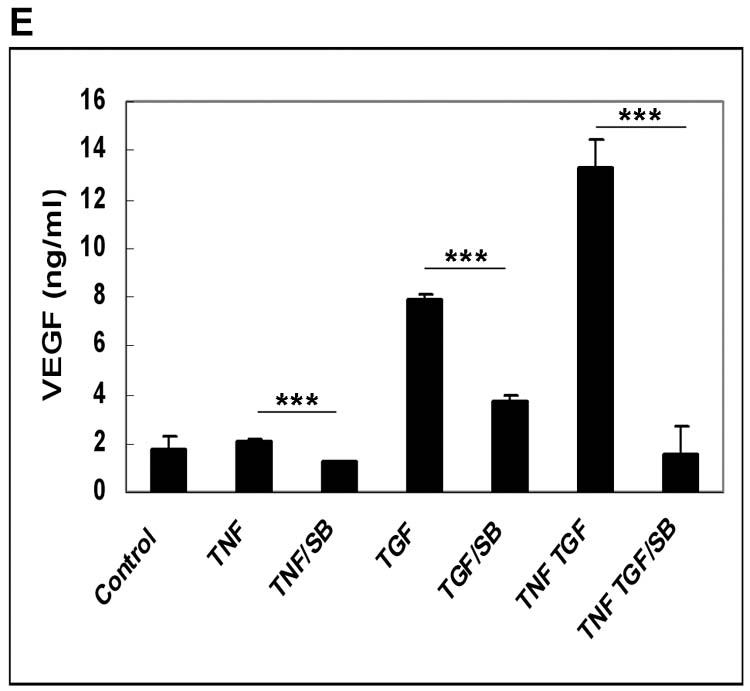
Inflammatory mediators and angiogenic cytokines are known to co-exist during retinal and choroidal neovascularization. We first evaluated effects on hRPE VEGF secretion by individual proinflammatory factors, IL-1β, TNF-α, and angiogenic factors, PDGF, bFGF, as well as lipopolysaccharide (LPS). IL-1β at 2 ng/ml and TNF-α at 20 ng/ml induced small but statistically significant increases in hRPE VEGF production (Fig. 3A). PDGF and bFGF at 10ng/ml only slightly stimulated hRPE VEGF production, and LPS at concentrations as high as 1 μg/ml was ineffective (Fig. 3 A, B). To assess the role of TGF-β2 in combination with these proangiogenic factors, we examined VEGF induction by TGF-β2 in combination with each of these factors. Of these factors only TNF-α and bFGF synergized with TGF-β2 inducing hRPE VEGF. As shown in Fig. 3B, both TGF-β2 plus TNF-α and TGF-β2 plus bFGF caused increases in hRPE VEGF protein production that were higher than the sum of each factor alone. As measured by quantitative real-time PCR, TGF-β2, TNF-α and bFGF alone increased hRPE VEGF mRNA synthesis by 14-, 2.7- and 3.2-fold, respectively, whereas combinations of TGF-β2 plus TNF-α and TGF-β2 plus bFGF increased hRPE VEGF mRNA by 29- and 22-fold, respectively (Fig. 3C).
Figure 3.
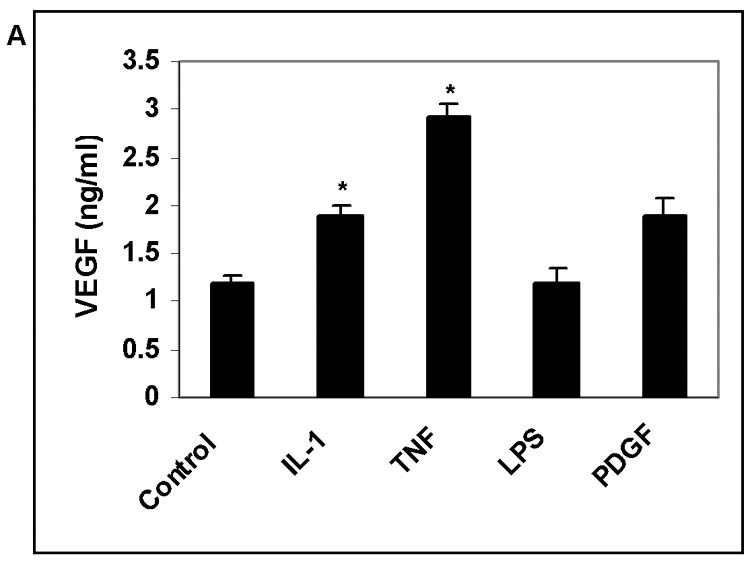
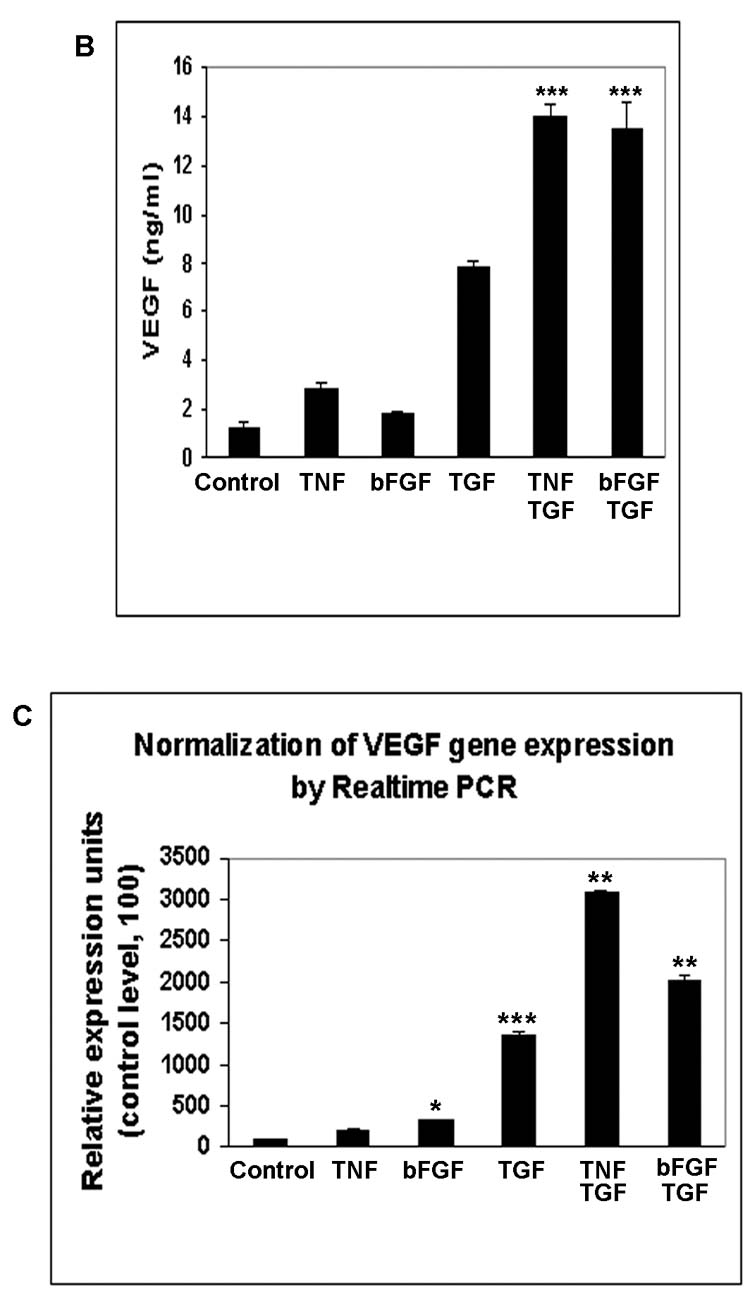
The effects of proinflammatory cytokines and relative growth factors on hRPE VEGF mRNA synthesis and protein secretion. The hRPE cells were incubated with IL-1β (IL-1), TNF-α (TNF), LPS, PDGF (A), and TNF-α, bFGF, TGF-β2 (TGF), TGF-β2 plus TNF-α or bFGF (B) for 24 hr. The unstimulated hRPE cells were used as control. The conditioned media were assayed for VEGF using ELISA. Values represent means ± SEM (n=3) in ELISA assay. The significance of increases in VEGF secretion is presented by p values (*p<0.05, **p<0.01, ***p<0.001) as compared to unstimulated cells (A) or compared to stimulated hRPE cells by TGF-β2 (10 ng/ml) alone (B). C, Quantitative RT-PCR analysis of the hRPE VEGF mRNA expression induced by TGF-β2 or co-administration of TGF-β2 with TNF-α or bFGF. The cells were incubated with TGF-β2 (10 ng/ml), TNF-α (TNF) (20 ng/ ml), bFGF (10 ng/ ml), TGF-β2 plus TNF-α and TGF-β2 plus bFGF for 3 hr. The total RNA was isolated, reverse transcribed, and subjected to real time PCR. The relative expression units were calculated by their cycle differences from control (unstimulated cells). The control level of VEGF expression was arbitrarily assigned as a value of 100. The data shown were means from two independent experiments.
