

Abstract
Reactive oxygen species (ROS) generated during inflammation are believed to play critical roles in various ocular diseases. However, the underlying mechanisms remain poorly understood. We investigated if pro-inflammatory cytokines, tumor necrosis factor (TNF)-α, interleukin-1β (IL-1β), and interferon-γ (IFN-γ), induce ROS in human retinal pigment epithelial (RPE) cells. TNF-α, IL-1β and IFN-γ increased both intracellular and extracellular ROS production in a time- and dose-dependent manner. Thenoyltrifluoroacetone (TTFA), an inhibitor of mitochondrial respiratory chain, blocked TNF-α- and IFN-γ-, but not IL-1β-induced ROS, whereas other two mitochondrial respiratory chain inhibitors, rotenone and antimycin A, had no effect. NADPH oxidase inhibitor (diphenylene iodinium) abolished the ROS production induced by IL-1β or IFN-γ, but not by TNF-α, whereas 6-aminonicotinamide (6AN), an inhibitor of the hexose monophosphate shunt (HMS), had no significant effects on the ROS induced by all three cytokines. ROS scavengers, pyrrolidinedithiocarbamate (PDTC) and N-acetyl-cysteine (NAC), reduced the levels of ROS induced by TNF-α, IL-1β and IFN-γ (P < 0.05). Collectively, these results demonstrate that TNF-α, IL-1β and IFN-γ increase mitochondrial- and NADPH oxidase-generated ROS in human RPE cells.
Keywords: reactive oxygen species, human retinal pigment epithelium, TNF-α, IL-1β, IFN-γ
1. Introduction
Reactive oxygen species (ROS) are ubiquitous, highly reactive, diffusible molecules, including superoxide anions, hydrogen peroxide, hydroxyl radical, and nitric oxide (Fridovich, 1997). Cells generate ROS intracellularly and may release them extracellularly (Karlsson and Dahlgren, 2002; Kopprasch et al., 2003). While intracellular ROS serve mainly for host defense against infectious agents, redox-sensitive signal transduction, and other cellular processes, the extracellular release of ROS may damage surrounding tissues, potentially promoting inflammatory processes (Duval et al., 2003; Kopprasch et al., 2003). ROS are involved in aging and many diseases such as atherosclerosis, angiogenesis, cancer, diabetes mellitus, neurological degeneration, and tumor invasion (Harris and Shi, 2003; Wu, 2004). ROS also promote oxidative damage in eye disorders including age-related macular degeneration (AMD), cataracts, and uveitis (Rao, 1990; Winkler et al., 1999; Beatty et al., 2000; Cai et al., 2000; Truscott, 2000).
The retinal pigment epithelium (RPE) separates the neural retina from its blood supply in the choroid. In this strategic position, the RPE helps maintain an appropriate environment for photoreceptor function by transporting fluid, ions, and metabolites into and out of the space surrounding the photoreceptor outer segments. The RPE is an important factor in the development of AMD; central vision decreases when RPE cells cease to function properly, causing photoreceptor degeneration or damage in the macula, the portion of the retina used for central vision (Green et al., 1985; Young et al., 1987; Hageman et al, 2001; Penfold et al, 2001). Several studies have indicated that RPE cells are capable of producing ROS under certain conditions (Dorey et al., 1989; Miceli et al., 1994; Tate et al., 1995a; Wu and Rao, 1999; Yoshida et al., 2003; Kannan et al., 2004; Kindzelskii et al., 2004). RPE cells may therefore be an important source of ROS in the eye. Indeed, focused light (Dorey et al., 1990), high oxygen tension in the macula (Alder and Cringle, 1985), and photoreceptor outer segment phagocytosis and degradation (Tate et al., 1995b; Higgins et al., 2003) all promote RPE oxidative stress (Tso, 1987; Gottsch et al, 1990; Cai et al, 1999; Wassell et al., 1999; Higgins et al., 2003 Kindzelskii et al., 2004). Godley et al. (2005) showed that ROS levels were strikingly higher in macular RPE compared to peripheral RPE. Usually, RPE oxidative stress is reduced by endogenous RPE antioxidants and antioxidant enzymes, such as superoxide dismutase (SOD), catalase and glutathione peroxidase (GPX) (Newsome et al., 1990; Tate et al., 1993, 1995a; Edge et al, 1997; Fukuzawa et al., 1998; Jarrett et al., 2006). With aging, however, these RPE defenses decrease (Tate et al., 1993; Liles et al, 1991; Samiec et al, 1998), permitting ROS to ultimately overwhelm RPE cell defenses, leading to apoptotic damage (Ballinger et al., 1999).
AMD is a degenerative eye disease, common in people over 65 years of age, in which low-grade inflammation is now recognized to play a role. The inflammatory response in AMD lesions is characterized by an infiltration of the blood-retina barrier including RPE layer, by macrophages and lymphocytes (Seregard et al., 1994; Reddy et al., 1995; Penfold et al., 2001; Grossniklaus et al., 2002). In AMD, reactive, migrating or proliferating RPE cells are found adjacent to newly formed vessels in the subretinal space of wet AMD lesions (Miller et al., 1986; Sakamoto et al., 1995). Activation of RPE, inflammatory and endothelial cells may result in the release of a plethora of inflammatory mediators that individually or in concert may induce pathological changes in the retina. Pro-inflammatory cytokines such as tumor necrosis factor (TNF)- α, interleukin-1β (IL-1β), and interferon-γ (IFN-γ) may be among the primary components responsible for the inflammatory response observed in the AMD, as TNF-α and IL-1β are secreted by macrophages and vescular endothelial cells while IFN-γ is secreted by lymphocytes (Oh et al., 1999). Furthermore, TNF-α, IL-1β, and IFN-γ are linked with pro-inflammatory RPE cell functions (Elner et al., 1990, 1991, 1992, 1997; Hollborn et al., 2001).
TNF-α, IL-1β, and IFN-γare polypeptides that exert pleiotropic actions on multiple cell functions regulating gene expression, host defense reactions, and the immune response. TNF-α increases mitochondrial ROS production in tumor cells, endothelial cells, and hepatocytes (Schulze-Osthoff, 1993; Corda et al., 2001). IL-1β stimulates ROS production in various cell types (Mendes et al., 2003; Brigelius-Flohe et al., 2004; Hwang et al., 2004; Kaur et al., 2004). IFN-γ induces an immediate and marked augmentation of intracellular ROS in transformed lymphoblast cell lines (HSC536/N and PD149L) (Pearl-Yafe et al., 2003, 2004). IFN-γ also induces ROS and endoplasmic reticulum stress during IFN-γ-induced apoptosis of hepatocytes (Watanabe et al., 2003). However, the abilities of TNF-α, IL-1β, or IFN-γ to stimulate ROS production in RPE cells have not yet been reported.
In this study, therefore, we assessed: 1) whether human RPE cells produce ROS when stimulated by pro-inflammatory cytokines, TNF-α, IL-1β, or IFN-γ ; and 2) if so, the cellular source of ROS production induced by these cytokines.
2. Materials and Methods
2.1. Materials
Recombinant human TNF-α, IL-1β, and IFN-γ were purchased from R&D Systems, Inc. (Minneapolis, MN) and PeproTech, Inc. (Rocky Hill, NJ). MitoTracker Red CMXRos and 5-(and 6)-chloromethyl-2′,7′-dichlorodihydrofluorescence diacetate, acetyl ester (CM-H2DCFDA) were purchased from Molecular Probes (Eugene, OR). Poly-D-lysine coated 96-well plates and Hoechst 33342 were purchased from Sigma-Aldrich (St. Louis, MO). All other reagents were purchased from Sigma-Aldrich or Gibco BRL Life Technologies (Gaithersburg, MD).
2.2. Human RPE Cell Culture
Human RPE cells were isolated from donor eyes by enzymatic digestion as previously described by Elner et al. (1991). In brief, the sensory retina was separated gently from the RPE monolayer, and the RPE cells were removed from Bruch’s membrane using one hour incubation with papain (Sigma-Aldrich). Isolated RPE cells were grown into Falcon Primaria flasks (Becton-Dickinson Inc., Lincoln Park, NJ) in Dulbecco’s modified essential medium (DMEM)/F12 (Sigma-Aldrich) containing 10% fetal bovine serum (Sigma-Aldrich), penicillin G (100 U ml−1; Sigma-Aldrich), streptomycin sulfate (100 μg ml−1; Sigma-Aldrich), and amphotericin B (0.25 μg ml−1; Gibco BRL Life Technologies) at 37 °C in a humidified incubator under 5% CO2. In all experiments, simultaneous, parallel assays were performed on second to sixth passaged RPE cells seeded at the same time and density from the same parent cultures. All experiments were repeated at least three times on different RPE cell lines.
