

SUMMARY
Diapedesis is critical for immune system function and inflammatory responses. This occurs by migration of blood leukocytes either directly through individual microvascular endothelial cells (the “transcellular” route) or between them (the “paracellular” route). Mechanisms for transcellular pore formation in endothelium remain unknown. Here we demonstrate that lymphocytes used podosomes and extended “invasive podosomes” to palpate the surface of, and ultimately form transcellular pores through, the endothelium. In lymphocytes, these structures were dependent on Src kinase and the actin regulatory protein WASP; inhibition of podosome formation selectively blocked the transcellular route of diapedesis. In endothelium, membrane fusion events dependent on the SNARE-containing membrane fusion complex and intracellular calcium were required for efficient transcellular pore formation in response to podosomes. These findings provide insights into basic mechanisms for leukocyte trafficking and the functions of podosomes.
INTRODUCTION
Whereas functions for most cells of the body are coupled to their organization in tissues, functions for blood leukocytes are uniquely centered on their ability to efficiently cross tissue barriers, i.e., invasiveness. In order to conduct immune surveillance, respond to infection, and undergo differentiation, leukocytes must traffic throughout the body, moving in and out of the bone marrow, lymphoid organs, and inflamed tissues (von Andrian and Mackay, 2000). A central component to this process is the repeated crossing of vascular and lymphatic endothelial barriers, i.e., diapedesis, as leukocytes leave and re-enter the circulation (von Andrian and Mackay, 2000). Fundamental aspects of the mechanisms for this barrier-crossing function remain unknown.
Diapedesis occurs in vivo either directly through individual endothelial cells (the transcellular route) or between them at interendothelial cell junctions (the paracellular route) (Feng et al., 1998; Greenwood et al., 1994; Lossinsky and Shivers, 2004; Marchesi, 1961; Marchesi and Gowans, 1963). In both cases, the process begins with the accumulation of leukocytes on the luminal surface of the endothelium through a well-characterized sequence of rolling, activation, and firm adhesion events (Springer, 1994). Subsequent integrin-dependent lateral migration is an essential next step that seems to allow leukocytes to search out sites permissive for endothelial barrier penetration (Schenkel et al., 2004). The final and critical steps of identifying such sites and then formally breaching the endothelium are incompletely understood.
Interendothelial cell junctions provide a defined and intuitive site for paracellular diapedesis, and mechanistic models for this process have been proposed (Muller, 2003). In contrast, neither an intuitive locus nor an obvious mechanism for transcellular diapedesis exists. Whatever the mechanism, transcellular diapedesis requires, at minimum, local displacement of endothelial cytoplasmic organelles and fusion of the apical and basal plasma membranes. Formally, this process could be driven by the leukocyte, by the endothelium, or by contributions from both.
On the part of the leukocyte, actin-dependent protrusive structures might be able to both displace endothelial organelles and facilitate membrane fusion by forcing apical and basal membranes into close opposition. Candidate protrusive structures include the well-characterized filopodia, lamellipodia, and related pseudopodia (Ridley et al., 2003), along with more recently discovered protrusive organelles termed podosomes (“foot-protrusions”) and invadopodia (Buccione et al., 2004; Linder and Aepfelbacher, 2003; Yamaguchi et al., 2005b).
Podosomes and invadopodia are micron-scale cylindrical protrusions formed on the ventral aspect of adherent cells with the defining architecture of a peripheral ring of β2 or β3 integrin, talin, and vinculin, and a central core of F-actin (Buccione et al., 2004; Linder and Aepfelbacher, 2003). These protrusive structures share a strong dependence on Src kinase and the cytoskeletal regulator Wiskott-Aldrich syndrome (WAS) protein (WASP) or N-WASP in nonhematopoietic cells (Calle et al., 2004; Linder and Aepfelbacher, 2003). Distinction between podosomes and invadopodia are seen in their size (with substantially greater protrusion depths observed in invadopodia), dynamics, and apparent functions (Buccione et al., 2004; Yamaguchi et al., 2005b). Podosomes have been described in a wide range of migratory cell types, most notably leukocytes, where they form dynamic clustered arrays or “rosettes” (Buccione et al., 2004; Evans et al., 2003; Linder and Aepfelbacher, 2003). However, their function is unclear, though generally linked to adhesion and migration. In contrast, invadopodia, which have been described only in transformed cell types, have a more defined function in burrowing across matrix barriers, thereby conferring invasiveness to metastatic tumor cells (Buccione et al., 2004; Yamaguchi et al., 2005a).
An active role for the endothelium during diapedesis has been widely demonstrated through, for example, leukocyte-triggered calcium and Rho signaling (Adamson et al., 1999; Carman and Springer, 2004; Etienne-Manneville et al., 2000; Huang et al., 1993; Muller, 2003). We and others have previously hypothesized that such intraendothelial signals could potentially trigger membrane fusion events that facilitate transcellular pore formation (Carman and Springer, 2004; Feng et al., 2002; Lossinsky and Shivers, 2004; Olah and Glick, 1985).
Here we demonstrate that leukocytes dynamically inserted numerous podosomes and extended “invasive podosomes,” the latter of which exhibit similarities to invadopodia, into the endothelial surface both in vitro and in vivo. This serves to probe for sites permissive to transcellular pore formation and to trigger endothelial fusion activity that supports this process. This previously unrecognized palpation behavior was critical for transcellular diapedesis and may also play broader roles in leukocyte path finding during trafficking.