The synergistic increase in VEGF mRNA production by co-administration of TGF-β2 and TNF-α might have resulted from increased VEGF mRNA transcription and/or increased VEGF mRNA stability. To address the latter possibility we measured VEGF mRNA stability following stimulation by TGF-β2 alone and in combination with TNF-α. In the presence or absence of TNF-α, the half-live of the TGF-β2-induced hRPE VEGF mRNA were close, measuring 5.2 and 4.5 hr, respectively (data not shown). These results suggest that the synergy between TGF-β2 and TNF-α was mainly at transcription level and not the result of increased VEGF mRNA stability.
3.4. Multiple signal transduction pathways involved in hRPE VEGF mRNA expression.
A valuable method used to detect the signal pathways responsible for inducing the expression of a gene is to examine effects of the inhibitors specific for individual signal pathway or molecule (Table 1). Activation of mitogen-activated protein kinase (MAPK) pathways [signal-regulated kinase (ERK), p38 and c-Jun NH2-terminal kinase (JNK)], nuclear factor-κB (NF-κB), and phosphatidylinositol 3-kinase (PI3K) pathways are known to be involved in hRPE chemokine mRNA expression (Bian et al., 2003; 2004). Therefore, we investigated whether these pathways are also required for TGF-β-induced hRPE VEGF mRNA expression. Ly294002 (50, 10 μM), a PI3K inhibitor, U0126 (20, 10, 2 μM), an inhibitor of MEK (the signal molecule upstream from ERK), SB202190 (30, 10, 3 μM), a P38 inhibitor, Sp600125 (20 μM), a JNK inhibitor, and CAPE (25, 10, 5μg/μl), a NF-κB inhibitor, were added to human RPE cells before and during 24 hr stimulation with 10 ng/ml TGF-β2. ELISA assays of the culture media showed that the TGF-β-induced human RPE VEGF protein production was strongly reduced by Ly294002 at 50 μM, U0126 at 10 μM, SB202190 at 10 μM and CAPE at 25 μg/ml by 68, 78, 70, and 98%, respectively, and the inhibition was dose dependent (Fig. 4 A, B, C). In addition, a moderate inhibition (30%) was observed with Sp600125 (Fig. 4 D). These data suggest that PI3K, NF-κB and MAPK pathways are involved in regulation of VEGF expression by TGF-β2. The TGF-β2-stimulated human RPE VEGF protein production was also very sensitive to genistein, a protein-tyrosine kinase (PTK) inhibitor, at both 12.5 and 25μg/ml, Ro318220, a PKC inhibitor, at 10 μM, N-acetyl-cysteine (Nac) and diphenyleneiodonium chloride (DPI), both inhibitors of reactive oxygen species, exhibiting inhibition by 60 to 70, 90, 85 and 95%, respectively. Ro 318220 at 5 and 10 μM, and genistein at 12.5 and 25 μg/ml showed dose-dependent inhibition (Fig. 4 B, C and D). These results suggest that activation of PTK, PKC, and reactive oxygen species are also required for up-regulation of VEGF expression by TGF-β2. By contrast, AG490, a specific Janus kinase (jak)2 inhibitor, was ineffective, although this pathway has been shown involved in MCP-1 expression in hRPE cells in our previous study (Fig. 4 D) (Bian et al. 2001).
Figure 4.
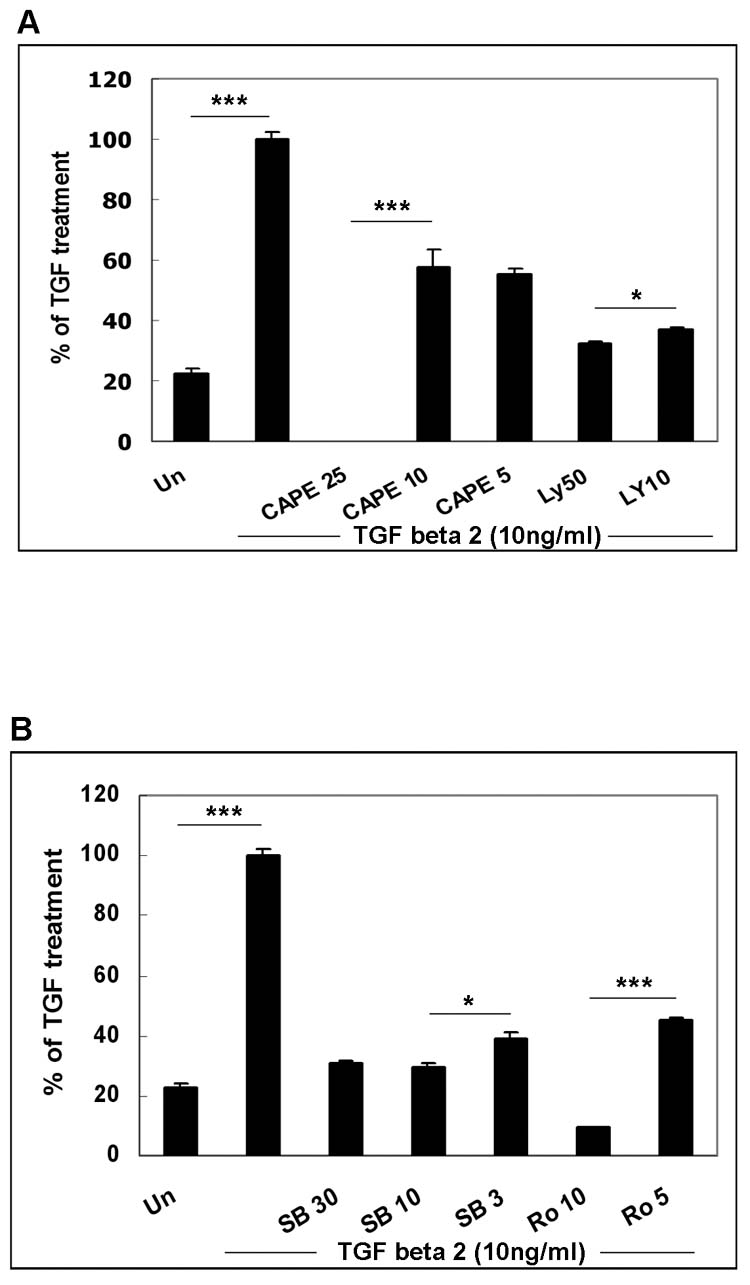
