

Abstract
Acute motor axonal neuropathy (AMAN) variant of Guillain-Barré syndrome is often associated with IgG anti-GM1 and -GD1a antibodies. The pathophysiological basis of antibody-mediated selective motor nerve dysfunction remains unclear. We investigated the effects of IgG anti-GM1 and -GD1a monoclonal antibodies (mAbs) on neuromuscular transmission and calcium influx in hemidiaphragm preparations and in cultured neurons, respectively, to elucidate mechanisms of Ab-mediated muscle weakness. Anti-GM1 and -GD1a mAbs depressed evoked quantal release to a significant yet different extent, without affecting postsynaptic currents. At equivalent concentrations, anti-GD1b, -GT1b, or sham mAbs did not affect neuromuscular transmission. At fourfold higher concentration, an anti-GD1b mAb (specificity described in immune sensory neuropathies) induced completely reversible blockade. In neuronal cultures, anti-GM1 and -GD1a mAbs significantly reduced depolarization-induced calcium influx. In conclusion, different anti-gangliosde mAbs induce distinct effects on presynaptic transmitter release by reducing calcium influx, suggesting that this is one mechanism of antibody-mediated muscle weakness in AMAN.
Keywords: Guillain-Barré syndrome, AMAN, anti-ganglioside antibodies, gangliosides, immune neuropathies, neuromuscular junction, calcium influx
Introduction
Anti-ganglioside antibodies (Abs) are implicated as primary immune effectors in acute motor axonal neuropathy (AMAN) and Fisher syndrome (FS) variants of Guillain-Barré syndrome (GBS). Recent studies show that IgG anti-GQ1b Abs in FS and IgG Abs to GD1a, GM1, and structurally related gangliosides are strongly associated with AMAN (Hughes et al., 1999; Latov., 1990; Willison and Yuki., 2002; Yuki., 2001). Passive and active immunization models indicate that anti-GD1a and -GM1 antibodies produce axonal neuropathy (Sheikh et al., 2004; Yuki et al., 2001). In contrast to AMAN, acute and chronic sensory ataxic neuropathies are associated with Abs to GD1b, with or without cross-reactivity to other gangliosides containing disialyosyl moieties (Miyazaki et al., 2001; Willison et al., 1994). Understanding the mechanism by which anti-ganglioside Abs with varied specificities produce distinct clinical features associated with different forms of GBS is an important pathobiologic issue relevant to several autoimmune neurological disorders.
Although, in AMAN muscle weakness is the predominant clinical finding (McKhann et al., 1991; McKhann et al., 1993) its pathophysiology remains incompletely defined. The following observations suggest that blockade of axonal conduction, in addition to motor axonal degeneration, contributes significantly to clinical weakness in AMAN. First, many patients with AMAN recover quite quickly, with or without intravenous immunoglobulin (IVIg) treatment, and their time course of recovery is incompatible with degeneration and regeneration of nerve fibers (Ho et al., 1997; Kuwabara et al., 1998). Second, pathologic studies on AMAN patients and its rabbit model indicate that some subjects, despite severe flaccid paralysis and abnormal nerve conductions, do not show nerve fiber degeneration, raising the possibility that axonal degeneration is a late but not essential event in the pathogenesis of acute muscle weakness in AMAN (Griffin et al., 1996; Susuki et al., 2003). Third, serial clinical electrophysiological observations in GBS cases with IgG anti-GM1 Abs (Kuwabara et al., 1998) show rapid recovery of CMAP amplitudes and motor conduction slowing without features of remyelination suggesting a reversible conduction failure at the level of axon. Whether anti-ganglioside Abs associated with AMAN can induce physiological blockade of conduction is a matter of debate.
Electrophysiological studies at the mouse neuromuscular junction (NMJ) have demonstrated that serum and IgG fractions from patients with GBS and its variants which contain antibodies to various gangliosides block neuromuscular transmission (Buchwald et al., 1998a; Buchwald et al., 2001). We asked whether application of anti-GM1 and -GD1a monoclonal Abs (mAbs) by the perfused macro-patch clamp electrode can induce neuromuscular blockade without complement-mediated structural damage. Because calcium homeostasis is pivotal for presynaptic transmitter release and normal motor nerve terminal function, we investigated the effects of anti-ganglioside Abs on depolarization-induced calcium influx by calcium imaging in cultured olfactory bulb neurons, which is a convenient model because: a) olfactory neurons are known to express P/Q type calcium channels (Isaacson and Strowbridge., 1998; Takahashi and Nagasu., 2005), which are also present at the NMJ (Santafe et al., 2005); and b) lack of glia in these cultures allows study of the direct effects of mAbs on neuronal calcium channels.
Materials and Methods
Monoclonal Abs
Anti-ganglioside mAbs used in this study are designated according to their ganglioside specificity and IgG isotype (1, 2a, 2b); for example, GD1a/GT1b-2b refers to mAb with GD1a and GT1b specificity and IgG2b isotype and GM1-2b refers to mAb with GM1 specificity and IgG2b isotype. The following five mAbs were examined: GM1-2b, GD1a-2a, GD1a/GT1b-2b, GD1b-1, and GT1b-2b. mAb GD1b-1 was selected for comparison because this specificity is particularly relevant for ataxic sensory neuropathies. These mAbs have distinct specificity for major nervous system gangliosides and binding patterns to peripheral nervous system (Gong et al., 2002). The generation, specificity, and purification of anti-ganglioside and control mAbs used in this study were reported previously (Gong et al., 2002; Lunn et al., 2000; Schnaar et al., 2002). These mAbs were used for immunohistochemistry, electrophysiology, and calcium imaging studies. A mouse monoclonal IgG Ab, HB-94, with specificity for a combinatorial determinant of the human HLA A,B,C-beta-2-microglobulin complex was used as control (Brodsky and Parham., 1982).
Immunostaining of NMJs
Diaphragms were harvested from 12- to- 16-week-old Balb-c mice and fixed in 2% paraformaldehyde for 2 h at 4°C. Diaphragms were immunostained with anti-ganglioside mAbs (50–100 μg/ml in blocking buffer containing 0.1% Triton overnight) and developed with anti-mouse IgG conjugated to FITC (1:100, 90 min at room temperature; Vector Laboratory, Burlingame, CA) and motor end plates were simultaneously labeled with α-bungarotoxin conjugated to Alexa Fluor® 594 (2 μg/ml; Molecular Probes, Eugene, OR). Whole muscles were mounted and examined by epi-fluorescent and confocal microscopy. To determine whether anti-GM1 mAb binds presynaptic Schwann cells, immunostaining was done with GM1-2b mAb (100 μg/ml) and rabbit anti-S100 (Sigma-Aldich; 1:200) in blocking buffer containing 0.05% Triton overnight on 100 μm cryostat sections of the diaphragm. These sections were developed with specific anti-mouse (1:200) and -rabbit (1:200) secondary antibodies conjugated to different fluorophores (Vector Laboratory) and α-bungarotoxin conjugated to Alexa Fluor® 594 and examined by confocal microscopy.