2.3. Measurement of Reactive Oxygen Species Production by RPE Cells
ROS production was assessed both intracellularly and extracellularly.
2.3.1. Intracellular ROS Production
Intracellular ROS production by human RPE cells in response to TNF-α, IL-1β, or IFN-γ treatment was measured by a cell-based fluorometric assay based on deacetylation and oxidation of non-fluorescent reduced CM-H2DCFDA (Molecular Probes) into fluorescent CM-DCF. The reliability and specificity of the probe have been established in various cells including cultured human RPE cells (Deshpande et al, 2000; Nishikawa et al., 2000; Touyz et al., 2001; Yamagishi et al., 2001; Higgins et al., 2003; Li et al., 2003; Fukami et al., 2004; Hermann et al., 2004; Hwang et al., 2004; Kannan et al., 2004; Shanker et al., 2004; Kim et al., 2005).
RPE cells were seeded into 96-well plates pre-coated with poly-D-lysine (Sigma-Aldrich) and cultured for 1 day or 7days. RPE cells in 96-well plates were washed with Hanks’ balanced salt solution (HBSS), containing Ca2+ and Mg2+, without phenol red. Subsequently, the medium was replaced with HBSS containing 5 μM CM-H2DCFDA and incubated for 1 hr at 37 °C in the dark. Plates were then washed, and pre-incubated for 30 min with or without different inhibitors [mitochondrial respiratory chain inhibitors: rotenone (2.5 μM), thenoyltrifluoroacetone (TTFA; 10 μM), and antimycin A (100 ng/ml); NADPH oxidase inhibitor: diphenylene iodinium (DPI; 5 μM), hexose monophosphate shunt (HMS) inhibitor: 6-aminonicotinamide (6AN; 2 μM); or ROS scavengers: pyrrolidinedithiocarbamate (PDTC; 50 μM) or N-acetyl-cysteine (NAC; 1 mM)], followed by stimulation with TNF-α (20 ng/ml), IL-1β (0.02 ng/ml), or IFN-γ (2 units/ml) in the presence and absence of the same inhibitor used during pre-incubation. Fluorescence was measured over a period of 60 min with a FlexStation fluorescence plate reader (Molecular Devices, Sunnyvale, CA). Excitation and emission wavelengths were set to 495 nm and 528 nm, respectively, with a cutoff of 515 nm.
2.3.2. Extracellular Release of Hydrogen Peroxide
Human RPE cells were cultured to near confluence in 35 mm diameter tissue culture wells and stimulated with TNF-α (0–50 ng/ml), IL-1β (0–40 ng/ml), IFN-γ (0–20 units/ml) or left unstimulated. Following incubation at 37°C for 0–60 min, hydrogen peroxide (H2O2) levels in the RPE media were measured using a fluorometric assay based on the H2O2-dependent oxidation of homovanillic acid (Till et al., 1987, 1991).
2.4. Fluorescence Microscopy
The RPE cells were grown on coverslips, stimulated with TNFα, IFN-γ or left unstimulated. After treatment, RPE cells were simultaneously incubated with 5 μM CM-H2DCFDA which detects ROS production (green) and 300 nM mitochondria-specific dye, MitoTracker Red CMXRos for 1 hr at 37 °C in the dark, washed, and then stained Hoechst 33342 for 10 min at room temperature. The coverslips were then placed on glass slides and cells were observed using fluorescent microscope and images were collected as described previously (Yang et al., 2003). Non-fluorescent reduced CM-H2DCFDA was deacetylated and then oxidized by ROS into green fluorescent CM-DCF within the cells. MitoTracker Red CMXRos stains mitochondria red and Hoechst 33342 stains the cell nuclei of healthy cells faintly blue and those of the apoptotic cells bluish-white. A yellow color of merged images indicates that accumulated ROS (green) are colocalized in the mitochondria (red).
2.5. Viability Assay
RPE cells were stimulated with TNF-α (20 ng/ml), IL-1β (0.02 ng/ml), or IFN-γ (2 units/ml) in the presence or in the absence of different inhibitors, as described above, for 60 min. Cells were washed with PBS and dissociated with 0.05% trypsin/0.5 mM EDTA. Cell viability was determined with a trypan blue exclusion assay. Live cells that excluded 0.2% trypan blue dye were counted with an improved Neubauer hemocytometer (American Optical Corp., Buffalo, NY).
2.6. Statistical Analysis
Data are expressed as means ± standard error (SEM) and evaluated by Student’s unpaired t test or one-way analysis of variance (ANOVA) followed by a Student–Newman–Keul’s post hoc test. P < 0.05 is considered statistically significant.
3. Results
3.1. RPE ROS Production Is Induced by TNF-α, IL-1β or IFN-γ
ROS play an important role in the pathogenesis of various forms of inflammatory ocular injury. Cells generate ROS intracellularly and may release them extracellularly (Karlsson and Dahlgren, 2002; Kopprasch et al., 2003). Therefore, we examined both intracellular and extracellular ROS production in response to cytokines (TNF-α, IL-1β and IFN-γ) in cultured human RPE cells.
As shown in Fig. 1A, TNF-α-induced RPE intracellular ROS levels in a dose-dependent manner with maximal stimulation was achieved at 20 ng/ml (P < 0.05). RPE intracellular ROS production induced by TNF-α was also time-dependent, being significantly higher than that of control by 30 min, with continued increases to 60 min (P < 0.05; Fig. 1B). Maximal TNF-α-induced extracellular ROS production was also observed at 20 ng/ml (P < 0.01; Fig. 1C). RPE ROS release induced by TNF-α was also time-dependent, peaking after 40 min of stimulation (P < 0.001; Fig. 1D). As the intracellular accumulation of ROS in endothelial cells peaked at 2–3 hrs after TNF-α treatment (Corda et al., 2001), we tested whether longer treatment would be associated with more ROS accumulation in the RPE cells. By comparing ROS accumulation in the RPE cells stimulated by TNF-α at 0, 1, 2, 4, and 24 hr, we found that, unlike endothelial cells, there were no further increases in the intracellular ROS accumulation in RPE cells in response to TNF-α at 2, 4, or 24 hr, compared to the ROS accumulation at 1 hr. Compared to unstimulated RPE cells, TNF-α again significantly increased the intracellular ROS accumulation in the RPE cells at 1hr. We also compared TNF-α induced ROS accumulation in the RPE cells 1 day and 7 days after plating, and found that there was no significant difference between the two groups. Please note that there were no significant changes in the control values (without cytokine) between 0 and 60 min. The released H2O2 in unstimulated control cells from three experiments were 2.25 ± 0.07 nanomoles H2O2 per million cells at 0 min, and 2.29 ± 0.14 nanomoles H2O2 per million cells at 60 min. The baseline intracellular ROS (H2O2) concentrations in the RPE cells were estimated to be around 75 nanomoles ml−1, comparable to the baseline intracellular ROS concentration (52 nanomoles ml−1) in bovine aortic endothelial cells (Nishikawa et al., 2000). Like TNF-α, IL-1β increased both intracellular and extracellular ROS production in time- and dose-dependent manners with significant differences compared to unstimulated cells. IL-1β-induced intracellular ROS production peaked at lower concentration (0.02 ng/ml) and sooner (5 min) (Fig. 2A, 2B). RPE H2O2 release also continued to increase with IL-1β higher concentrations (20–50 ng/ml) and maximal extracellular H2O2 levels were attained by 30 min (Fig. 2C, 2D). In a similar manner, IFN-γ induced both intracellular and extracellular ROS production in time- and dose-dependent manners (Fig. 3A, 3B). The maximal induction of intracellular ROS was achieved by a relatively low concentration of 2 units/ml (Fig. 3A). At this concentration of IFN-γ, the maximal induction of intracellular and extracellular RPE ROS occurs by 5 min (Fig. 3B, 3D).
Figure 1. Dose and time course of ROS production induced by TNF-α in human RPE cells.