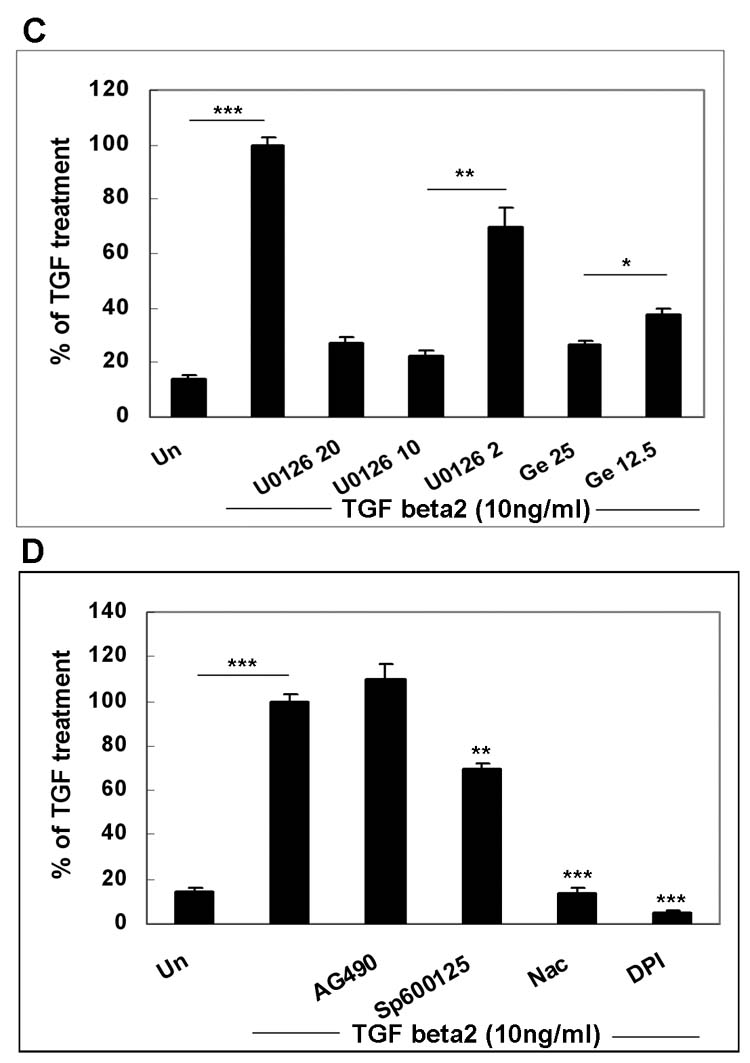
Inhibition of TGF-β2- and TGF-β2 plus TNFα-stimulated VEGF production. The cells were pretreated with CAPE (25, 10, 5 μg/ml) and Ly294002 (Ly) (50,10μM) (Fig. 4A), SB202190 (SB) (30, 10, 3μM) and Ro318220 (Ro) (10, 5μM) (Fig. 4B, E), U0126 (20, 10, 2μM) and genistein (Ge) (25, 12.5 μg/ml) (Fig. 4C), Sp600125 (20 μM), AG490 (50μM), Nac (25mM), and DPI (25 μM) (Fig. 4D), and then stimulated with or without TGF-β (10 ng/ ml), TNF-α (20ng/ml), TGF-β 2 plus TNF-α (Fig. 4) for 24 hr in the continued presence or absence of the inhibitors. Secretion of VEGF was determined by ELISA of the medium. Media from unstimulated hRPE cell cultures were used as control (Un or control). Values represent are means ± SEM (n=3).
To further investigate the role of p38 and PKC signaling in hRPE VEGF mRNA expression, activation of p38 and PKC proteins (phosphorylation) was assessed by Western blotting using antibodies against the active (phospho) forms of p38 and PKC proteins. As shown in Fig. 5A and Fig 6B, TGF-β2 (10 and 20 ng/ ml) induced dose- and time-dependent increases in phosphorylation of p38 protein and PKC up to 3 hr. The phosphorylation of p38 and PKC was detected at 0.5 hr and peaked at 1 to 1.5 and 1 hr for p38 and PKC, respectively. At a high TGF-β2 concentration (20ng/ml), the levels of phosphorylated p38 showed further increases, while only slight additional increase was observed for phosphorylated PKC. Moreover, the enhanced phosphorylation decreased at 3 hr after the stimulation, indicating that activation of p38 and PKC was transient. Consistent with synergistic induction of hRPE VEGF, TGF-β2 in combination with cytokines bFGF or TNF-α also synergized p38 phosphorylation with increases higher than the sum by each factor alone (Fig. 5B). While p38 inhibitor SB202190 partly (around half) inhibited VEGF production when stimulated by TNF or TGF-β2 alone, it inhibited almost 90% of VEGF protein production when stimulated by TNF plus TGF-β2 (Fig. 4 E). In addition, Ro318220, an inhibitor of the four PKC isozymes, PKC β, PKCα, PKCγ and PKCε, blocked not only VEGF protein production, but also VEGF mRNA synthesis (Fig. 6A). Multiple isozymes have been identified in RPE cells (Wood et al, 1998). Western blots using antibody against phosphorylated PKC β1 revealed that TGF-β2 induced PKC β1 phosphorylation, and the induction of which was blocked by the PKC inhibitor Ro318220 (Fig. 6 C).
Figure 5.
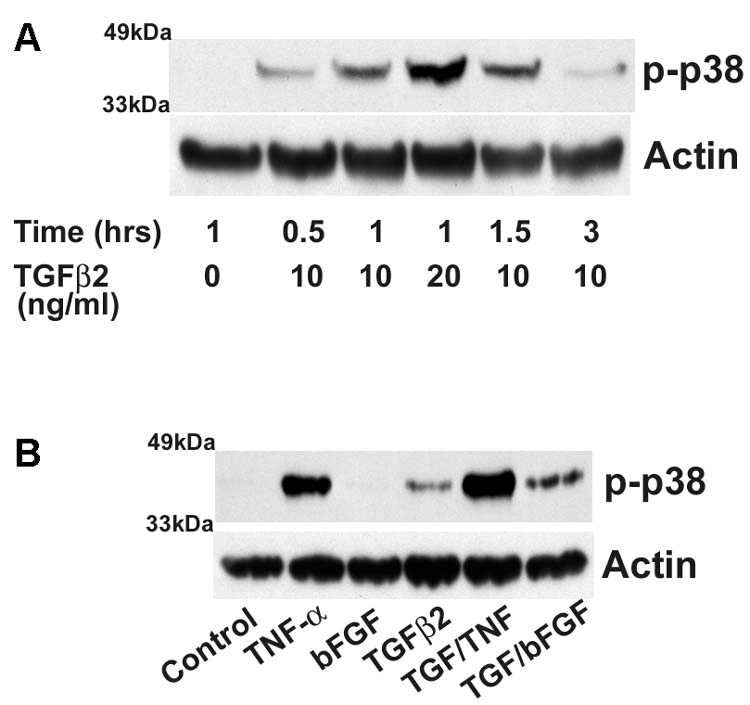
Western blotting analysis of p38 phosphorylation by TGF-β2, TGF-β2 plus TNF-α, and TGF-β2 plus bFGF. The whole-cell lysates were obtained from the hRPE cell cultures unstimulated (control), exposed to TGF-β2 (10 or 20 ng/ ml) for 0.5, 1, 1.5, 3 hr. (A), and incubated with TGF-β2 (10 ng/ ml), TNF-α (TNF) (20 ng/ ml), bFGF (10 ng/ml), TGF-β2 plus TNF-α or TGF-β2 plus bFGF for 30 min (B). The levels of p38 activation were detected by probing with anti-phosphorylated p38 antibody. Anti-actin antibody was used as the control probe to monitor equal lysate loading in each lane.