Perfused macro-patch clamp technique
Experiments were performed on hemidiaphragms of 12-week-old male BALB/c mice at 20 +/− 0.5°C as previously described (Buchwald et al., 1998a). Endplate currents were recorded by means of a perfused macro-patch-clamp electrode, a type of extracellular voltage clamp recording, as described (Buchwald et al., 1998a; Dudel., 1989). In brief, the electrode with a 10-μm wide opening covers part of the pre- and postsynaptic membrane with its adjacent Schwann cell. In addition, the electrode is equipped with a fine tube that enables perfusion of the tip of the electrode with pressurized solution. With this set-up it is possible to record quantal excitatory postsynaptic currents corresponding to the release of one acetylcholine-containing presynaptic vesicle and to apply Abs directly to the nerve terminal while recording form the synaptic site. To prevent twitches of the diaphragm, tetrodotoxin (2 × 10−7 M), a sodium channel blocker was added to the bath solution and to the solution perfusing the electrode.
At the beginning of each experiment the electrode was perfused with buffer, the position of the electrode was then optimized and qEPSC were recorded and counted. When recording conditions were stable, mAbs (diluted in HEPES buffer) at a defined concentration were added to the perfusate of the electrode while recording from the selected nerve terminal upon stimulating with a fixed depolarizing current pulse (−0.3 to −06 pA, 0.5 ms, 5 Hz). All mAbs were investigated at 50–100 μg/ml for examining concentration-dependence of physiologic effects; if no physiologic effect was found at 50–100 μg/ml higher concentrations (up to 200 μg/ml) were used. Similar concentrations of mAbs have been used by other investigators with the in vitro mouse hemidiaphragm models (Goodyear et al., 1999; Halstead et al., 2005; O’Hanlon et al., 2003).
For measurement of quantal release, a parameter for presynaptic function, the effects of n=28 to 212 pulses were evaluated as described previously (Buchwald et al., 1998b). An automatic discriminator determined whether any release occurred during a time window of 4 msec after each pulse, and counted the number of failures (n0). The quantal content m was calculated from the proportion of failures, n0/n, by using the Poisson formula m = ln (n/n0). In the figures the quantal content is shown in absolute values for the depicted experiment at the given time point. In Table 1 the quantal content is given relative to the control, normalized at 1, as mean +/− SD for a number of independent experiments. For the postsynaptic analysis, recordings were filtered at 50 kHz, digitized, and stored for later analysis (ISO, MfK software). qEPSC amplitudes, rise times, decays and delay times were analyzed with ISO, MfK software. Results were expressed as mean +/− SE values of data from the number (n) of qEPSCs evaluated. Statistical analysis was done with a commercially available computer program (Origin, Microcal Software, Northampton, MA). p values were calculated by using Student’s t-test for grouped data after determining that the data were normally distributed.
Table 1.
Changes in quantal content and response to washout with different mAbs used in this study
| mAbs | Ab concentration (μg/ml) | Normalized quantal content m* (Mean +/− SD) | Reversibility after washout |
|---|---|---|---|
| GM1-2b | 50–100 | 0.20+/−0.08 | Partly reversible |
| GD1a-1 | 50–100 | 0.28+/−0.06 | Partly reversible |
| 200 | 0.09+/−0.05 | Irreversible | |
| GD1a-2a | 50–100 | 0.11+/−0.07 | Partly reversible |
| GD1a/GT1b-2b | 100 | 0.25+/−0.04 | Partly reversible |
| GD1b-1 | 100 | 0.95+/−0.09 | No blockade |
| 200 | 0.31+/−0.07 | Fully reversible | |
| GT1b-2b | 100–200 | 1.02+/−0.16 | No blockade |
| HB94 | 100 | 1.17+/−0.22 | No blockade |
Quantal content m is given relative to the control normalized at 1 as mean +/− SD for at least 6 to 10 independent experiments.
Ca2+ imaging
These studies were done on olfactory bulb neurons, because it has been shown that calcium channels of the P/Q class play a dominant role in synaptic transmission in these neurons (Isaacson and Strowbridge., 1998). Olfactory bulb neurons were prepared as previously described (Brunig et al., 1999). Briefly, olfactory bulbs were obtained from neonatal C57Bl/6 mice (P1–P3) and digested in trypsin for 20 minutes at 37°C, washed twice in L15, and dissociated mechanically by using a fire-polished Pasteur pipette. Cells were resuspended in Neurobasal Medium (Invitrogen) containing 2 ml of B27 (Invitrogen) and 500 μl of Glutamax (Invitrogen) per 100 ml, seeded onto polylysine-coated (mol. wt. 70 000–150 000, Sigma) cell culture dishes (Nunclon) at a density of 200,000 cells/cm2, and cultured for 7–10 days (cultures are practically devoid of glia under these serum-free conditions (Brewer et al., 1993). Then, medium was removed from neuronal cultures and replaced with the Ca-sensitive dye Fura-2/AM (3 μM) (Molecular Probes) in extracellular solution containing 140 mM NaCl, 5 mM KCl, 2 mM MgCl2, 2 mM CaCl2, 10 mM HEPES, 10 mM glucose, pH 7.4. Cells were loaded for 30 min at room temperature and then incubated for 30 min with different Abs at a final concentration of 10 μg/ml. The cells were examined with a Zeiss IM100 inverted microscope equipped for ratiometric imaging (Tillvision) with 32× magnification. All cells in a field of view were illuminated every second for 75 msec at 340 nm and 75 msec at 380 nm. The average pixel intensity within the user-selected regions of interest, corresponding to the individual cells, was digitized and stored. The Ca2+-dependent fluorescence signal at 510 nm was expressed as the F340/F380 ratio and viewed as a function of time.