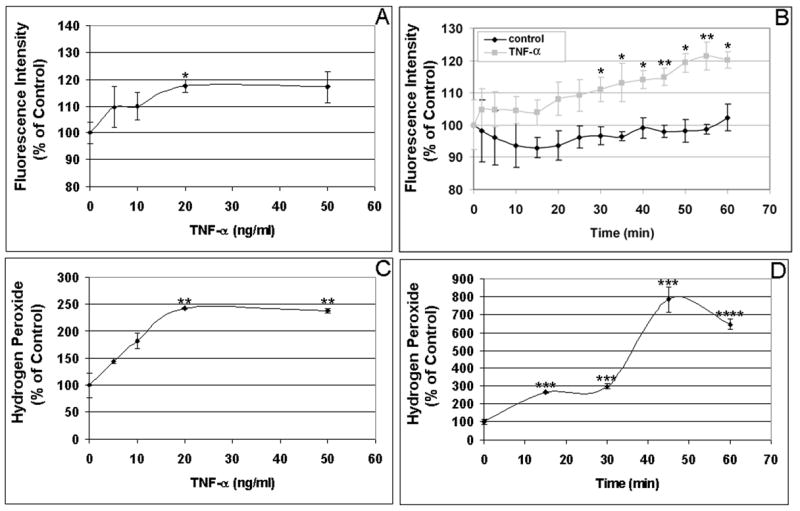
(A) Dose dependent induction of RPE intracellular ROS production by TNF-α. Cells were incubated with the indicated concentrations of TNF-α for 60 min, and intracellular ROS levels were determined by oxidative conversion of nonfluorescent reduced DCF to fluorescent DCF. (B) Time course of TNF-α-induced intracellular ROS production. RPE cells were incubated with 20 ng/ml TNF-α for 0-60 min, and intracellular ROS levels were determined by DCF fluorescence. (C) Dose response of TNF-α-induced extracellular ROS production. RPE cells were incubated with the indicated concentrations of TNF-α for 60 min, and H2O2 released by RPE cells was detected by oxidation of homovanillic acid into its fluorescent dimer. (D) Time course of TNF-α-induced extracellular ROS production. RPE cells were incubated with 20 ng/ml TNF-α for 0-60 min, and H2O2 released by RPE was detected by monitoring formation of the fluorescent homovanillic acid dimer. The released H2O2 in unstimulated control cells from three experiments were 2.25 ± 0.07 nanomoles H2O2 per million cells at 0 min, and 2.29 ± 0.14 nanomoles H2O2 per million cells at 60 min. The baseline intracellular ROS (H2O2) concentrations in the RPE cells were estimated to be around 75 nanomoles ml−1. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001, compared to unstimulated cells.
Figure 2. Dose and time course of ROS production induced by IL-1β in human RPE cells.
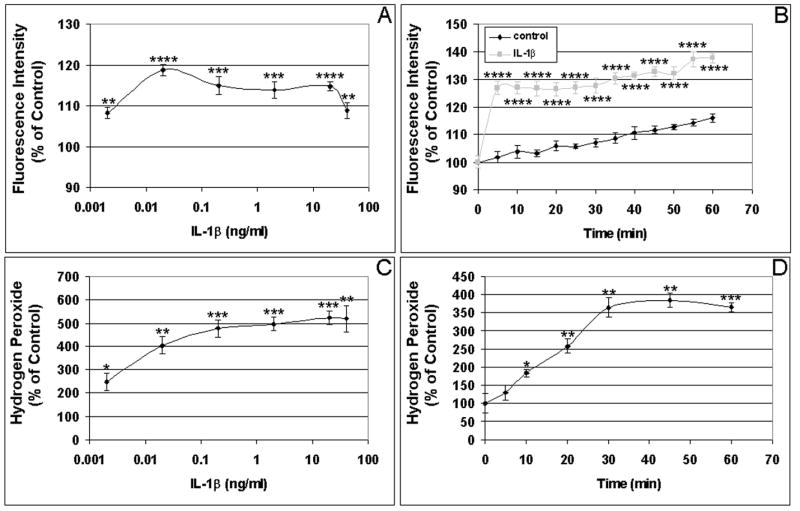
(A) Dose response of IL-1β-induced intracellular ROS production. RPE cells were incubated with the indicated concentrations of IL-1β for 60 min, and intracellular ROS levels were determined by DCF fluorescence. (B) Time course of IL-1β-induced intracellular ROS production. RPE cells were incubated with 0.02 ng/ml IL-1β for 0-60 min, and intracellular ROS levels were determined by detecting DCF fluorescence. (C) Dose response of IL-1β-induced extracellular ROS production. RPE cells were incubated with the indicated concentrations of IL-1β for 60 min, and H2O2 released by RPE was detected by measuring oxidized homovanillic dimer. (D) Time course of IL-1β-induced extracellular ROS production. RPE cells were incubated with 20 ng/ml IL-1β for 0-60 min, and H2O2 released by RPE was detected by monitoring of fluorescent homovanillic acid dimer. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001, compared with unstimulated cells.
Figure 3. Dose and time course of ROS production induced by IFN-γ in human RPE cells.

(A) Dose response of RPE intracellular ROS production due to IFN-γ stimulation. Cells were incubated with the indicated concentrations of IFN-γ for 60 min, and intracellular ROS levels were determined by measuring oxidized fluorescent DCF. (B) Time course of IFN-γ-induced intracellular ROS production. RPE cells were incubated with 2 units/ml IFN-γ for 0-60 min, and intracellular ROS levels were determined by monitoring fluorescent DCF. (C) Dose response of IFN-γ-induced extracellular ROS production. RPE cells were incubated with the indicated concentrations of IFN-γ for 60 min, and H2O2 released by RPE was detected by oxidation of homovanillic acid into its fluorescent dimer. (D) Time course of IFN-γ-induced extracellular ROS production. RPE cells were incubated with 2 units/ml IFN-γ for 0-60 min, and H2O2 released by RPE was detected monitoring oxidized homovanillic acid dimes. *P < 0.05; **P < 0.01; ***P < 0.001, compared with unstimulated cells.
Compared to unstimulated cells, TNF-α, IL-1β, or IFN-γ, resulted in higher extracellular ROS accumulation than intracellular ROS production (Figs 1–3; also see Figs 4–6). TNF-α increased intracellular and extracellular ROS by 11.2% – 48.4% (Fig. 1A, 1B) and 142% – 680% (Fig. 1C, 1D), respectively. IL-1β induced ROS by 18.7% – 37.7% intracellularly (Fig. 2A, 2B) and 285% – 422% extracellularly (Fig. 2C, 2D). IFN-γ stimulation resulted in 13.0% – 39.6% increases in intracellular ROS (Fig. 3A, 3B) and 69.0% – 70.5% increases in extracellular ROS (Fig. 3C, 3D). Thus, our results suggest that the RPE cells have the ability to prevent the accumulation of high concentrations of ROS in the cells by converting ROS to H2O2 and releasing it into the extracellular environment.
Figure 4. Effects of mitochondrial electron transport inhibitors on intracellular ROS production by RPE cells.

(A) Effects of the mitochondrial electron transport inhibitors (2.5 μM rotenone; 10 μM TTFA; 100 ng/ml antimycin A) on TNF-α-, IL-1β-or IFN-γ-induced intracellular ROS by RPE cells. RPE cells were preincubated with or without either rotenone, TTFA or antimycin A for 30 min and then treated with or without cytokine. After 60 min, ROS were quantitated. Data are presented as mean ± SEM. *P < 0.001; **P < 0.00001, compared to unstimulated cells. #P < 0.05, compared to TNF-α-stimulated cells; ##P < 0.00001, compared to IFN-γ-stimulated cells. (B) Localization of TNF-α- or IFN-γ-induced ROS in the mitochondria of human RPE cells. Human RPE cells were triple labeled with CM-H2DCFDA to detect ROS (green), MitoTracker Red CMXRos to stain mitochondria (red), and Hoechst 33342 to stain cell nuclei (blue). A yellow color of merged images of ROS (green), mitochondrial (red), and cell nuclear (blue) fluorescence indicates that accumulated ROS (green) are colocalized in the mitochondria (red). Scale bar, 10 μm.
Figure 6. Effects of ROS scavengers on intracellular ROS production by RPE cells.

Effect of ROS scavengers, N-acetyl-cysteine (NAC; 1mM) and pyrrolidinedithiocarbamate (PDTC; 50 μM) on TNF-α-, IL-1β- or IFN-γ-induced intracellular ROS production. RPE cells were preincubated with or without either NAC or PDTC for 30 min and then treated with or without TNF-α, IL-1β or IFN-γ. After 60 min, ROS were quantitated. Data are presented as mean ± SEM.
*P < 0.05, compared with unstimulated cells. #P < 0.05, compared with IFN-γ-stimulated cells; ##P < 0.01, ###P < 0.001, compared with TNF-α- or IL-1β-stimulated cells.
Of note, the concentration of IL-1β (0.02 ng/ml) capable of inducing maximal intracellular RPE ROS levels is below the concentration (20 ng/ml) that maximally induces extracellular ROS; it is also lower than the concentrations found to maximally induced intracellular ROS in other cell types (Hwang et al, 2004), suggesting the potential importance of ROS in IL-1β-mediated signal transduction in the RPE.