Figure 6.

Involvement of PKC in TGF-β2-induced VEGF expression in hRPE cells. A, Quantitative RT-PCR analysis of hRPE VEGF mRNA expression that was induced by TGF-β2 in the presence of PKC inhibitor Ro31−8220. The cells were exposed to TGF-β2 (10 ng/ ml) alone or pretreated and co-incubated with Ro31−8220 (10 μM) for 3 hr. The total RNA was isolated, reverse transcribed, and subjected to real time PCR. The relative expression under various conditions was calculated by the threshold cycle differences from control (unstimulated cells), the level of which was arbitrarily assigned as a value of 100. The data were means from two independent experiments. B, Western blotting analysis of phosphorylation of PKC. C, Western blotting analysis of phosphorylation of PKCβ1. The whole-cell lysates were made from the unstimulated hRPE cells (Control), the cells exposed to TGF-β2 (10 or 20 ng/ ml) for 0.5, 1, 1.5, 3 hr, and the cells pre-treated with Ro31−8220 (10 μM), and then exposed to TGF-β2 in the presence of the inhibitor for an additional 30 min. The blots were probed with specific antibodies to anti-phosphorylated PKC β1 and phosphorylated PKC to determine the levels of PKC activation. Anti-actin antibody was used as the control probe to monitor equal loading of each lane with cell lysates.
4. Discussion
In this study, we showed that TGF-β2 strongly induced VEGF mRNA expression and protein secretion in hRPE cells, while growth factors bFGF and TNF-α stimulated VEGF very weakly. The latter results were consistent with previous observations in hRPE cells (Nagineni et al., 2003). However, our data also demonstrated that these two angiogenic factors synergized TGF-β2-induced VEGF mRNA expression and protein secretion. Synergies between TGF-β plus TNF-α and TGF-β plus bFGF have been reported for other cellular functions. For example, bFGF synergizes TGF-β2-stimulated cell proliferation of articular chondrocytes (Okazaki et al., 1996). TNF-α also synergizes TGF-β1-induced apoptosis of human umbilical vein endothelial cells (Emmanuel et al., 2002).
Numerous cytokines and growth factors participate in ocular diseases. These factors may derive from the serum as a consequence of blood-ocular barrier breakdown, or from infiltrating leucocytes and resident cells in diseased ocular tissue. Several different cell types are found in active preretinal neovascular membranes and choroidal subretinal membranes, e.g. macrophages, choroidal fibroblasts, RPE cells and vascular endothelial cells. These cells serve as sources of cytokines and growth factors such as VEGF, TNF-α, and bFGF. VEGF plays a pivotal role in both nonproliferative and proliferative retinopathy (Chiarelli et al., 2000b; Katsura et al., 1998). Although LPS, PDGF and bFGF have been shown to stimulate VEGF expression in other cell types (Morozumi et al., 2001; Finkenzeller et al., 1997; Hata et al., 1999), they have little function on hRPE VEGF expression. Several angiogenic factors have been known co-existing within vitreous, retina, preretinal and choroidal neovascular membranes. TNF-α has been detected in the vitreous of patients with diabetic retinopathy (Spranger et al., 1995), infiltrating leukocytes, and extracellular matrix of PDR fibrovascular membranes in human eyes (Limb et al., 1996). TNF-α is the major cytokine produced by macrophages which are often present at the sites of active neovascularization (Penfold et al., 1984). In addition to TNF-α, various growth factors are produced at onset of ocular neovascularization and have been detected in vitreous (Sivalingam et al., 1990) and epiretinal membranes (Malecaze et al., 1994; Armstrong et al., 1998; Ambati et al., 1997). Human RPE cells are strongly immunoreactive for aFGF/bFGF and TGF-β in choroidal neovascular membranes of age-related macular degeneration (AMD), but not in normal human eyes (Amin et al., 1994). bFGF is believed to play an important role in the development of choroidal neovascularization (Ogata et al., 1996) and increased in hyperglycemia, suggesting its role in PDR (Nakamura et al., 1993). All these proangiogenic cytokines, TGF-β, bFGF and TNF-α have been shown to be produced by RPE cells and macrophages (Sternfeld et al., 1989; Amin et al., 1994). Our current results showed that bFGF and TNF-α potentiated TGF-β–stimulated hRPE VEGF secretion and may, therefore, be pathophysiologicaly relevant in the neovascularization of retina and choroids.
The RPE-Bruch's membrane-choroid complex contains approximately 10 times more TGF-β than the neurosensory retina. TGF-β2 is the predominant isoform of TGF-β in the neural retina, RPE-choroid and vitreous with β2:β1 ratio of 6:1 in the retina-choroid and 425:1 in vitreous of the monkey eye (Pfeffer et al., 1994). TGF-β2, but not TGF-β1, accumulates over time in conditioned media of first passaged hRPE cultures. TGF-β2 is the isoform detected immunohistochemically in photoreceptors and RPE cells in tissue sections (Pfeffer et al., 1994). Therefore, TGF-β 2 is likely to be the isoform involved in the site-specific retinal and choroidal responses (Pfeffer et al., 1994). In addition, RPE cells produce high levels of TGF-β2, the expression of which is up-regulated by serum-free culture conditions and altered as function of RPE proliferation rate in cultured human RPE cells and monkey eye (Kvanta, 1994, Pfeffer et al., 1994). It is thus possible that RPE cells under metabolic stress in diseased states, such as AMD, may produce high levels of TGF-β2. Sequentially, the enhanced TGF-β2 expression stimulates RPE VEGF production in autocrine and paracrine fashions, triggering angiogenic events.
RPE TGF-β2 expression and TGF-β2 responses have been shown in experimental models of retinal disease. TGF-β2 has been detected in RPE cells in regions of neovascularization during experimental choroidal neovascularization (Ogata et al., 1997). While TGF-β2 promotes neovascularization by stimulating VEGF, TGF-β1 itself has both pro- and anti-angiogenic effects (Bein et al., 2004). TGF-β1 promotes angiogenesis in vivo and endothelial tube formation in vitro, while inhibiting endothelial cell proliferation in vitro. Retinal endothelial cells proliferation has been shown significantly inhibited by TGF-β2 that is released from Müller cells (Eichler et al., 2004). The pro-angiogenic role of TGF-β could be direct and indirect. The direct differential pro-angiogenic effects of TGF-β1 and TGF-β2 may depend on two distinct receptors (Goumans et al., 2002). The possible indirect effects of TGF-β on neovascularization may involve inflammatory cell-induced neovascularization since TGF-β is one of the most potent chemotactic agents for monocytes. Monocytes are required for in vivo neovascularization of many tissues including retinal tissue (Wahl et al., 1987). Furthermore, TGF-β also has other effects by stimulating hRPE phagocytosis (Sheu et al., 1994) and contraction of collagen gels (Choudhury et al., 1997).
Seven VEGF splice variants have been reported by GenBank (NM_001025366, NM_003376, NM_001025367, NM_001025368, NM_001025369, NM_001025370 and NM_0011033756). The primers used in our study are expected to be able to detect all variants except for variant 6 (NM_001025370). However, consistent with previous notion that these primer pairs only produce predominate 424 nucleotide (VEGF165) species (Kanuga et al., 2002). It has been known that there are two isofoms of VEGF165, VEGF165a and VEGF165b. VEGF165a has potent angiogenic properties, whereas VEGF165b has anti-angiogenic properties (Churchill et al., 2006). Therefore, it is interesting to further study which isoform of VEGF165 is induced by TGF-β2 in hRPE cells.