For each recording, cells were exposed to three pulses of high potassium extracellular solution (containing NaCl 110 mM, KCl 60 mM, MgCl2 1 mM, CaCl2 5 mM, HEPES 10 mM, glucose 10 mM, pH 7.4) to open voltage-gated calcium channels. Each pulse lasted for 7 sec and the pulse-to-pulse interval was 100 sec. An application system was used that could transiently superfuse all the cells in the field of view from one of seven capillary tubes. Switching time between high potassium and background solution and the delay of onset after switching were essentially instantaneous because of the close proximity of the tube tips to the optical field. A constant stream of extracellular solution superfused all the cells in the dish between applications to minimize the accumulation of potassium in the bath and to exclude the possibility that the observed effects arose as a result of mechanical stimulation from the solution flow.
Results
Anti-ganglioside mAbs bind terminal motor axons at NMJ
As shown by immunocytochemistry all anti-ganglioside mAbs examined in the present study stained motor nerve terminals and terminal axonal sprouts (Fig. 1). Some of the mAbs appeared to co-localize with motor end plate staining in 2-D reconstructions; however, 3-D confocal reconstructions of NMJ (Z series) showed that α-bungarotoxin and anti-ganglioside staining did not co-localize precisely. These studies suggest that, under immunolabeling conditions, mAbs preferentially deposit on the presynaptic terminals and not on the postsynaptic membrane where α-bungarotoxin binds exclusively. Co-localization studies with GM1-2b and anti-S100 antibodies showed that this mAb had no detectable binding to presynaptic Schwann cells on 3-D confocal reconstructions of NMJ (Z series) (Fig. 2). In the 2-D merged figure (bottom right panel) few spots appear to colocalize but this is likely due to close apposition of Schwann cell processes and axons.
Figure 1.

Neuromuscular junctions double labeled with anti-ganglioside mAbs (green) and α-bungarotoxin (red). 2D co-localization is also shown (Merge).
Figure 2.
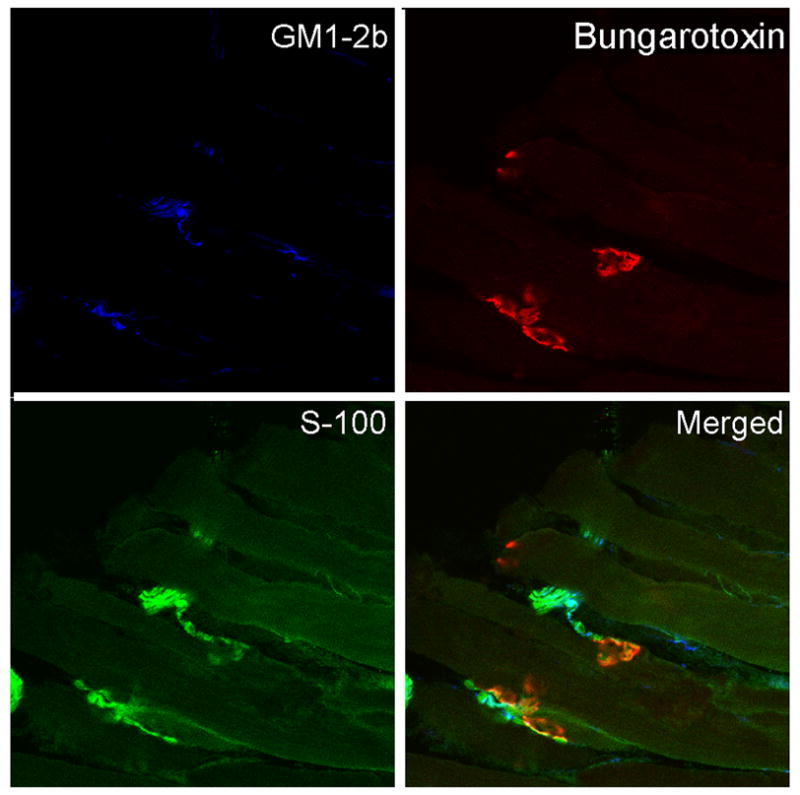
Neuromuscular junctions labeled with anti-ganglioside mAbs (blue), anti-S100 (green) and α-bungarotoxin (red). 2D co-localization is also shown (Merge).
Distinct blocking effects of anti-ganglioside mAbs on presynaptic transmitter release
First, we investigated the effects of the anti-GM1 mAbs on presynaptic transmitter release by means of the perfused macro-patch-clamp electrode in mouse hemidiaphragms. Application of GM1-2b directly to the nerve terminals significantly depressed evoked quantal release within minutes (Fig. 3A). This blockade was not reversed after washout with control buffer. GD1a-1 also caused a partially reversible blockade of presynaptic transmitter release (Fig. 3B) at low mAb concentrations (50–100 μg/ml), whereas higher mAb concentration (200 μg/ml) caused irreversible blockade of presynaptic transmitter release (Fig. 3C). GD1a-2a induced a pronounced blockade of presynaptic transmitter release, which, in most of the experiments, could be partly reversed after washout (Fig. 3D). GD1a/GT1b-2b mAb depressed evoked quantal release in a partly reversible manner (Fig. 3E). In comparison, mAb GD1b-1, an Ab specificity associated with immune-mediated sensory neuropathies, did not interfere with presynaptic transmitter release at concentrations of 50–100 μg/ml (a concentration range that clearly produced presynaptic dysfunction with GM1- and GD1a-reactive mAbs) (Fig. 3F). A higher concentration of GD1b-1 (200 μg/ml) induced a significant blockade of presynaptic transmitter release; this blockade was completely reversible after washout (Fig. 3G). Sham Ab (Hb94; data not shown) and GT1b-2b (at concentrations up to 200 μg/ml) did not significantly affect presynaptic transmitter release and quantal release remained stable over 90 minutes (Fig. 3H). That inactivity of GT1b-2b mAb was not due to degradation was confirmed by using three different batches of GT1b-2b mAb that specifically bound to GT1b ganglioside by ELISA according to previously published methods (Zhang et al., 2004). We observed that anti-ganglioside Ab-mediated blockade was partially overcome by stronger depolarization of the nerve terminal, confirming the presynaptic nature of the blockade (data not shown), similar to experiments with IgG fractions from GBS patients reported previously (Buchwald et al., 1998b). Table 1 summarizes the distinct effects of different Abs used in this study on presynaptic transmitter release.
Figure 3.