3.2. TNF-α- or IFN-γ-, but not IL-1β-Induced ROS Is Produced within the Mitochondrial Electron Transport Chain
As all three cytokines induced ROS production, we next examined the source of ROS accumulation within RPE cells. Mitochondria have been identified as a major source of TNF-α-induced ROS production in other cell types (Schulze-Osthoff et al, 1993; Corda S et al., 2001). To determine whether mitochondria are the source of TNF-α-induced ROS generation in RPE cells, we used the mitochondrial complex inhibitors, rotenone, thenoyltrifluoroacetone (TTFA), and antimycin A, that block mitochondrial electron transport at various sites that may be associated with ROS production, while monitoring the formation of ROS in human RPE cells. In the presence of ROS, especially H2O2, nonfluorescent reduced DCF is oxidized to fluorescent DCF (Cai et al, 2000; Higgins et al, 2003).
As seen in Fig. 4A, TTFA (10 μM), an inhibitor of complex II, resulted in 43.0 ± 5.2% (n=4) decrease in TNF-α-induced-DCF fluorescence, compared to RPE cells exposed to TNF-α alone (P < 0.05), while rotenone (an inhibitor of complex I; 2.5 μμM) and antimycin A (an inhibitor of complex III;100 ng/ml) had no significant effect (P > 0.05). Since TTFA inhibited less than half of the RPE intracellular ROS induced by TNF-α, we conclude that only a portion of the TNF-α-induced intracellular ROS occurs in mitochondria.
To evaluate whether mitochondria were also involved in the production of ROS induced by two other inflammatory mediators, IL-1β and IFN-γ, we used the same mitochondrial respiration chain inhibitors. The level of stimulated ROS production in presence of rotenone, TTFA, or antimycin A was not significantly different from IL-1β alone (Fig. 4A). This would suggest that the intracellular signaling pathway of IL-1β is different from that of TNF-α and not mediated through mitochondrial transport. IFN-γ-induced RPE intracellular ROS production, however, was almost completely blocked TTFA (10 μM) (Fig. 4A; P < 0.00001), while rotenone and antimycin A had no significant effect on IFN-γ-induced intracellular ROS, indicating that selective mitochondrial secretion of ROS via the action of succinate dehydrogenase is required for its action.
To confirm the mitochondrial source of ROS production after TNFα or IFN-γ stimulation, we performed triple-labeling experiments using CM-H2DCFDA to detect ROS, MitoTracker Red CMXRos to stain mitochondria and Hoechst 33342 to stain cell nuclei. Figure 4B shows fluorescence images of human RPE cells triple labeled with the three dyes. Unstimulated control cells showed baseline levels of ROS. Treatment with TNF-α or IFN-γ increased the intracellular accumulation of ROS that were found to localize in part in mitochondria as demonstrated by a yellow color of the merged images. These results further confirmed that the portion of ROS was produced within the mitochondria of TNF-α- or IFN-γ-stimulated RPE cells.
3.3. IL-1β- or IFN-γ-, but not TNF-α-Induced ROS Production Is Abolished by NADPH Oxidase Inhibitor
To evaluate whether DPI, which inhibits NADPH oxidase, the enzyme directly responsible for ROS generation, would inhibit TNF-α, IL-1β or IFN-γ induction of ROS, we pre-treated RPE cells with 5 μM DPI or vehicle for 30 min, followed by stimulation with each cytokine in the presence and absence of DPI. As shown in Fig. 5, the data indicate that DPI abolished IL-1β- or IFN-γ-, but not TNF-α-induced RPE ROS that was measured after 1 hr of cytokine stimulation.
Figure 5. Effects of NADPH oxidase inhibitor on intracellular ROS production by RPE cells.
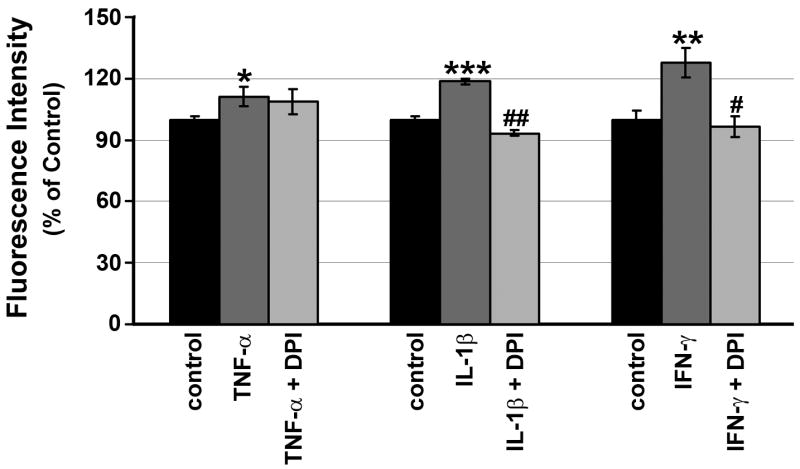
RPE cells were preincubated with or without 5 μM DPI for 30 min, and then treated with or without 20 ng/ml TNF-α, 0.02 ng/ml IL-1β or 2 units/ml IFN-γ. After 60 min, ROS were quantitated. Data are presented as mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.0001, compared with unstimulated cells. #P < 0.01, compared to IFN-γ-stimulated cells; ##P < 0.00001, compared to IL-1β-stimulated cells.
Previous studies have associated ROS production by leukocytes is associated with activation of the hexose monophosphate shunt (HMS) (Chanock et al., 1994). To test whether cytokine-induced ROS production involves HMS activation, human RPE cells were stimulated with TNF-α, IL-1β or IFN-γ in the presence of 2 μM 6-aminonicotinamide, an inhibitor of the HMS. However, 6-aminonicotinamide had no effect on TNF-α, IL-1β or IFN-γ-induced ROS production (data not shown).
3.4. TNF-α-, IL-1β- or IFN-γ-Induced ROS Production Is Abolished by ROS Scavenger
ROS levels in other cell types have been shown to be decreased by ROS scavengers, pyrrolidinedithiocarbamate (PDTC) and N-acetyl-cysteine (NAC) (Hwang et al., 2004). We evaluated whether ROS scavengers can modulate TNF-α-, IL-1β- or IFN-γ-induced ROS. As expected, PDTC (50 μM) and NAC (1 mM) each reduced levels of ROS stimulation by TNF-α-, IL-1β- or IFN-γ-ROS to baseline levels in RPE cells (Fig. 6) without affecting cell viability at the concentrations used (data not shown).
4. Discussion
This study demonstrates that TNF-α, IL-1β and IFN-γ induce ROS production by human RPE cells in comparison to unstimulated RPE cells. TNF-α increases mitochondrial ROS production in RPE cells; IL-1β induces ROS production via NADPH oxidase; and IFN-γ induces ROS through both mitochondrial ROS and ROS via NADPH oxidase. Furthermore, ROS induced by each cytokine are readily abolished by two ROS scavengers, NAC and PDTC.
Although cytokine-induced intracellular ROS levels are low in RPE, trace levels of ROS, such as superoxide or H2O2, may act to mediate signaling in various signal transduction cascades (Duranteau et al., 1998; Duval et al., 2003; Kopprasch et al., 2003). Indeed, ROS are implicated as participants in many intracellular signaling pathways, including activation of stress- and mitogen-activated protein kinases and the nuclear transcription factors c-Jun and NF-κB in other cell types (Guyton et al, 1996; Laderoute et al, 1997; Flohe et al, 1997; Lander, 1997; Hwang et al., 2004; Pearl-Yafe et al., 2004). RPE cells were less susceptible to death induced by H2O2 or paraquat, and had greater CuZnSOD, catalase and GPX enzymatic activity, compared to other cell types (Jarrett et al., 2006; Lu et al., 2006). Therefore, low levels of intracellular ROS induced by the three cytokines may be due to the good endogenous antioxidant defense system in the RPE.