Various signal transduction pathways including TGF-β-specific Smad pathway and pathways such as ERK, p38, PI3K, PKC, NFkB, ROS, JNK and jak3 for TGF-β have been reported either induced or cross-talked with TGF-β signaling in various cell types (Yue & Mulder, 2001; Ten Dijke & Hill, 2004; Massague et al., 2000), but little is known about these pathways in hRPE cells while stimulation by TGF-β2 (Osusky et al., 1994; Nagineni, et al., 2003; Kimoto et al., 2004). The cross-talk of Smad signaling pathway with other signaling pathways remains poorly understood (Kim et al., 2006). Recently, the signal transduction pathways for TGF-β-induced VEGF mRNA expression have been reported in many cell types (Chua et al., 2000; Nagineni et al., 2003; Kim et al., 2002). Upon binding to its receptors, TGF-β activates Smad proteins. The activated Smad proteins form complexes that are translocated to the nucleus and interact with other nuclear molecules to regulate expression of the target genes (Moustakas et al., 2001). We found that treatment of hRPE cells with TGF-β2 resulted in phosphorylation of Smad 2 and 3 (data not shown). However, the synergistic stimulation of VEGF by TGF-β2 plus TNF-α or plus bFGF did not further enhance Smad2/3 phosphorylation, suggesting that other pathways may mediate the synergistic enhancement of TNF-α and bFGF stimulation by TGF-β2.
A series of previous reports have indicated that TGF-β not only actives Smads, but also trigers other signal pathways. For example, MAP kinases can be rapidly activated by TGF-β in a manner that is highly dependent on the cell type and conditions (Massague et al., 2000). In fact, TGF-β/Smad pathway is integrated into a signaling network, a variety of components function at different levels of the pathway. It is unclear if Smad pathway cross interacts with the signal pathways studied by the present study. In fact the downstream effector VEGF by TGF-β2 have been well known upregulated by various signal pathways such as PTK, PKC, PI3K and NF-κB. In this study, p38 appeared to be an important pathway for hRPE VEGF expression. We found that p38 pathway is required for induction of hRPE VEGF expression either by TGF-β2 alone or in combination with TNF-α. These results were consistent with previous observation for TGF-β2-induced expression of type I collagen (COL1) and TGF-β1-induced expression of VEGF (Kimoto et al., 2004; Nagineni et al., 2003) with some important differences. For example, the previous study by Nagineni et al (2003), showed that TGF-β1-induced VEGF expression does not require activation of the ERK MAPK pathway, thus in contrast to our present study. The discrepancies could be a result thatTGF-β1 activates VEGF differently from that by TGF-β2.
Activation of NF-kB or PKC has also been investigated for VEGF induction by TGF-β1 in hRPE cells (Nagineni et al., 2003), however, PKC and NF-κB pathways appear not involved in VEGF expression by TGF-β1. In contrast to this report, we found that activation of NF-kB and PKC is important for TGF-β2 induction of VEGF in hRPE cells, suggesting that TGF-β2 regulates VEGF expression differently from TGF-β1. It should be noted that TGF-β may either increase or decrease NF-κB activity, depending on the cell type (Yue & Mulder, 2001). In addition, we found that ROS and other cytoplasmic signaling proteins, such as PTK and PI3K, are involved in TGF-β2 stimulation of VEGF in hRPE cells. The involvement of ROS, PTK, and PI3K in TGF-β signaling in human RPE cells has been variably reported in other cell types (Jardine et al., 2002; Vinals & Pouyssegur, 2001; Kucich et al., 2001).
Dexamethasone and cyclosporine A are well known anti-inflamma tory agents. The concentrations used in this study were 10−6 M and 10−7 M for dexamethasone, and 3 and 30 ng/ml for cyclosporine A that are comparable to the intraocular concentrations achieved with systemic and topical administration of these drugs (Kurtz et al., 1997). Inhibition of VEGF production by dexamethasone has been reported in mononuclear phagocytes (Li et al., 2002), astrocytes (Yoshida et al., 2002), pituitary tumor cells (Lohrer et al., 2001), brain vascular endothelial cells (Fischer et al., 2001), and chondrocytes (Koedam et al., 2002). The inhibitory effect is at both transcriptional and post-transcriptional levels. Dexamethasone suppresses neovascularization and VEGF expression in the rat cornea (Edelman et al., 1999). Laser-induced choroidal neovascularization, in which enhanced expression of VEGF and its receptors occurs, is inhibited in a dose-dependent fashion by dexamethasone (Edelman and Castro, 2000). Our results also support the therapeutic usefulness of corticosteroids for posterior ocular neovascularization by inhibiting VEGF production. In contrast, inhibition of VEGF expression by cyclosporine A has been less well characterized, but supported by our data. In addition to its immunomodulating effect, cyclosporine A has been demonstrated to have anti-angiogenic ability (Oliver et al., 1995; Hernandez et al., 2001). Specifically, cyclosporine A was found to inhibit human fibroblast VEGF expression induced by TGF-β (Cho et al., 2002). Further investigation of hRPE signal pathways are likely to provide a means of suppressing VEGF and neovascularization in retinal disease by specific drug intervention.
Acknowledgements
This study was supported by NIH Grants EY-09441, EY007003, and Research to Prevent Research to Prevent Blindness, Senior Scientific Investigator Award.
The authors wish to thank Margarete G Hamlett for excellent technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adamis AP, Miller JW, Bernal MT, D'Amico DJ, Folkman J, Yeo TK, Yeo KT. Increased vascular endothelial growth factor levels in the vitreous of eyes with proliferative diabetic retinopathy. Am. J. Ophthalmol. 1994;118:445–450. doi: 10.1016/s0002-9394(14)75794-0. [DOI] [PubMed] [Google Scholar]
- Adamis AP, Shima DT, Yeo KT, Yeo TK, Brown LF, Berse B, D'Amore PA, Folkman J. Synthesis and secretion of vascular permeability factor/vascular endothelial growth factor by human retinal pigment epithelial cells. Biochem. Biophys. Res. Commun. 1993;193:631–638. doi: 10.1006/bbrc.1993.1671. [DOI] [PubMed] [Google Scholar]
- Aiello LP, Avery RL, Arrigg PG, Keyt BA, Jampel HD, Shah ST, Pasquale LR, Thieme H, Iwamoto MA, Park JE, et al. Vascular endothelial growth factor in ocular fluid of patients with diabetic retinopathy and other retinal disorders. N. Engl. J. Med. 1994;331:1480–1487. doi: 10.1056/NEJM199412013312203. [DOI] [PubMed] [Google Scholar]