Effect of anti-GM1 and -GD1a mAbs on presynaptic transmitter release. The time course of evoked quantal release after application of the mAb is shown along with the quanta per pulse, m (logarithmic ordinate scale) versus time in minutes (abscissa). Each point of the curve was determined from the results of at least 256 stimuli; the quantal content (m) was calculated using the Poisson formula. Arrows represent the instant of solution change in the electrode. Evoked quantal release was depressed by GM1-2b (A). mAb GD1a-1 induced a partly reversible block at low concentration (B) and an irreversible blockade at high concentration (C). Different degrees of partly reversible blockade of presynaptic transmitter release occur after application of mAbs GD1a-2a (D) and GD1a/GT1b-2b (E). GD1b-1 at 50–100 μg/ml did not induce any blocking effect (F), but 200 μg/ml induced a completely reversible blockade of evoked quantal release (G). In comparison, presynaptic quantal release was not affected by mAbGT1b-2b (H).
Anti-ganglioside mAbs do not affect the amplitude of postsynaptic currents
To address whether the different mAbs may also interfere with postsynaptic currents as were seen with IgG fractions from some patients with FS and GBS (Buchwald et al., 1998b; Buchwald et al., 2001; Krampfl et al., 2003), we analyzed the amplitude of quantal excitatory postsynaptic currents (qEPSC). Figure 4 depicts the amplitude distribution under control settings and before and during application of the mAb GD1a/GT1b-2b (50μg/ml). The histogram shows the number of qEPSCs observed at each amplitude. The peaks of the histogram occur at one and two times the mean amplitude of qEPSCs, reflecting the release of single and double quanta. The solid line represents the distribution for single quanta with a mean of 1164 +/− 370 pA under control conditions (Fig. 4A). In the presence of GD1a/GT1b-2b (Fig. 4B), the mean amplitude of single quanta (1106 +/− 239 pA) was not significantly (p=1) changed, indicating that the mAb did not affect postsynaptic AChR channels. The observation that during application of the GD1a/GT1b-2b the number of double quanta was reduced is in accord with the observed presynaptic blockade. None of the mAbs investigated in the present study significantly reduced the amplitude of qEPSCs, indicating that these mouse mAbs do not affect the amplitude of postsynaptic currents. The absence of a postsynaptic blocking effect is in agreement with the 3D-confocal observation that the anti-ganglioside mAbs did not stain the postsynaptic membranes.
Figure 4.
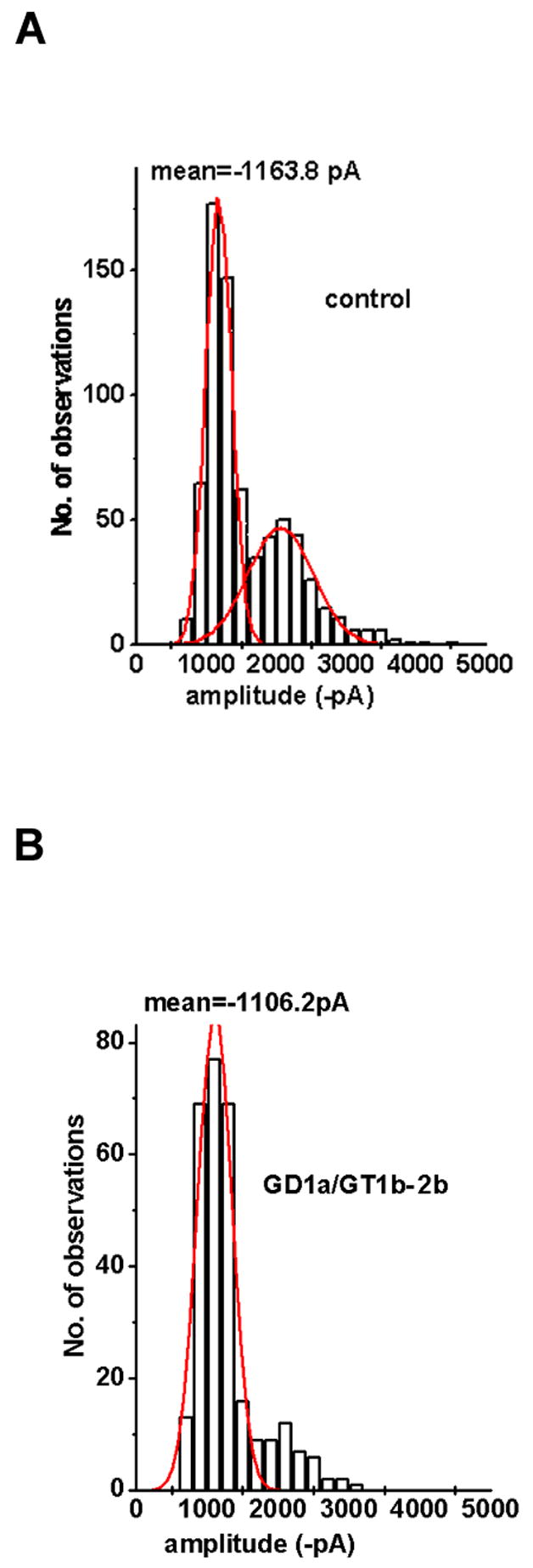
Amplitude distribution of quantal excitatory postsynaptic currents (qEPSCs) before and during application of mAb GD1a/GT1b-2b Ab. Abscissa: amplitude of qEPSCs, binwidth 200 pA. (A) Control solution. (B) mAb GD1a/GT1b-2b. Data from A and B were recorded at the same nerve terminal. The number of recordings evaluated for each distribution was 1,500 to 3,000 depending on quantal content.
Anti-ganglioside mAbs reduce calcium influx
Calcium imaging was performed in primary neuronal cultures to investigate whether the anti-ganglioside Ab-mediated presynaptic blockade is due to modulation of calcium influx. Neurons were superfused with pulses of a high potassium extracellular solution, which causes a shift in the membrane potential sufficient to open voltage-gated calcium channels. The dynamic changes in the intracellular calcium concentration were evaluated for the cells in the field of view. After incubation with the anti-ganglioside mAbs, the average amplitudes of increase in Ca(i) upon the first depolarization were not significantly changed. With subsequent pulses of high potassium extracellular solution, the respective amplitudes of Ca(i) were markedly reduced relative to the first amplitude after incubation with anti-GM1 and -GD1a mAbs (10 μg/ml) (Fig. 5A). Application of control mAb results in a stable train of responses and the amplitude of Ca(i) remained unchanged (Fig. 5B). The coefficient of 2nd to 1st amplitude was reduced from 1.3 (±0.8) to 0.5 (±1.2) and 0.7 (±0.2), p< 0.01 with GM1-2b and GD1a-2a mAbs, respectively (Fig. 5C). There was no reduction in amplitude of Ca (i) (coefficient of 2nd to 1st amplitude 1.1) with GT1b-2b or control mAbs (Fig. 5C).