Our results show that TNF-α-induced ROS are generated by mitochondria and are not abolished by the NADPH oxidase inhibitor DPI. These results for TNF-α were consistent with previous studies in endothelial cells (Corda et al. 2001). At the concentration of TNF-α (20 ng/ml) used for present study, there was no evidence of RPE cytotoxicity, as evaluated by trypan blue exclusion assays. This is consistent with observations by us and others who reported that human RPE cells are resistant to TNF-α-induced cell death at 6, 12, 24, 48 and 72 hr (Elner et al., 1990, 1991; Yang et al., 2004). Although the increase in fluorescence induced by TNF-α required more than 30 min to reach maximal stimulation (Fig. 1), the time required to induce ROS in RPE cells was shorter than that reported for endothelial cells (Corda et al., 2001) and tumor cells (Goosens et al., 1995) where ROS production required at least 1 hr of exposure to TNF-α. The time required for IL-1β- and IFN-γ-induced intracellular and extracellular RPE ROS production was less than for TNF-α. The involvement of the mitochondrial electron transport in TNF-α-induced ROS production has been demonstrated in several other cell types (Schulze-Osthoff et al., 1992, 1993; Goosens et al., 1995; Corda et al., 2001). In hepatocytes and endothelial cells, two sites of the respiratory chain, complex I and complex III, were responsible for TNF-α-induced ROS (Corda et al., 2001). However, our study indicates that mitochondrial complex II is responsible for TNF-α-induced ROS production in the RPE cells. The reason for this could be cell type-specific differences. Interestingly, mitochondrial complex II is involved in hyperglycaemia-induced and leptin-induced intracellular ROS in cultured bovine aortic endothelial cells (Nishikawa et al., 2000; Yamagishi et al., 2001). In the present study, as TTFA only partially blocked TNF-α-induced intracellular ROS production, it is likely that additional sources contribute to intracellular ROS production induced by TNF-α.
ROS are formed by several different mechanisms like unavoidable byproducts of cellular respiration. ROS are also synthesized by NADPH oxidase in phagocytic cells such as neutrophils and macrophages. RPE cells possess NADPH oxidase (Miceli et al., 1994) and phagocytic function.
Cells normally have a variety of defenses against the harmful effects of ROS and activate a diverse array of protective mechanisms in response to various stimuli-induced ROS. The defense system in the RPE includes enzymatic antioxidants, nonenzymatic antioxidants and oxidative repair mechanisms. Protection against superoxide anion is provided by SOD which catalyzes the dismutation of superoxide anion to H2O2. SOD has three isoforms: SOD1 (CuZnSOD) is localized in the cytoplasm; SOD2 (MnSOD) is localized in mitochondria (Newsome et al., 1990; Lu et al, 2006); and SOD3 (extracellular SOD) is secreted into the extracellular space (Lu et al, 2006). Catalase and GPX remove H2O2 formed after the SOD-catalyzed dismutation reaction (Miceli et al., 1994; Lu et al, 2006; Tate et al., 2006).
One of the important functions of the RPE cell is the phagocytosis and degradation of photoreceptor outer segments. Phagocytosis of latex beads stimulates extracellular superoxide anion production in porcine RPE cells in the first 15 min and declined thereafter (Dorey et al., 1989). Phagocytosis of latex beads also stimulates extracellular H2O2 production twofold in human RPE cells (Tate et al., 1995b). Phagocytosis of photoreceptor outer segments increased extracellular H2O2 production ninefold (Tate et al., 1995b). The cytokine-induced accumulation of both intracellular and extracellular H2O2 in our system suggests that H2O2 is generated within cells and released extracellularly. However, it is also possible that cytokines induce superoxide anion production through mitochondria or activate NADPH oxidase producing superoxide anions. The induced superoxide anions would be quickly dismutated to H2O2 by SOD1 and SOD2 inside the RPE cells or released into extracellular environment. The released superoxide anions might be quickly dismutated to H2O2 by SOD3.
It is well known that IL-1β upregulates IL-8 expression in various cells including RPE cells (Elner et al., 1990; Baggiolini et al., 1994; Bian et al., 2001, 2004; Jung et al., 2002; Hwang et al., 2004). In this study we provide the first evidence that IL-1β induces RPE production of ROS intracellularly and extracellularly. It is likely that intracellular generation of H2O2 is important to signaling mediating the induction of IL-8 expression in RPE cells (Elner et al., 1990; Bian et al., 2001, 2004) as NAC has been shown to prevent the IL-1β-induced ROS production and IL-8 expression in other cell types.
To our knowledge, the mechanisms that mediate ROS generation after membrane receptor binding of TNF-α, IL-1β or IFN-γ are not yet characterized in RPE cells. In hepatocytes (Garcia-Ruiz et al., 1997; Fernandez-Checa et al., 1997) and RPE cells (Kannan et al., 2004), ceramide has been shown to cause an increase in ROS generation in mitochondria. In Jurkat T lymphocytes, binding of TNF-α to its receptor causes rapid activation of an acidic sphingomyelinase (ASMase), followed by sphingomyelin hydrolysis and ceramide production within 3 min (Schutze et al., 1991). Furthermore, Corda et al. (2001) showed that mitochondrial ROS generation induced by TNF-α may be inhibited by the ASMase inhibitor, desipramine and by the ceramide-activated protein kinase (CAPK) inhibitor, dimethylaminopurine (DMAP). Protein secretion of TNF-α, IL-β, or IFN-γ has not been demonstrated in RPE cells in vitro (Jaffe et al, 1992; Yoshida et al., 2001). However, descriptive studies of AMD lesions confirm the presence of macrophages expressing TNF-α and IL-1β (Oh et al, 1999), both of which we have shown to be potent inducers of RPE IL-8 and MCP-l (Elner et al, 1991, 1997; Bian et al., 2004). We have also found that TNF-α and IL-1β cause phosphorylation of Akt and its downstream targets, glycogen synthase kinase (GSK) and forkhead transcription factor (FKHR) (Bian et al., 2004). Recently, others have shown that exogenous H2O2 results in Akt phosphorylation in human RPE cells (Yang et al., 2006). Taken together, these results lead to the speculation that binding of TNF-α or IL-1β (even IFN-γ) in RPE cells activates ROS generation, participating in the phosphorylation of Akt and its downstream targets (GSK and FKHR), which lead to IL-8 and MCP-l gene transcription.
Since RPE cells in vivo likely encounter a complex and varying mix of cytokines, we will investigate whether the three cytokines and other cytokines have additive and/or synergistic effects on ROS production in the RPE. Further in vivo studies using animal model or freshly isolated human RPE cells are required to characterize the mechanisms by which ROS are generated, released and regulated.
In conclusion, the results of this study provide the first evidence that TNF-α, IL-1β and IFN-γ induce ROS production in RPE cells. The TNF-α-induced ROS are produced in mitochondria, IL-1β-induced ROS are produced via NADPH oxidase, and IFN-γ-induced ROS are generated by both mechanisms. These findings may help to elucidate the possible roles of ROS in the RPE responses to cytokines in AMD.