- Aiello LP, Northrup JM, Keyt BA, Takagi H, Iwamoto MA. Hypoxic regulation of vascular endothelial growth factor in retinal cells. Arch. Ophthalmol. 1995;113:1538–1544. doi: 10.1001/archopht.1995.01100120068012. [DOI] [PubMed] [Google Scholar]
- Ambati J, Chalam KV, Chawla DK, D'Angio CT, Guillet EG, Rose SJ, Vanderlinde RE, Ambati BK. Elevated gamma-aminobutyric acid, glutamate, and vascular endothelial growth factor levels in the vitreous of patients with proliferative diabetic retinopathy. Arch. Ophthalmol. 1997;115:1161–1166. doi: 10.1001/archopht.1997.01100160331011. [DOI] [PubMed] [Google Scholar]
- Amin R, Puklin JE, Frank RN. Growth factor localization in choroidal neovascular membranes of age-related macular degeneration. Invest. Ophthalmol. Vis. Sci. 1994;35:3178–3188. [PubMed] [Google Scholar]
- Armstrong D, Augustin AJ, Spengler R, Al-Jada A, Nickola T, Grus F, Koch F. Detection of vascular endothelial growth factor and tumor necrosis factor alpha in epiretinal membranes of proliferative diabetic retinopathy, proliferative vitreoretinopathy and macular pucker. Ophthalmologica. 1998;212:410–414. doi: 10.1159/000027378. [DOI] [PubMed] [Google Scholar]
- Bein K, Odell-Fiddler ET, Drinane M. Role of TGF-beta1 and JNK signaling in capillary tube patterning. Am. J. Physiol. Cell. Physiol. 2004;287:C1012–C1022. doi: 10.1152/ajpcell.00101.2004. [DOI] [PubMed] [Google Scholar]
- Bian ZM, Elner SG, Yoshida A, Elner VM. Differential involvement of phosphoinositide 3-kinase/Akt in human RPE MCP-1 and IL-8 expression. Invest. Ophthalmol. Vis. Sci. 2004;45:1887–1896. doi: 10.1167/iovs.03-0608. [DOI] [PubMed] [Google Scholar]
- Bian ZM, Elner SG, Yoshida A, Elner VM. Human RPE-monocyte co-culture induces chemokine gene expression through activation of MAPK and NIK cascade. Exp. Eye. Res. 2003;76:573–583. doi: 10.1016/s0014-4835(03)00029-0. [DOI] [PubMed] [Google Scholar]
- Bian ZM, Elner VM, Yoshida A, Kunkel SL, Elner SG. Signaling pathways for glycated human serum albumin-induced IL-8 and MCP-1 secretion in human RPE cells. Invest. Ophthalmol. Vis. Sci. 2001;42:1660–1668. [PubMed] [Google Scholar]
- Chiarelli F, Spagnoli A, Basciani F, Tumini S, Mezzetti A, Cipollone F, Cuccurullo F, Morgese G, Verrotti A. Vascular endothelial growth factor (VEGF) in children, adolescents and young adults with Type 1 diabetes mellitus: relation to glycaemic control and microvascular complications. Diabet. Med. 2000 a;17:650–656. doi: 10.1046/j.1464-5491.2000.00350.x. [DOI] [PubMed] [Google Scholar]
- Chiarelli F, Santilli F, Mohn A. Role of growth factors in the development of diabetic complications. Horm. Res. 2000 b;53:53–67. doi: 10.1159/000023515. [DOI] [PubMed] [Google Scholar]
- Churhill AJ, Carter JG, Lovell HC, Ramsden C, Turner SJ, Yeung A, Escardo J, Atan D. VEGF polymorphisms are associated with neovascular age-related macular degeneration. Hum Mol Genet. 2006;15:2953–2961. doi: 10.1093/hmg/ddl238. [DOI] [PubMed] [Google Scholar]
- Choudhury P, Chen W, Hunt RC. Production of platelet-derived growth factor by interleukin-1 beta and transforming growth factor-beta-stimulated retinal pigment epithelial cells leads to contraction of collagen gels. Invest. Ophthalmol. Vis. Sci. 1997;38:824–833. [PubMed] [Google Scholar]
- Cho ML, Cho CS, Min SY, Kim SH, Lee SS, Kim WU, Min DJ, Min JK, Youn J, Hwang SY, Park SH, Kim HY. Cyclosporine inhibition of vascular endothelial growth factor production in rheumatoid synovial fibroblasts. Arthritis Rheum. 2002;46:1202–1209. doi: 10.1002/art.10215. [DOI] [PubMed] [Google Scholar]
- Chua CC, Hamdy RC, Chua BH. Mechanism of transforming growth factor-beta1-induced expression of vascular endothelial growth factor in murine osteoblastic MC3T3-E1 cells. Biochim. Biophys. Acta. 2000;1497:69–76. doi: 10.1016/s0167-4889(00)00040-9. [DOI] [PubMed] [Google Scholar]
- Connolly DT, Heuvelman DM, Nelson R, Olander JV, Eppley BL, Delfino JJ, Siegel NR, Leimgruber RM, Feder J. Tumor vascular permeability factor stimulates endothelial cell growth and angiogenesis. J. Clin. Invest. 1989;84:1470–1478. doi: 10.1172/JCI114322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhury P, Chen W, Hunt RC. Production of platelet-derived growth factor by interleukin-1 beta and transforming growth factor-beta-stimulated retinal pigment epithelial cells leads to contraction of collagen gels. Invest. Ophthalmol. Vis. Sci. 1997;38:824–833. [PubMed] [Google Scholar]
- Edelman JL, Castro MR. Quantitative image analysis of laser-induced choroidal neovascularization in rat. Exp. Eye. Res. 2000;71:523–533. doi: 10.1006/exer.2000.0907. [DOI] [PubMed] [Google Scholar]
- Edelman JL, Castro MR, Wen Y. Lipocortin V may function as a signaling protein for vascular endothelial growth factor receptor-2/Flk-1. Biochem. Biophys. Res. Commun. 1999;258:713–721. doi: 10.1006/bbrc.1999.0678. [DOI] [PubMed] [Google Scholar]
- Eichler W, Yafai Y, Wiedemann P, Reichenbach A. Angiogenesis-related factors derived from retinal glial (Muller) cells in hypoxia. Neuroreport. 2004;15:1633–1637. doi: 10.1097/01.wnr.0000133071.00786.a4. [DOI] [PubMed] [Google Scholar]
- Elner SG, Elner VM, Pavilack MA, Todd RF, 3rd, Mayo-Bond L, Franklin WA, Strieter RM, Kunkel SL. Huber AR. Modulation and function of intercellular adhesion molecule-1 (CD54) on human retinal pigment epithelial cells. Lab. Invest. 1992;66:200–211. [PubMed] [Google Scholar]
- Emmanuel C, Foo E, Medbury HJ, Matthews J, Comis A, Zoellner H. Synergistic induction of apoptosis in human endothelial cells by tumour necrosis factor-alpha and transforming growth factor-beta. Cytokine. 2002;18:237–241. doi: 10.1006/cyto.2002.1042. [DOI] [PubMed] [Google Scholar]
- Finkenzeller G, Sparacio A, Technau A, Marme D, Siemeister G. Sp1 recognition sites in the proximal promoter of the human vascular endothelial growth factor gene are essential for platelet-derived growth factor-induced gene expression. Oncogene. 1997;15:669–676. doi: 10.1038/sj.onc.1201219. [DOI] [PubMed] [Google Scholar]
- Fischer S, Renz D, Schaper W, Karliczek GF. In vitro effects of dexamethasone on hypoxia-induced hyperpermeability and expression of vascular endothelial growth factor. Eur J Pharmacol. 2001;411:231–243. doi: 10.1016/s0014-2999(00)00915-8. [DOI] [PubMed] [Google Scholar]
- Goumans MJ, Valdimarsdottir G, Itoh S, Rosendahl A, Sideras P, ten Dijke P. Balancing the activation state of the endothelium via two distinct TGF-beta type I receptors. EMBO. J. 2002;21:1743–1753. doi: 10.1093/emboj/21.7.1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hata Y, Rook SL, Aiello LP. Basic fibroblast growth factor induces expression of VEGF receptor KDR through a protein kinase C and p44/p42 mitogen-activated protein kinase-dependent pathway. Diabetes. 1999;48:1145–1155. doi: 10.2337/diabetes.48.5.1145. [DOI] [PubMed] [Google Scholar]