Figure 5.
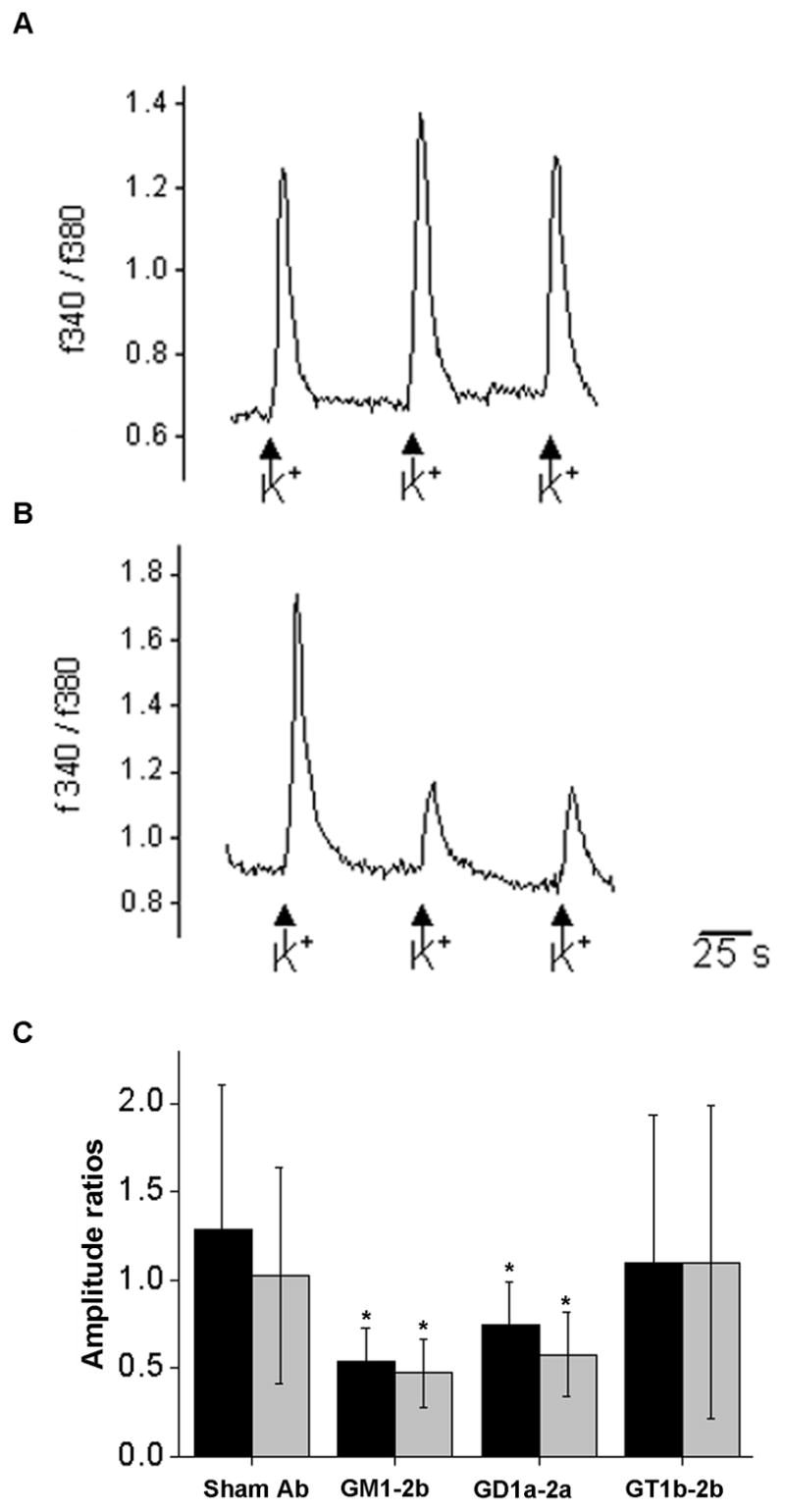
Repetitive application of high potassium extracellular solution induces transient increases of intracellular calcium in mouse olfactory bulb neurons. In neurons incubated with sham Ab or GT1b-2b, the average peak remains similar during three applications, whereas incubation with GM1-2b and GD1a-2a results in a decrease in amplitude during successive stimulations/applications. A) Representative trace of the fura 2-AM fluorescence ratio at 340 and 380 nm of a neuron incubated with sham Ab. B) Representative trace of a neuron incubated with GM1-2b. C) Coefficient of 2–1 (black bars) and 3–1 (gray bars) amplitude for neurons incubated with sham Ab, GM1-2b, GD1a-2a, or GT1b-2b. At least 50 neurons were measured from at least 3 different dishes for each condition.
Discussion
Our results establish that IgG anti-ganglioside mAbs with different specificities induce distinct pathophysiologically relevant effects on evoked quantal release in motor nerve terminals. The mAb-mediated decrease in presynaptic transmitter release is most likely to be due to reduction of depolarization-induced calcium influx and these effects are complement-independent. Anti-ganglioside mAbs associated with AMAN were more efficient blockers of presynaptic transmitter release than were Abs associated with immune sensory neuropathies. These findings support the concept that: a) functional blockade of motor nerve terminals by anti-GM1 and -GD1a Abs is one mechanism of muscle weakness in patients with AMAN; and b) clinicopathological heterogeneity in GBS may be due in part to differences in the specificities and thus functional effects of anti-ganglioside Abs that are present in individual patients.
Anti-ganglioside mAbs-mediated presynaptic (motor nerve terminal) blockade
In the present study, the neuromuscular transmission defect induced by anti-ganglioside Abs was exclusively due to a presynaptic blockade; the amplitudes of postsynaptic currents were not affected. Immunocytochemical studies indicated that anti-ganglioside Abs did not truly co-localize with the postsynaptic marker α-bungarotoxin (also reported by other investigators (Santafe et al., 2005)), thus providing one explanation for the lack of postsynaptic effects. The presynaptic blockade is due to direct effects of mAbs on motor nerve terminals and not indirectly via presynaptic Schwann cells, because GM1-2b mAb did not stain these glial cells. This is in line with the results of our calcium imaging showing activity of these mAbs on neurons in the absence of glial cells. Further, our previous studies indicate that anti-GD1a mAbs used in this study do not cause presynaptic Schwann cell injury (Goodfellow et al., 2005). Moreover, structural injury of axons is unlikely as the basis for presynaptic motor nerve dysfunction because the anti-GM1 and anti-GD1a mAb induced functional effects in our model were examined in the absence of complement and these effects could be partially reversed by antibody washout or by increasing depolarization currents.