Acknowledgments
This research was supported by the National Institutes of Health grants EY09441 (V.M. Elner) and EY07003 (core). V.M. Elner is a recipient of Lew R. Wasserman Merit Award from Research to Prevent Blindness.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alder VA, Cringle SJ. The effect of the retinal circulation on vitreal oxygen tension. Curr Eye Res. 1985;4:121–129. doi: 10.3109/02713688508999977. [DOI] [PubMed] [Google Scholar]
- Baggiolini M, Dewald B, Moser B. Interleukin-8 and related chemotactic Cytokines-CXC and CC chemokines. Adv Immunol. 1994;55:97–179. [PubMed] [Google Scholar]
- Ballinger SW, Van Houten B, Jin GF, Conklin CA, Godley BF. Hydrogen peroxide causes significant mitochondrial DNA damage in human RPE cells. Exp Eye Res. 1999;68:765–772. doi: 10.1006/exer.1998.0661. [DOI] [PubMed] [Google Scholar]
- Beatty S, Koh H, Phil M, Henson D, Boulton M. The role of oxidative stress in the pathogenesis of age-related macular degeneration. Surv Ophthalmol. 2000;45:115–134. doi: 10.1016/s0039-6257(00)00140-5. [DOI] [PubMed] [Google Scholar]
- Bian ZM, Elner SG, Yoshida A, Kunkel SL, Su J, Elner VM. Activation of p38, ERK1/2 and NIK pathways is required for IL-1beta and TNF-alpha-induced chemokine expression in human retinal pigment epithelial cells. Exp Eye Res. 2001;73:111–121. doi: 10.1006/exer.2001.1019. [DOI] [PubMed] [Google Scholar]
- Bian ZM, Elner SG, Yoshida A, Elner VM. Differential involvement of phosphoinositide 3-kinase/Akt in human RPE MCP-1 and IL-8 expression. Invest Ophthalmol Vis Sci. 2004;45:1887–1896. doi: 10.1167/iovs.03-0608. [DOI] [PubMed] [Google Scholar]
- Brigelius-Flohe R, Banning A, Kny M, Bol GF. Redox events in interleukin-1 signaling. Arch Biochem Biophys. 2004;423:66–73. doi: 10.1016/j.abb.2003.12.008. [DOI] [PubMed] [Google Scholar]
- Cai J, Wu M, Nelson KC, Sternberg P, Jr, Jones DP. Oxidant-induced apoptosis in cultured human retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. 1999;40:959–966. [PubMed] [Google Scholar]
- Cai J, Nelson KC, Mei W, Sternberg P, Jones DP. Oxidative damage and protection of the RPE. Prog Retinal Eye Res. 2000;19:205–221. doi: 10.1016/s1350-9462(99)00009-9. [DOI] [PubMed] [Google Scholar]
- Chanock SJ, el Benna J, Smith RM, Babior BM. The respiratory burst oxidase. J Biol Chem. 1994;269:24519–24522. [PubMed] [Google Scholar]
- Corda S, Laplace C, Vicaut E, Duranteau J. Rapid reactive oxygen species production by mitochondria in endothelial cells exposed to tumor necrosis factor-alpha is mediated by ceramide. Am J Respir Cell Mol Biol. 2001;24:762–768. doi: 10.1165/ajrcmb.24.6.4228. [DOI] [PubMed] [Google Scholar]
- Deshpande SS, Angkeow P, Huang J, Ozaki M, Irani K. Rac1 inhibits TNF-alpha-induced endothelial cell apoptosis: dual regulation by reactive oxygen species. FASEB J. 2000;14:1705–1714. doi: 10.1096/fj.99-0910com. [DOI] [PubMed] [Google Scholar]
- Dorey CK, Khouri GG, Syniuta LA, Curran SA, Weiter JJ. Superoxide production by porcine retinal pigment epithelium in vitro. Invest Ophthalmol Vis Sci. 1989;30:1047–1054. [PubMed] [Google Scholar]
- Dorey CK, Delori FC, Akeo K. Growth of cultured RPE and endothelial cells is inhibited by blue light but not green or red light. Curr Eye Res. 1990;9:549–559. doi: 10.3109/02713689008999595. [DOI] [PubMed] [Google Scholar]
- Duranteau J, Chandel NS, Kulisz A, Shao Z, Schumacker PT. Intracellular signaling by reactive oxygen species during hypoxia in cardiomyocytes. J Biol Chem. 1998;273:11619–11624. doi: 10.1074/jbc.273.19.11619. [DOI] [PubMed] [Google Scholar]
- Duval C, Cantero AV, Auge N, Mabile L, Thiers JC, Negre-Salvayre A, Salvayre R. Proliferation and wound healing of vascular cells trigger the generation of extracellular reactive oxygen species and LDL oxidation. Free Radic Biol Med. 2003;35:1589–1598. doi: 10.1016/j.freeradbiomed.2003.09.008. [DOI] [PubMed] [Google Scholar]
- Edge R, McGarvey DJ, Truscott TG. The carotenoids as anti-oxidants-a review. J Photochem Photobiol B. 1997;41:189–200. doi: 10.1016/s1011-1344(97)00092-4. [DOI] [PubMed] [Google Scholar]
- Elner VM, Strieter RM, Elner SG, Baggiolini M, Lindley I, Kunkel SL. Neutrophil chemotactic factor (IL-8) gene expression by cytokine-treated retinal pigment epithelial cells. Am J Pathol. 1990;136:745–750. [PMC free article] [PubMed] [Google Scholar]
- Elner SG, Strieter RM, Elner VM, Rollins BJ, Del Monte MA, Kunkel SL. Monocyte chemotactic protein gene expression by cytokine-treated human retinal pigment epithelial cells. Lab Invest. 1991;64:819–825. [PubMed] [Google Scholar]
- Elner SG, Elner VM, Pavilack MA, Todd RF, 3rd, Mayo-Bond L, Franklin WA, Strieter RM, Kunkel SL, Huber AR. Modulation and function of intercellular adhesion molecule-1 (CD54) on human retinal pigment epithelial cells. Lab Invest. 1992;66:200–211. [PubMed] [Google Scholar]
- Elner VM, Burnstine MA, Strieter RM, Kunkel SL, Elner SG. Cell-associated human retinal pigment epithelium interleukin-8 and monocyte chemotactic protein-1: immunochemical and in-situ hybridization analyses. Exp Eye Res. 1997;65:781–789. doi: 10.1006/exer.1997.0380. [DOI] [PubMed] [Google Scholar]
- Fernandez-Checa JC, Kaplowitz N, Garcia-Ruiz C, Colell A, Miranda M, Mari M, Ardite E, Morales A. GSH transport in mitochondria: defense against TNF-induced oxidative stress and alcohol-induced defect. Am J Physiol. 1997;273(1 Pt 1):G7–17. doi: 10.1152/ajpgi.1997.273.1.G7. [DOI] [PubMed] [Google Scholar]
- Flohe L, Brigelius-Flohe R, Saliou C, Traber MG, Packer L. Redox regulation of NF-kappa B activation. Free Radic Biol Med. 1997;22:1115–1126. doi: 10.1016/s0891-5849(96)00501-1. [DOI] [PubMed] [Google Scholar]
- Fridovich I. Superoxide anion radical (O2−·), superoxide dismutases, and related matters. J Biol Chem. 1997;272:18515–18517. doi: 10.1074/jbc.272.30.18515. [DOI] [PubMed] [Google Scholar]
- Fukami K, Ueda S, Yamagishi S, Kato S, Inagaki Y, Takeuchi M, Motomiya Y, Bucala R, Iida S, Tamaki K, Imaizumi T, Cooper ME, Okuda S. AGEs activate mesangial TGF-beta-Smad signaling via an angiotensin II type I receptor interaction. Kidney Int. 2004;66:2137–2147. doi: 10.1111/j.1523-1755.2004.66004.x. [DOI] [PubMed] [Google Scholar]
- Fukuzawa K, Inokami Y, Tokumura A, Terao J, Suzuki A. Rate constants for quenching singlet oxygen and activities for inhibiting lipid peroxidation of carotenoids and alpha-tocopherol in liposomes. Lipids. 1998;33:751–756. doi: 10.1007/s11745-998-0266-y. [DOI] [PubMed] [Google Scholar]
- Garcia-Ruiz C, Colell A, Mari M, Morales A, Fernandez-Checa JC. Direct effect of ceramide on the mitochondrial electron transport chain leads to generation of reactive oxygen species. Role of mitochondrial glutathione. J Biol Chem. 1997;272:11369–11377. doi: 10.1074/jbc.272.17.11369. [DOI] [PubMed] [Google Scholar]
- Godley BF, Alsaadi R, Liang F. Increased oxidative stress in human macular RPE cells. Invest Ophthalmol Vis Sci. 2005;46 ARVO E-Abstract 5288. [Google Scholar]
- Goossens V, Grooten J, De Vos K, Fiers W. Direct evidence for tumor necrosis factor-induced mitochondrial reactive oxygen intermediates and their involvement in cytotoxicity. Proc Natl Acad Sci U S A. 1995;92:8115–8119. doi: 10.1073/pnas.92.18.8115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottsch JD, Pou S, Bynoe LA, Rosen GM. Hematogenous photosensitization. A mechanism for the development of age-related macular degeneration. Invest Ophthalmol Vis Sci. 1990;31:1674–1682. [PubMed] [Google Scholar]