- Hernandez GL, Volpert OV, Iniguez MA, Lorenzo E, Martinez-Martinez S, Grau R, Fresno M, Redondo JM. Selective inhibition of vascular endothelial growth factor-mediated angiogenesis by cyclosporin A: roles of the nuclear factor of activated T cells and cyclooxygenase 2. J. Exp. Med. 2001;193:607–620. doi: 10.1084/jem.193.5.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houck KA, Leung DW, Rowland AM, Winer J, Ferrara N. Dual regulation of vascular endothelial growth factor bioavailability by genetic and proteolytic mechanisms. J. Biol. Chem. 1992;267:26031–26037. [PubMed] [Google Scholar]
- Jardine H, MacNee W, Donaldson K, Rahman I. Molecular mechanism of transforming growth factor (TGF)-beta1-induced glutathione depletion in alveolar epithelial cells. Involvement of AP-1/ARE and Fra-1. J. Biol. Chem. 2002;277:21158–21166. doi: 10.1074/jbc.M112145200. [DOI] [PubMed] [Google Scholar]
- Kanuga N, Winton HL, Beauchene L, Koman A, Zerbib A, Halford S, Couraud PO, Keegan D, Coffey P, Lund RD, Adamson P, Greenwood J. Characterization of genetically modified human retinal pigment epithelial cells developed for in vitro and transplantation studies. Invest. Ophthalmol. Vis. Sci. 2002;43:546–555. [PubMed] [Google Scholar]
- Katsura Y, Okano T, Noritake M, Kosano H, Nishigori H, Kado S, Matsuoka T. Hepatocyte growth factor in vitreous fluid of patients with proliferative diabetic retinopathy and other retinal disorders. Diabetes. Care. 1998;21:1759–1763. doi: 10.2337/diacare.21.10.1759. [DOI] [PubMed] [Google Scholar]
- Keck PJ, Hauser SD, Krivi G, Sanzo K, Warren T, Feder J, Connolly DT. Vascular permeability factor, an endothelial cell mitogen related to PDGF. Science. 1989;246:1309–1312. doi: 10.1126/science.2479987. [DOI] [PubMed] [Google Scholar]
- Kim HH, Lee SE, Chung WJ, Choi Y, Kwack K, Kim SW, Kim MS, Park H, Lee ZH. Stabilization of hypoxia-inducible factor-1alpha is involved in the hypoxic stimuli-induced expression of vascular endothelial growth factor in osteoblastic cells. Cytokine. 2002;17:14–27. doi: 10.1006/cyto.2001.0985. [DOI] [PubMed] [Google Scholar]
- Kim YK, Bae GU, Kang JK, Park JW, Lee EK, Lee HY, Choi WS, Lee HW, Han JW. Cooperation of H2O2-mediated ERK activation with Smad pathway in TGF-beta1 induction of p21WAF1/Cip1. Cell Signalling. 2006;18:236–243. doi: 10.1016/j.cellsig.2005.04.008. [DOI] [PubMed] [Google Scholar]
- Kimoto K, Nakatsuka K, Matsuo N, Yoshioka H. p38 MAPK mediates the expression of type I collagen induced by TGF-beta 2 in human retinal pigment epithelial cells ARPE-19. Invest. Ophthalmol. Vis. Sci. 2004;45:2431–2437. doi: 10.1167/iovs.03-1276. [DOI] [PubMed] [Google Scholar]
- Koedam JA, Smink JJ, van Buul-Offers SC. Glucocorticoids inhibit vascular endothelial growth factor expression in growth plate chondrocytes. Mol. Cell. Endocrinol. 2002;197:35–44. doi: 10.1016/s0303-7207(02)00276-9. [DOI] [PubMed] [Google Scholar]
- Kucich U, Rosenbloom JC, Herrick DJ, Abrams WR, Hamilton AD, Sebti SM, Rosenbloom J. Signaling events required for transforming growth factor-beta stimulation of connective tissue growth factor expression by cultured human lung fibroblasts. Arch Biochem Biophys. 2001;395:103–112. doi: 10.1006/abbi.2001.2571. [DOI] [PubMed] [Google Scholar]
- Kurtz RM, Elner VM, Bian ZM, Striete RM, Kunkel SL, Elner SG. Dexamethasone and cyclosporin A modulation of human retinal pigment epithelial cell monocyte chemotactic protein-1 and interleukin-8. Invest. Ophthalmol. Vis. Sci. 1997;38:436–445. [PubMed] [Google Scholar]
- Kvanta A. Expression and secretion of transforming growth factor-beta in transformed and nontransformed retinal pigment epithelial cells. Ophthalmic. Res. 1994;26:361–367. doi: 10.1159/000267502. [DOI] [PubMed] [Google Scholar]
- Li YH, Brauner A, Jensen JS, Tullus K. Induction of human macrophage vascular endothelial growth factor and intercellular adhesion molecule-1 by Ureaplasma urealyticum and downregulation by steroids. Biol. Neonate. 2002;82:22–28. doi: 10.1159/000064148. [DOI] [PubMed] [Google Scholar]
- Limb GA, Chignell AH, Green W, LeRoy F, Dumonde DC. Distribution of TNF alpha and its reactive vascular adhesion molecules in fibrovascular membranes of proliferative diabetic retinopathy. Br J Ophthalmol. 1996;80:168–173. doi: 10.1136/bjo.80.2.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohrer P, Gloddek J, Hopfner U, Losa M, Uhl E, Pagotto U, Stalla GK, Renner U. Vascular endothelial growth factor production and regulation in rodent and human pituitary tumor cells in vitro. Neuroendocrinology. 2001;74:95–105. doi: 10.1159/000054675. [DOI] [PubMed] [Google Scholar]
- Lutty GA, McLeod DS, Merges C, Diggs A, Plouet J. Localization of vascular endothelial growth factor in human retina and choroid. Arch. Ophthalmol. 1996;114:971–977. doi: 10.1001/archopht.1996.01100140179011. [DOI] [PubMed] [Google Scholar]
- Malecaze F, Clamens S, Simorre-Pinatel V, Mathis A, Chollet P, Favard C, Bayard F, Plouet J. Detection of vascular endothelial growth factor messenger RNA and vascular endothelial growth factor-like activity in proliferative diabetic retinopathy. Arch. Ophthalmol. 1994;112:1476–1482. doi: 10.1001/archopht.1994.01090230090028. [DOI] [PubMed] [Google Scholar]
- Merwin JR, Newman W, Beall LD, Tucker A, Madri J. Vascular cells respond differentially to transforming growth factors beta 1 and beta 2 in vitro. Am. J. Pathol. 1991a;138:37–51. [PMC free article] [PubMed] [Google Scholar]
- Merwin JR, Roberts A, Kondaiah P, Tucker A, Madri J. Vascular cell responses to TGF-beta 3 mimic those of TGF-beta 1 in vitro. Growth Factors. 1991b;5:149–158. doi: 10.3109/08977199109000279. [DOI] [PubMed] [Google Scholar]
- Miller JW, Adamis AP, Shima DT, D'Amore PA, Moulton RS, O'Reilly MS, Folkman J, Dvorak HF, Brown LF, Berse B. Vascular endothelial growth factor/vascular permeability factor is temporally and spatially correlated with ocular angiogenesis in a primate model. Am. J. Pathol. 1994;145:574–584. [PMC free article] [PubMed] [Google Scholar]
- Massague J, blain SW, lo RS. TGFβ signaling in growth control, cancer, and heritable disorders. Cell. 2000;103:295–309. doi: 10.1016/s0092-8674(00)00121-5. [DOI] [PubMed] [Google Scholar]
- Morozumi T, Kubota T, Sugita N, Ohsawa Y, Yamazaki K, Yoshie H. Elevated mRNA expression for supervillin and vascular endothelial growth factor in human neutrophils stimulated with lipopolysaccharide from Porphyromonas gingivalis. J. Periodontal. Res. 2001;36:160–168. doi: 10.1034/j.1600-0765.2001.360304.x. [DOI] [PubMed] [Google Scholar]