Extent, time course, and reversibility of presynaptic blockade depended on the specificity of the mAb. The strongest blockade of evoked quantal release was induced by IgG anti-GM1 and -GD1a mAbs, reactivities often associated with the AMAN variant of GBS. In contrast, at equivalent concentrations, an anti-GD1b mAb, a specificity typically associated with sensory immune neuropathies, produced presynaptic blockade only at a fourfold higher concentration and with a distinc Pattern. Differences in mAb affinities is unlikely to be related to their distinct effects because our preliminary studies indicate that the affinities of the mAbs used in this study are similar (Lopez P, Sheik K, unpublished observations). These effects are specific; we found no functional blockade with anti-GT1b mAb despite the demonstration of its binding to motor nerve terminals as shown by immunohistochemistry. A limitation of the immunostaining studies is that these cannot distinguish differences in the subcellular distribution of gangliosides at the motor nerve terminal. It is possible that distinct pathophysiologic effects induced by different mAbs are due to the distribution of various gangliosides in distinct membrane raft fractions of terminal motor axons. This issue requires further studies.
mAbs-mediated decrease in calcium influx – Mechanism of presynaptic blockade
The voltage-dependence of the preynaptic blockade (see also (Buchwald et al., 1998b)) and calcium imaging results support the hypothesis that anti-ganglioside mAbs block calcium influx via P/Q type calcium channels. Notably, reduction in depolarization-induced calcium influx upon incubation with anti-GM1 and -GD1a mAbs was only seen in trains of responses, while first amplitudes remained unchanged. This is best explained by a mechanism that is based on the activation of a voltage-gated calcium channel, i.e., the channel has to be in an activated or open state before an anti-ganglioside mAb can bind and block. Our results support the notion that the alteration of calcium influx is a primary event, and may occur independent of motor nerve terminal degeneration, and that it involves activation-dependent block of P/Q type channels that consequently decreases the efficiency of neuromuscular transmission. Suppression of sodium currents does not contribute to presynaptic blockade, because all experiments were performed in the presence of tetrodotoxin, a sodium channel blocker.
Anti-ganglioside Ab-induced blockade of motor nerve terminal is a mechanism of muscle weakness in AMAN
Our results indicate that anti-ganglioside Ab-mediated presynaptic block may be an important step in the pathophysiology of early muscle weakness in GBS, especially in AMAN. A presynaptic block of calcium influx is known to reduce the safety margin of neuromuscular transmission, leading to muscle weakness. An Ab-mediated presynaptic blockade could also explain the impaired refractory period of transmission as measured electrophysiologically in AMAN patients (Kuwabara et al., 2002; Kuwabara et al., 2004). The reduced safety factor in distal nerves could well account for both conduction block in some fibers and the prolonged refractory period of transmission in others (Kuwabara et al., 2004). Similarly, the rapid recovery of the safety factor could account for parallel recovery of CMAP amplitude and reduction in refractoriness documented in AMAN patients (Kuwabara et al., 2002). These electrophysiological changes would best explain rapid disease evolution and recovery, independent of axon regeneration, which is not uncommon in patients with GBS (Griffin et al., 1995; Hartung et al., 1995; Ropper et al., 1991). That axonal dysfunction precedes the onset of axonal degeneration in axonal forms of GBS is supported by human and animal studies showing severe muscle weakness and changes in axonal physiology, but lack of significant axonal degeneration in autopsied tissues (Griffin et al., 1995; Griffin et al., 1996).
The complement-independent blocking activity of some anti-ganglioside mAbs on the presynaptic motor nerve terminal does not preclude the possibility that complement and other proinflammatory factors are critically involved in the ultimate structural damage to axons in patients with GBS and, more specifically, with AMAN (Hafer-Macko et al., 1996a; Hafer-Macko et al., 1996b). In clinical settings, the pathogenic processes acting in concert are typically much more complex than in our experimental model: even if only humorally-mediated mechanisms are considered, multiple factors are likely to contribute to the underlying pathophysiology, including the fine specificity, affinity/avidity, titers, and complement-fixing and/or nonfixing isotype(s) of anti-ganglioside Abs, and a balance between complement activation and inhibitory factors in the milieu surrounding NMJs, and even at other sites proximal to the nerve terminal along the peripheral nerve and spinal root.
In summary, our results provide direct experimental evidence for the hypothesis that anti-ganglioside Abs induce paralysis without complement-mediated nerve fiber destruction and that their fine specificity is crucial. Thus, one may propose that differences in disease expression and pattern of paralysis could partly be due to differences in antigen distribution and Ab reactivities in individual patients.