- Green WR, McDonnell PJ, Yeo JH. Pathologic features of senile macular degeneration. Ophthalmology. 1985;92:615–627. [PubMed] [Google Scholar]
- Grossniklaus HE, Ling JX, Wallace TM, Dithmar S, Lawson DH, Cohen C, Elner VM, Elner SG, Sternberg P., Jr Macrophage and retinal pigment epithelium expression of angiogenic cytokines in choroidal neovascularization. Mol Vis. 2002;8:119–126. [PubMed] [Google Scholar]
- Guyton KZ, Liu Y, Gorospe M, Xu Q, Holbrook NJ. Activation of mitogen-activated protein kinase by H2O2. Role in cell survival following oxidant injury. J Biol Chem. 1996;271:4138–4142. doi: 10.1074/jbc.271.8.4138. [DOI] [PubMed] [Google Scholar]
- Hageman GS, Luthert PJ, Victor Chong NH, Johnson LV, Anderson DH, Mullins RF. An integrated hypothesis that considers drusen as biomarkers of immune-mediated processes at the RPE-Bruch’s membrane interface in aging and age related macular degeneration. Prog Retin Eye Res. 2001;20:705–732. doi: 10.1016/s1350-9462(01)00010-6. [DOI] [PubMed] [Google Scholar]
- Harris GK, Shi X. Signaling by carcinogenic metals and metal-induced reactive oxygen species. Mutat Res. 2003;533:183–200. doi: 10.1016/j.mrfmmm.2003.08.025. [DOI] [PubMed] [Google Scholar]
- Hermann AC, Millard PJ, Blake SL, Kim CH. Development of a respiratory burst assay using zebrafish kidneys and embryos. J Immunol Methods. 2004;292:119–129. doi: 10.1016/j.jim.2004.06.016. [DOI] [PubMed] [Google Scholar]
- Higgins GT, Wang JH, Dockery P, Cleary PE, Redmond HP. Induction of angiogenic cytokine expression in cultured RPE by ingestion of oxidized photoreceptor outer segments. Invest Ophthalmol Vis Sci. 2003;44:1775–1782. doi: 10.1167/iovs.02-0742. [DOI] [PubMed] [Google Scholar]
- Hollborn M, Kohen L, Wiedemann P, Enzmann V. The influence of pro-inflammatory cytokines on human retinal pigment epithelium cell receptors. Graefes Arch Clin Exp Ophthalmol. 2001;239:294–301. doi: 10.1007/s004170100263. [DOI] [PubMed] [Google Scholar]
- Hwang YS, Jeong M, Park JS, Kim MH, Lee DB, Shin BA, Mukaida N, Ellis LM, Kim HR, Ahn BW, Jung YD. Interleukin-1beta stimulates IL-8 expression through MAP kinase and ROS signaling in human gastric carcinoma cells. Oncogene. 2004;23:6603–6611. doi: 10.1038/sj.onc.1207867. [DOI] [PubMed] [Google Scholar]
- Jaffe GJ, Van Le L, Valea F, Haskill S, Roberts W, Arend WP, Stuart A, Peters WP. Expression of interleukin-1 alpha, interleukin-1 beta, and an interleukin-1 receptor antagonist in human retinal pigment epithelial cells. Exp Eye Res. 1992;55:325–335. doi: 10.1016/0014-4835(92)90197-z. [DOI] [PubMed] [Google Scholar]
- Jarrett SG, Albon J, Boulton M. The contribution of DNA repair and antioxidants in determining cell type-specific resistance to oxidative stress. Free Radic Res. 2006;40:1155–1165. doi: 10.1080/10715760600876613. [DOI] [PubMed] [Google Scholar]
- Jung YD, Fan F, McConkey DJ, Jean ME, Liu W, Reinmuth N, Stoeltzing O, Ahmad SA, Parikh AA, Mukaida N, Ellis LM. Role of P38 MAPK, AP-1, and NF-kappaB in interleukin-1beta-induced IL-8 expression in human vascular smooth muscle cells. Cytokine. 2002;18:206–213. doi: 10.1006/cyto.2002.1034. [DOI] [PubMed] [Google Scholar]
- Kannan R, Jin M, Gamulescu MA, Hinton DR. Ceramide-induced apoptosis: role of catalase and hepatocyte growth factor. Free Radic Biol Med. 2004;37:166–175. doi: 10.1016/j.freeradbiomed.2004.04.011. [DOI] [PubMed] [Google Scholar]
- Karlsson A, Dahlgren C. Assembly and activation of the neutrophil NADPH oxidase in granule membranes. Antioxid Redox Signal. 2002;4:49–60. doi: 10.1089/152308602753625852. [DOI] [PubMed] [Google Scholar]
- Kaur J, Dhaunsi GS, Turner RB. Interleukin-1 and nitric oxide increase NADPH oxidase activity in human coronary artery smooth muscle cells. Med Princ Pract. 2004;13:26–29. doi: 10.1159/000074047. [DOI] [PubMed] [Google Scholar]
- Kim JH, Lee BC, Kim JH, Sim GS, Lee DH, Lee KE, Yun YP, Pyo HB. The isolation and antioxidative effects of vitexin from Acer palmatum. Arch Pharm Res. 2005;28:195–202. doi: 10.1007/BF02977715. [DOI] [PubMed] [Google Scholar]
- Kindzelskii AL, Elner VM, Elner SG, Yang D, Hughes BA, Petty HR. Human, but not bovine, photoreceptor outer segments prime human retinal pigment epithelial cells for metabolic activation and massive oxidant release in response to lipopolysaccharide and interferon-gamma. Exp Eye Res. 2004;79:431–435. doi: 10.1016/j.exer.2004.06.005. [DOI] [PubMed] [Google Scholar]
- Kopprasch S, Pietzsch J, Graessler J. Validation of different chemilumigenic substrates for detecting extracellular generation of reactive oxygen species by phagocytes and endothelial cells. Luminescence. 2003;18:268–273. doi: 10.1002/bio.737. [DOI] [PubMed] [Google Scholar]
- Laderoute KR, Webster KA. Hypoxia/reoxygenation stimulates Jun kinase activity through redox signaling in cardiac myocytes. Circ Res. 1997;80:336–344. doi: 10.1161/01.res.80.3.336. [DOI] [PubMed] [Google Scholar]
- Lander HM. An essential role for free radicals and derived species in signal transduction. FASEB J. 1997;11:118–124. [PubMed] [Google Scholar]
- Li H, Burkhardt C, Heinrich UR, Brausch I, Xia N, Forstermann U. Histamine upregulates gene expression of endothelial nitric oxide synthase in human vascular endothelial cells. Circulation. 2003;107:2348–2354. doi: 10.1161/01.CIR.0000066697.19571.AF. [DOI] [PubMed] [Google Scholar]
- Liles MR, Newsome DA, Oliver PD. Antioxidant enzymes in the aging human retinal pigment epithelium. Arch Ophthalmol. 1991;109:1285–1288. doi: 10.1001/archopht.1991.01080090111033. [DOI] [PubMed] [Google Scholar]
- Lu L, Hackett SF, Mincey A, Lai H, Campochiaro PA. Effects of different types of oxidative stress in RPE cells. J Cell Physiol. 2006;206:119–125. doi: 10.1002/jcp.20439. [DOI] [PubMed] [Google Scholar]
- Mendes AF, Caramona MM, Carvalho AP, Lopes MC. Hydrogen peroxide mediates interleukin-1beta-induced AP-1 activation in articular chondrocytes: implications for the regulation of iNOS expression. Cell Biol Toxicol. 2003;19:203–214. doi: 10.1023/b:cbto.0000003730.21261.fa. [DOI] [PubMed] [Google Scholar]
- Miceli MV, Liles MR, Newsome DA. Evaluation of oxidative processes in human pigment epithelial cells associated with retinal outer segment phagocytosis. Exp Cell Res. 1994;214:242–249. doi: 10.1006/excr.1994.1254. [DOI] [PubMed] [Google Scholar]
- Miller H, Miller B, Ryan SJ. The role of retinal pigment epithelium in the involution of subretinal neovascularization. Invest Ophthalmol Vis Sci. 1986;27:1644–1652. [PubMed] [Google Scholar]
- Newsome DA, Dobard EP, Liles MR, Oliver PD. Human retinal pigment epithelium contains two distinct species of superoxide dismutase. Invest Ophthalmol Vis Sci. 1990;31:2508–2513. [PubMed] [Google Scholar]
- Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP, Giardino I, Brownlee M. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404:787–790. doi: 10.1038/35008121. [DOI] [PubMed] [Google Scholar]
- Oh H, Takagi H, Takagi C, Suzuma K, Otani A, Ishida K, Matsumura M, Ogura Y, Honda Y. The potential angiogenic role of macrophages in the formation of choroidal neovascular membranes. Invest Ophthalmol Vis Sci. 1999;40:1891–1898. [PubMed] [Google Scholar]
- Pearl-Yafe M, Halperin D, Halevy A, Kalir H, Bielorai B, Fabian I. An oxidative mechanism of interferon induced priming of the Fas pathway in Fanconi anemia cells. Biochem Pharmacol. 2003;65:833–842. doi: 10.1016/s0006-2952(02)01620-9. [DOI] [PubMed] [Google Scholar]