- Moustakas A, Souchelnytskyi S, Heldin CH. Smad regulation in TGF-beta signal transduction. J.Cell. Sci. 2001;114(Pt 24):4359–4369. doi: 10.1242/jcs.114.24.4359. [DOI] [PubMed] [Google Scholar]
- Nagineni CN, Samuel W, Nagineni S, Pardhasaradhi K, Wiggert B, Detrick B, Hooks JJ. Transforming growth factor-beta induces expression of vascular endothelial growth factor in human retinal pigment epithelial cells: involvement of mitogen-activated protein kinases. J. Cell. Physiol. 2003;197:453–462. doi: 10.1002/jcp.10378. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Fukui M, Ebihara I, Osada S, Nagaoka I, Tomino Y, Koide H. mRNA expression of growth factors in glomeruli from diabetic rats. Diabetes. 1993;42:450–456. doi: 10.2337/diab.42.3.450. [DOI] [PubMed] [Google Scholar]
- Ogata N, Matsushima M, Takada Y, Tobe T, Takahashi K, Yi X, Yamamoto C, Yamada H, Uyama M. Expression of basic fibroblast growth factor mRNA in developing choroidal neovascularization. Curr Eye Res. 1996;15:1008–1018. doi: 10.3109/02713689609017649. [DOI] [PubMed] [Google Scholar]
- Ogata N, Yamamoto C, Miyashiro M, Yamada H, Matsushima M, Uyama M. Expression of transforming growth factor-beta mRNA in experimental choroidal neovascularization. Curr. Eye. Res. 1997;16:9–18. doi: 10.1076/ceyr.16.1.9.5121. [DOI] [PubMed] [Google Scholar]
- Okazaki R, Sakai A, Nakamura T, Kunugita N, Norimura T, Suzuki K. Effects of transforming growth factor-beta s and basic fibroblast growth factor on articular chondrocytes obtained from immobilised rabbit knees. Ann. Rheumatic Diseases. 1996;55:181–186. doi: 10.1136/ard.55.3.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver SJ, Cheng TP, Banquerigo ML, Brahn E. Suppression of collagen-induced arthritis by an angiogenesis inhibitor, AGM-1470, in combination with cyclosporin: reduction of vascular endothelial growth factor (VEGF) Cell. Immunol. 1995;166:196–206. doi: 10.1006/cimm.1995.9978. [DOI] [PubMed] [Google Scholar]
- Osusky R, Soriano D, Ye J, Ryan SJ. Cytokine effect on fibronectin release by retinal pigment epithelial cells. Curr. Eye. Res. 1994;13:569–574. doi: 10.3109/02713689408999890. [DOI] [PubMed] [Google Scholar]
- Penfold P, Killingsworth M, Sarks S. An ultrastructural study of the role of leucocytes and fibroblasts in the breakdown of Bruch's membrane. Aust. J. Ophthalmol. 1984;12:23–31. [PubMed] [Google Scholar]
- Pfeffer BA, Flanders KC, Guerin CJ, Danielpour D, Anderson DH. Transforming growth factor beta 2 is the predominant isoform in the neural retina, retinal pigment epithelium-choroid and vitreous of the monkey eye. Exp. Eye. Res. 1994;59:323–333. doi: 10.1006/exer.1994.1114. [DOI] [PubMed] [Google Scholar]
- Renner U, Lohrer P, Schaaf L, Feirer M, Schmitt K, Onofri C, Arzt E, Stalla GK. Transforming growth factor-beta stimulates vascular endothelial growth factor production by folliculostellate pituitary cells. Endocrinology. 2002;143:3759–3765. doi: 10.1210/en.2002-220283. [DOI] [PubMed] [Google Scholar]
- Sivalingam A, Kenney J, Brown GC, Benson WE, Donoso L. Basic fibroblast growth factor levels in the vitreous of patients with proliferative diabetic retinopathy. Arch. Ophthalmol. 1990;108:869–872. doi: 10.1001/archopht.1990.01070080113046. [DOI] [PubMed] [Google Scholar]
- Sheu SJ, Sakamoto T, Osusky R, Wang HM, Ogden TE, Ryan SJ, Hinton DR, Gopalakrishna R. Transforming growth factor-beta regulates human retinal pigment epithelial cell phagocytosis by influencing a protein kinase C-dependent pathway. Graefes. Arch. Clin. Exp. Ophthalmol. 1994;232:695–701. doi: 10.1007/BF00171387. [DOI] [PubMed] [Google Scholar]
- Sporn MB, Roberts AB. Transforming growth factor-beta: recent progress and new challenges. J. Cell. Biol. 1992;119:1017–1021. doi: 10.1083/jcb.119.5.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spranger J, Meyer-Schwickerath R, Klein M, Schatz H, Pfeiffer A. [TNF-alpha level in the vitreous body. Increase in neovascular eye diseases and proliferative diabetic retinopathy] Med Klin (Munich) 1995;90:134–137. [PubMed] [Google Scholar]
- Sternfeld MD, Robertson JE, Shiple GD, Tsai J, Rosenbaum JT. Cultured human retinal pigment epithelial cells express basic fibroblast growth factor and its receptor. Curr. Eye. Res. 1989;8:1029–1037. doi: 10.3109/02713688908997395. [DOI] [PubMed] [Google Scholar]
- Ten Dijke P, Hill CS. New insights into TGF-beta-Smad signalling. Trends. Biochem. Sci. 2004;29:265–273. doi: 10.1016/j.tibs.2004.03.008. [DOI] [PubMed] [Google Scholar]
- Tolentino MJ, Adamis AP. Angiogenic factors in the development of diabetic iris neovascularization and retinopathy. Int. Ophthalmol. Clin. 1998;38:77–94. doi: 10.1097/00004397-199803810-00007. [DOI] [PubMed] [Google Scholar]
- Vinals F, Pouyssegur J. Transforming growth factor beta1 (TGF-beta1) promotes endothelial cell survival during in vitro angiogenesis via an autocrine mechanism implicating TGF-alpha signaling. Mol. Cell. Biol. 2001;21:7218–7230. doi: 10.1128/MCB.21.21.7218-7230.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl SM, Hunt DA, Wakefield LM, McCartney-Francis N, Wahl LM, Roberts AB, Sporn MB. Transforming growth factor type beta induces monocyte chemotaxis and growth factor production. Proc. Natl. Acad. Sci. U S A. 1987;84:5788–5792. doi: 10.1073/pnas.84.16.5788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilting J, Christ B, Weich HA. The effects of growth factors on the day 13 chorioallantoic membrane (CAM): a study of VEGF165 and PDGF-BB. Anat. Embryol (Berl) 1992;186:251–257. doi: 10.1007/BF00174147. [DOI] [PubMed] [Google Scholar]
- Wood JP, Osborne NN. Expression of protein kinase C isoenzymes in cultured hooded rat retinal pigmented epithelial cells: comparison with dystrophic Royal College of Surgeons rat. Curr. Eye. Res. 1998;17:757–760. [PubMed] [Google Scholar]
- Yoshida H, Imaizumi T, Tanji K, Matsumiya T, Sakaki H, Kimura D, Cui XF, Kumagai M, Tamo W, Shibata T, Hatakeyama M, Sato Y, Satoh K. Platelet-activating factor enhances the expression of vascular endothelial growth factor in normal human astrocytes. Brain Res. 2002;944:65–72. doi: 10.1016/s0006-8993(02)02708-7. [DOI] [PubMed] [Google Scholar]
- Yu J, Farjo R, MacNee SP, Baehr W, Stambolian DE, Swaroop A. Annotation and analysis of 10,000 expressed sequence tags from developing mouse eye and adult retina. Genome. Biol. 2003;4:R65. doi: 10.1186/gb-2003-4-10-r65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue J, Mulder KM. Transforming growth factor-beta signal transduction in epithelial cells. Pharmacol. Ther. 2001;91:1–34. doi: 10.1016/s0163-7258(01)00143-7. [DOI] [PubMed] [Google Scholar]