Acknowledgments
This work was supported by grants from the Deutsche Forschungsgemeinschaft DFG grants TO 61/9-1 (KVT and BB), SFB 391 TP8 (BB), and the National Institute of Health (NS42888 and NS54962) and the GBS Foundation (KAS). We thank Dr. Pamela Talalay for editorial discussion.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Brewer GJ, Torricelli JR, Evege EK, Price PJ. Optimized Survival of Hippocampal-Neurons in B27-Supplemented Neurobasal(Tm), A New Serum-Free Medium Combination. J Neurosci Res. 1993;35:567–576. doi: 10.1002/jnr.490350513. [DOI] [PubMed] [Google Scholar]
- Brodsky FM, Parham P. Monomorphic anti-HLA-A,B,C monoclonal antibodies detecting molecular subunits and combinatorial determinants. J Immunol. 1982;128:129–135. [PubMed] [Google Scholar]
- Brunig I, Sommer M, Hatt H, Bormann J. Dopamine receptor subtypes modulate olfactory bulb gamma-aminobutyric acid type A receptors. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:2456–2460. doi: 10.1073/pnas.96.5.2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchwald B, Bufler J, Carpo M, Heidenreich F, Pitz R, Dudel J, Nobile-Orazio E, Toyka KV. Combined pre- and postsynaptic action of IgG antibodies in Miller Fisher syndrome. Neurology. 2001;56:67–74. doi: 10.1212/wnl.56.1.67. [DOI] [PubMed] [Google Scholar]
- Buchwald B, Toyka KV, Zielasek J, Weishaupt A, Schweiger S, Dudel J. Neuromuscular blockade by IgG antibodies from patients with Guillain-Barre syndrome: a macro-patch-clamp study. Ann Neurol. 1998a;44:913–922. doi: 10.1002/ana.410440610. [DOI] [PubMed] [Google Scholar]
- Buchwald B, Weishaupt A, Toyka KV, Dudel J. Pre- and postsynaptic blockade of neuromuscular transmission by Miller-Fisher syndrome IgG at mouse motor nerve terminals. Eur J Neurosci. 1998b;10:281–290. doi: 10.1046/j.1460-9568.1998.00053.x. [DOI] [PubMed] [Google Scholar]
- Dudel J. Calcium dependence of quantal release triggered by graded depolarization pulses to nerve terminals on crayfish and frog muscle. Pflugers Arch. 1989;415:289–298. doi: 10.1007/BF00370878. [DOI] [PubMed] [Google Scholar]
- Gong Y, Tagawa Y, Lunn MP, Laroy W, Heffer-Lauc M, Li CY, Griffin JW, Schnaar RL, Sheikh KA. Localization of major gangliosides in the PNS: implications for immune neuropathies. Brain. 2002;125:2491–2506. doi: 10.1093/brain/awf258. [DOI] [PubMed] [Google Scholar]
- Goodfellow JA, Bowes T, Sheikh K, Odaka M, Halstead SK, Humphreys PD, Wagner ER, Yuki N, Furukawa K, Furukawa K, Plomp JJ, Willison HJ. Overexpression of GD1a ganglioside sensitizes motor nerve terminals to anti-GD1a antibody-mediated injury in a model of acute motor axonal neuropathy. J Neurosci. 2005;25:1620–1628. doi: 10.1523/JNEUROSCI.4279-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodyear CS, O’Hanlon GM, Plomp JJ, Wagner ER, Morrison I, Veitch J, Cochrane L, Bullens RW, Molenaar PC, Conner J, Willison HJ. Monoclonal antibodies raised against Guillain-Barre syndrome- associated Campylobacter jejuni lipopolysaccharides react with neuronal gangliosides and paralyze muscle-nerve preparations. J Clin Invest. 1999;104:697–708. doi: 10.1172/JCI6837. [published erratum appears in J Clin Invest 1999 Dec;104(12):1771] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin JW, Li CY, Ho TW, Xue P, Macko C, Cornblath DR, Gao CY, Yang C, Tian M, Mishu B, McKhann GM, Asbury AK. Guillain-Barre syndrome in northern China: The spectrum of neuropathologic changes in clinically defined cases. Brain. 1995;118:577–595. doi: 10.1093/brain/118.3.577. [DOI] [PubMed] [Google Scholar]
- Griffin JW, Li CY, Macko C, Ho TW, Hsieh S-T, Xue P, Wang FA, Cornblath DR, McKhann GM, Asbury AK. Early nodal changes in the acute motor axonal neuropathy pattern of the Guillain-Barre syndrome. J Neurocytol. 1996;25:33–51. doi: 10.1007/BF02284784. [DOI] [PubMed] [Google Scholar]
- Hafer-Macko C, Hsieh S-T, Li CY, Ho TW, Sheikh K, Cornblath DR, McKhann GM, Asbury AK, Griffin JW. Acute motor axonal neuropathy: an antibody-mediated attack on axolemma. Ann Neurol. 1996a;40:635–644. doi: 10.1002/ana.410400414. [DOI] [PubMed] [Google Scholar]
- Hafer-Macko C, Sheikh KA, Li CY, Ho TW, Cornblath DR, McKhann GM, Asbury AK, Griffin JW. Immune attack on the Schwann cell surface in acute inflammatory demyelinating polyneuropathy. Ann Neurol. 1996b;39:625–635. doi: 10.1002/ana.410390512. [DOI] [PubMed] [Google Scholar]
- Halstead SK, Morrison I, O’Hanlon GM, Humphreys PD, Goodfellow JA, Plomp JJ, Willison HJ. Anti-disialosyl antibodies mediate selective neuronal or Schwann cell injury at mouse neuromuscular junctions. Glia. 2005;52:177–189. doi: 10.1002/glia.20228. [DOI] [PubMed] [Google Scholar]
- Hartung HP, Pollard JD, Harvey GK, Toyka KV. Immunopathogenesis and treatment of the Guillain-Barre syndrome--Part I. Muscle Nerve. 1995;18:137–153. doi: 10.1002/mus.880180202. [DOI] [PubMed] [Google Scholar]
- Ho TW, Hsieh ST, Nachamkin I, Willison HJ, Sheikh K, Kiehlbauch J, Flanigan K, McArthur JC, Cornblath DR, McKhann GM, Griffin JW. Motor nerve terminal degeneration provides a potential mechanism for rapid recovery in acute motor axonal neuropathy after Campylobacter infection. Neurology. 1997;48:717–724. doi: 10.1212/wnl.48.3.717. [see comments] [DOI] [PubMed] [Google Scholar]
- Hughes RA, Hadden RD, Gregson NA, Smith KJ. Pathogenesis of Guillain-Barre syndrome. J Neuroimmunol. 1999;100:74–97. doi: 10.1016/s0165-5728(99)00195-2. [DOI] [PubMed] [Google Scholar]