- Pearl-Yafe M, Halperin D, Scheuerman O, Fabian I. The p38 pathway partially mediates caspase-3 activation induced by reactive oxygen species in Fanconi anemia C cells. Biochem Pharmacol. 2004;67:539–546. doi: 10.1016/j.bcp.2003.09.024. [DOI] [PubMed] [Google Scholar]
- Penfold PL, Madigan MC, Gillies MC, Provis JM. Immunological and aetiological aspects of macular degeneration. Prog Retin Eye Res. 2001;20:385–414. doi: 10.1016/s1350-9462(00)00025-2. [DOI] [PubMed] [Google Scholar]
- Rao NA. Role of oxygen free radicals in retinal damage associated with experimental uveitis. Trans Am Ophthalmol Soc. 1990;88:797–850. [PMC free article] [PubMed] [Google Scholar]
- Reddy VM, Zamora RL, Kaplan HJ. Distribution of growth factors in subfoveal neovascular membranes in age-related macular degeneration and presumed ocular histoplasmosis syndrome. Am J Ophthalmol. 1995;120:291–301. doi: 10.1016/s0002-9394(14)72158-0. [DOI] [PubMed] [Google Scholar]
- Sakamoto T, Sakamoto H, Murphy TL, Spee C, Soriano D, Ishibashi T, Hinton DR, Ryan SJ. Vessel formation by choroidal endothelial cells in vitro is modulated by retinal pigment epithelial cells. Arch Ophthalmol. 1995;113:512–520. doi: 10.1001/archopht.1995.01100040134039. [DOI] [PubMed] [Google Scholar]
- Samiec PS, Drews-Botsch C, Flagg EW, Kurtz JC, Sternberg P, Jr, Reed RL, Jones DP. Glutathione in human plasma: decline in association with aging, age-related macular degeneration, and diabetes. Free Radic Biol Med. 1998;24:699–704. doi: 10.1016/s0891-5849(97)00286-4. [DOI] [PubMed] [Google Scholar]
- Schutze S, Berkovic D, Tomsing O, Unger C, Kronke M. Tumor necrosis factor induces rapid production of 1’2’diacylglycerol by a phosphatidylcholine-specific phospholipase C. J Exp Med. 1991;174:975–988. doi: 10.1084/jem.174.5.975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze-Osthoff K, Bakker AC, Vanhaesebroeck B, Beyaert R, Jacob WA, Fiers W. Cytotoxic activity of tumor necrosis factor is mediated by early damage of mitochondrial functions. Evidence for the involvement of mitochondrial radical generation. J Biol Chem. 1992;267:5317–5323. [PubMed] [Google Scholar]
- Schulze-Osthoff K, Beyaert R, Vandevoorde V, Haegeman G, Fiers W. Depletion of the mitochondrial electron transport abrogates the cytotoxic and gene inductive effects of TNF. EMBO J. 1993;12:3095–3104. doi: 10.1002/j.1460-2075.1993.tb05978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seregard S, Algvere PV, Berglin L. Immunohistochemical characterization of surgically removed subfoveal fibrovascular membranes. Graefes Arch Clin Exp Ophthalmol. 1994;232:325–329. doi: 10.1007/BF00175983. [DOI] [PubMed] [Google Scholar]
- Shanker G, Aschner JL, Syversen T, Aschner M. Free radical formation in cerebral cortical astrocytes in culture induced by methylmercury. Brain Res Mol Brain Res. 2004;128:48–57. doi: 10.1016/j.molbrainres.2004.05.022. [DOI] [PubMed] [Google Scholar]
- Tate DJ, Jr, Newsome DA, Oliver PD. Metallothionein shows an age-related decrease in human macular retinal pigment epithelium. Invest Ophthalmol Vis Sci. 1993;34:2348–2351. [PubMed] [Google Scholar]
- Tate DJ, Miceli MV, Newsome DA, Alcock NW, Oliver PD. Influence of zinc on selected cellular functions of cultured human retinal pigment epithelium. Curr Eye Res. 1995a;14:897–903. doi: 10.3109/02713689508995129. [DOI] [PubMed] [Google Scholar]
- Tate DJ, Jr, Miceli MV, Newsome DA. Phagocytosis and H2O2 induce catalase and metallothionein gene expression in human retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. 1995b;36:1271–1279. [PubMed] [Google Scholar]
- Tate DJ, Newsome DA. A novel zinc compound (zinc monocysteine) enhances the antioxidant capacity of human retinal pigment epithelial cells. Curr Eye Res. 2006;31:675–683. doi: 10.1080/02713680600801024. [DOI] [PubMed] [Google Scholar]
- Till GO, Lutz MJ, Ward PA. Hydroxy radical as autotoxin in chemotactically activated neutrophils. Biomed Pharmacother. 1987;41:349–354. [PubMed] [Google Scholar]
- Till GO, Mahrougui M, Elner VM, Marak GE, Elner SG. Inflammatory mediator-induced hydrogen peroxide production by retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. 1991;32:1187–1187. [Google Scholar]
- Touyz RM, Schiffrin EL. Increased generation of superoxide by angiotensin II in smooth muscle cells from resistance arteries of hypertensive patients: role of phospholipase D-dependent NAD(P)H oxidase-sensitive pathways. J Hypertens. 2001;19:1245–1254. doi: 10.1097/00004872-200107000-00009. [DOI] [PubMed] [Google Scholar]
- Tso MO. Retinal photic injury in normal and scorbutic monkeys. Trans Am Ophthalmol Soc. 1987;85:498–556. [PMC free article] [PubMed] [Google Scholar]
- Truscott RJ. Age-related nuclear cataract: a lens transport problem. Ophthalmic Res. 2000;32:185–194. doi: 10.1159/000055612. [DOI] [PubMed] [Google Scholar]
- Wassell J, Davies S, Bardsley W, Boulton M. The photoreactivity of the retinal age pigment lipofuscin. J Biol Chem. 1999;274:23828–23832. doi: 10.1074/jbc.274.34.23828. [DOI] [PubMed] [Google Scholar]
- Watanabe Y, Suzuki O, Haruyama T, Akaike T. Interferon-gamma induces reactive oxygen species and endoplasmic reticulum stress at the hepatic apoptosis. J Cell Biochem. 2003;89:244–253. doi: 10.1002/jcb.10501. [DOI] [PubMed] [Google Scholar]
- Winkler BS, Boulton ME, Gottsch JD, Sternberg P. Oxidative damage and age-related macular degeneration. Mol Vis. 1999;5:32. [PMC free article] [PubMed] [Google Scholar]
- Wu GS, Rao NA. Activation of NADPH oxidase by docosahexaenoic acid hydroperoxide and its inhibition by a novel retinal pigment epithelial protein. Invest Ophthalmol Vis Sci. 1999;40:831–839. [PubMed] [Google Scholar]
- Wu GS. The functional interactions between the p53 and MAPK signaling pathways. Cancer Biol Ther. 2004;3:156–161. doi: 10.4161/cbt.3.2.614. [DOI] [PubMed] [Google Scholar]
- Yamagishi SI, Edelstein D, Du XL, Kaneda Y, Guzman M, Brownlee M. Leptin induces mitochondrial superoxide production and monocyte chemoattractant protein-1 expression in aortic endothelial cells by increasing fatty acid oxidation via protein kinase A. J Biol Chem. 2001;276:25096–25100. doi: 10.1074/jbc.M007383200. [DOI] [PubMed] [Google Scholar]
- Yang D, Pan A, Swaminathan A, Kumar G, Hughes BA. Expression and localization of the inwardly rectifying potassium channel Kir7.1 in native bovine retinal pigment epithelium. Invest Ophthalmol Vis Sci. 2003;44:3178–3185. doi: 10.1167/iovs.02-1189. [DOI] [PubMed] [Google Scholar]
- Yang P, McKay BS, Allen JB, Jaffe GJ. Effect of NF-kappa B inhibition on TNF-alpha-induced apoptosis in human RPE cells. Invest Ophthalmol Vis Sci. 2004;45:2438–2446. doi: 10.1167/iovs.03-0805. [DOI] [PubMed] [Google Scholar]
- Yang P, Peairs JJ, Tano R, Jaffe GJ. Oxidant-mediated Akt activation in human RPE cells. Invest Ophthalmol Vis Sci. 2006;47:4598–4606. doi: 10.1167/iovs.06-0140. [DOI] [PubMed] [Google Scholar]
- Yoshida A, Elner SG, Bian ZM, Kunkel SL, Lukacs NW, Elner VM. Thrombin regulates chemokine induction during human retinal pigment epithelial cell/monocyte interaction. Am J Pathol. 2001;159:1171–1180. doi: 10.1016/S0002-9440(10)61793-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida A, Elner SG, Bian ZM, Kindezelskii AL, Petty HR, Elner VM. Activated monocytes induce human retinal pigment epithelial cell apoptosis through caspase-3 activation. Lab Invest. 2003;83:1117–1129. doi: 10.1097/01.lab.0000082393.02727.b5. [DOI] [PubMed] [Google Scholar]
- Young RW. Pathophysiology of age-related macular degeneration. Surv Ophthalmol. 1987;31:291–306. doi: 10.1016/0039-6257(87)90115-9. [DOI] [PubMed] [Google Scholar]