- Isaacson JS, Strowbridge BW. Olfactory reciprocal synapses: Dendritic signaling in the CNS. Neuron. 1998;20:749–761. doi: 10.1016/s0896-6273(00)81013-2. [DOI] [PubMed] [Google Scholar]
- Krampfl K, Mohammadi B, Buchwald B, Jahn K, Dengler R, Toyka KV, Bufler J. IgG from patients with Guillain-Barre syndrome interact with nicotinic acetylcholine receptor channels. Muscle Nerve. 2003;27:435–441. doi: 10.1002/mus.10349. [DOI] [PubMed] [Google Scholar]
- Kuwabara S, Ogawara K, Misawa S, Koga M, Mori M, Hiraga A, Kanesaka T, Hattori T, Yuki N. Does Campylobacter jejuni infection elicit “demyelinating” Guillain-Barre syndrome? Neurology. 2004;63:529–533. doi: 10.1212/01.wnl.0000133205.05169.04. [DOI] [PubMed] [Google Scholar]
- Kuwabara S, Ogawara K, Sung JY, Mori M, Kanai K, Hattori T, Yuki N, Lin CSY, Burke D, Bostock H. Differences in membrane properties of axonal and demyelinating Guillain-Barre syndromes. Ann Neurol. 2002;52:180–187. doi: 10.1002/ana.10275. [DOI] [PubMed] [Google Scholar]
- Kuwabara S, Yuki N, Koga M, Hattori T, Matsuura D, Miyake M, Noda M. IgG anti-GM1 antibody is associated with reversible conduction failure and axonal degeneration in Guillain-Barre syndrome. Ann Neurol. 1998;44:202–208. doi: 10.1002/ana.410440210. [DOI] [PubMed] [Google Scholar]
- Latov N. Neuropathy and anti-GM1 antibodies. Ann Neurol. 1990;27(Suppl):S41–S43. doi: 10.1002/ana.410270711. [DOI] [PubMed] [Google Scholar]
- Lunn MP, Johnson LA, Fromholt SE, Itonori S, Huang J, Vyas AA, Hildreth JE, Griffin JW, Schnaar RL, Sheikh KA. High-affinity anti-ganglioside IgG antibodies raised in complex ganglioside knockout mice: reexamination of GD1a immunolocalization. J Neurochem. 2000;75:404–412. doi: 10.1046/j.1471-4159.2000.0750404.x. [DOI] [PubMed] [Google Scholar]
- McKhann GM, Cornblath DR, Griffin JW, Ho TW, Li CY, Jiang Z, Wu HS, Zhaori G, Liu Y, Jou LP, Liu TC, Gao CY, Mao JY, Blaser MJ, Mishu B, Asbury AK. Acute motor axonal neuropathy: A frequent cause of acute flaccid paralysis in China. Ann Neurol. 1993;33:333–342. doi: 10.1002/ana.410330402. [DOI] [PubMed] [Google Scholar]
- McKhann GM, Cornblath DR, Ho TW, Li CY, Bai AY, Wu HS, Yei QF, Zhang WC, Zhaori Z, Jiang Z, Griffin JW, Asbury AK. Clinical and electrophysiological aspects of acute paralytic disease of children and young adults in northern China. Lancet. 1991;338:593–597. doi: 10.1016/0140-6736(91)90606-p. [DOI] [PubMed] [Google Scholar]
- Miyazaki T, Kusunoki S, Kaida K, Shiina M, Kanazawa I. Guillain-Barre syndrome associated with IgG monospecific to ganglioside GD1b. Neurology. 2001;56:1227–1229. doi: 10.1212/wnl.56.9.1227. [DOI] [PubMed] [Google Scholar]
- O’Hanlon GM, Humphreys PD, Goldman RS, Halstead SK, Bullens RW, Plomp JJ, Ushkaryov Y, Willison HJ. Calpain inhibitors protect against axonal degeneration in a model of anti-ganglioside antibody-mediated motor nerve terminal injury. Brain. 2003;126:2497–2509. doi: 10.1093/brain/awg254. [DOI] [PubMed] [Google Scholar]
- Ropper AH, Wijdicks EFM, Truax BT. Guillain-Barre Syndrome 1991 [Google Scholar]
- Santafe MM, Sabate MM, Garcia N, Ortiz N, Lanuza MA, Tomas J. Changes in the neuromuscular synapse induced by an antibody against gangliosides. Ann Neurol. 2005;57:396–407. doi: 10.1002/ana.20403. [DOI] [PubMed] [Google Scholar]
- Schnaar RL, Fromholt SE, Gong Y, Vyas AA, Laroy W, Wayman DM, Heffer-Lauc M, Ito H, Ishida H, Kiso M, Griffin JW, Sheikh KA. IgG-class mouse monoclonal antibodies to major brain gangliosides. Anal Biochem. 2002;302:276–284. doi: 10.1006/abio.2001.5540. [DOI] [PubMed] [Google Scholar]
- Sheikh KA, Zhang G, Gong Y, Schnaar RL, Griffin JW. An anti-ganglioside antibody-secreting hybridoma induces neuropathy in mice. Ann Neurol. 2004;56:228–239. doi: 10.1002/ana.20173. [DOI] [PubMed] [Google Scholar]
- Susuki K, Nishimoto Y, Yamada M, Baba M, Ueda S, Hirata K, Yuki N. Acute motor axonal neuropathy rabbit model: immune attack on nerve root axons. Ann Neurol. 2003;54:383–388. doi: 10.1002/ana.33333. [DOI] [PubMed] [Google Scholar]
- Takahashi E, Nagasu T. Genetic background influences P/Q-type Ca2+ channel alpha(1A) subunit mRNA expression in olfactory bulb and reproductive ability of N-type Ca2+ channel alpha(1B) subunit-deficient mice. Biochemical Genetics. 2005;43:287–298. doi: 10.1007/s10528-005-5220-9. [DOI] [PubMed] [Google Scholar]
- Willison HJ, Almemar A, Veitch J, Thrush D. Acute ataxic neuropathy with cross-reactive antibodies to GD1b and GD3 gangliosides. Neurology. 1994;44:2395–2397. doi: 10.1212/wnl.44.12.2395. [DOI] [PubMed] [Google Scholar]
- Willison HJ, Yuki N. Peripheral neuropathies and anti-glycolipid antibodies. Brain. 2002;125:2591–2625. doi: 10.1093/brain/awf272. [DOI] [PubMed] [Google Scholar]
- Yuki N. Infectious origins of, and molecular mimicry in, Guillain-Barre and Fisher syndromes. Lancet Infect Dis. 2001;1:29–37. doi: 10.1016/S1473-3099(01)00019-6. [DOI] [PubMed] [Google Scholar]
- Yuki N, Yamada M, Koga M, Odaka M, Susuki K, Tagawa Y, Ueda S, Kasama T, Ohnishi A, Hayashi S, Takahashi H, Kamijo M, Hirata K. Animal model of axonal Guillain-Barre syndrome induced by sensitization with GM1 ganglioside. Ann Neurol. 2001;49:712–20. [PubMed] [Google Scholar]
- Zhang G, Lopez PH, Li CY, Mehta NR, Griffin JW, Schnaar RL, Sheikh KA. Anti-ganglioside antibody-mediated neuronal cytotoxicity and its protection by intravenous immunoglobulin: implications for immune neuropathies. Brain. 2004;127:1085–1100. doi: 10.1093/brain/awh127. [DOI] [PubMed] [Google Scholar]