RESULTS
Transcellular Diapedesis Is Quantitatively Important in Microvascular Endothelium
Given that the vast majority of leukocyte traffic in vivo occurs through the microvasculature (von Andrian and Mackay, 2000), in this study we investigated migration on human dermal (HDMVEC) and lung (HLMVEC) microvascular endothelial monolayers. By using our previously established fixed-cell imaging approach (Carman and Springer, 2004) with IL-2-cultured lymphocytes, we demonstrate that approximately one-third of diapedesis events occurred via the transcellular route on tumor necrosis factor (TNF)-α-activated HDMVEC and HLMVEC (Figure 1A). We also examined and found comparable results for dermal lymphatic endothelium (Figure 1A). By contrast, approximately 10% of lymphocytes transmigrated via the transcellular route on HUVEC (human umbilical vein endothelial cells), a phenotypically distinct macrovascular endothelium (Figure 1A; Carman and Springer, 2004). Dynamic live-cell imaging experiments showed that the kinetic parameters of para- and transcellular diapedesis are largely indistinguishable (Figure 1B). This confirmed the validity of using a fixed-cell, single-time-point analysis for quantitative comparison of para-and transcellular diapedesis.
Figure 1. Transcellular Diapedesis on Microvascular Endothelium.
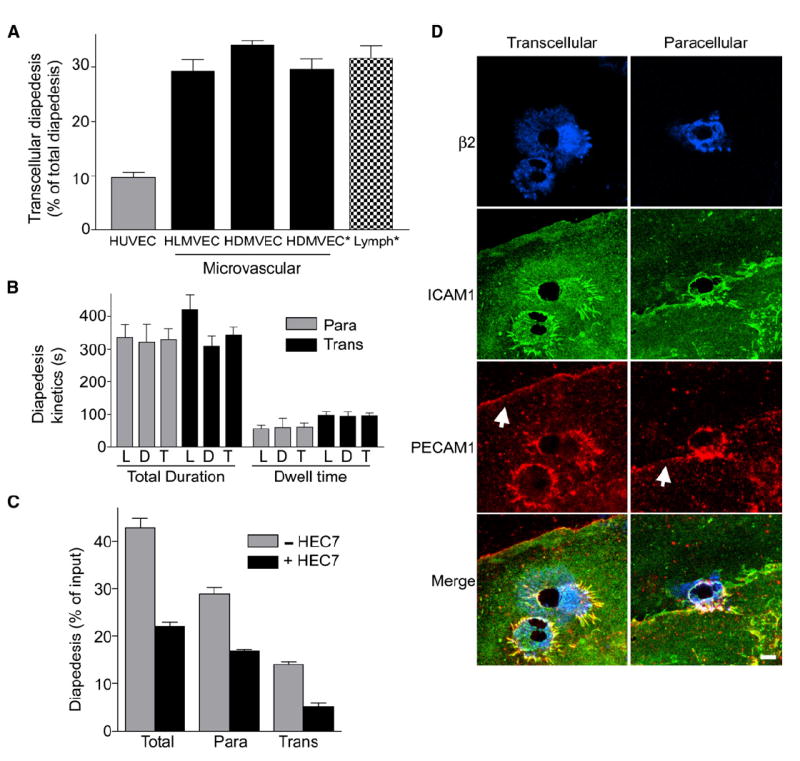
(A) IL-2-cultured lymphocytes were incubated with TNF-α-activated endothelial monolayers for 10 min followed by fixation, staining, and scoring as described in Experimental Procedures. Endothelial cells included HDMVEC and HLMVEC from Cambrex, as well as HDMVEC (HDMVEC*) and lymphatic ECs (Lymph*) selectively isolated from fresh neonatal foreskins. y axis represents the percentage of the total diapedesis that was transcellular. Values are mean ± SEM for at least three separate experiments.
(B) Videos from live-cell imaging of lymphocyte diapedesis on HDMVEC and HLMVEC (described in Figure 2, below) were analyzed to determine the total duration of diapedesis and the amount of time lymphocytes resided at a location before initiating diapedesis (dwell time) for transcellular (black bars) or paracellular (gray bars) events. Data for the HLMVECs (L) and HDMVECs (D) were analyzed both separately and together (T). Values are the mean ± SEM for at least seven measurements.
(C) Lymphocytes were incubated with activated HLMVEC as in (A) above, in the absence (gray bars) and presence (black bars) of PECAM-1 function-blocking mAb HEC7 (40 μg/ml). Graph depicts total diapedesis, as well as the separate the para- and transcellular diapedesis components, each as a percentage of total cells. Values are the mean ± SEM of three separate experiments. 40 or 120 μg/ml isotype control antibody had no effect on diapedesis (not shown).
(D) Lymphocytes were incubated with activated HLMVEC for 10 min followed by fixation and staining with fluorescent antibodies to integrin β2, ICAM-1, and PECAM-1 and subjected to confocal microscopy. Images are Z-stack projections of representative transcellular (left) and paracellular (right) diapedesis events. Note that under one of the two lymphocytes on the left, two transcellular pores have formed. Arrows indicate the PECAM-1-enriched interendothelial junctions. Scale bar represents 5 μm.
We attempted to selectively block paracellular diapedesis. Paracellular diapedesis is predicted to be uniquely dependent on molecules enriched at the endothelial junctions including platelet-endothelial cellular adhesion molecule-1 (PECAM-1), junctional adhesion molecule-1 (JAM-1), and CD99 (Muller, 2003). Thus, we employed a widely used PECAM-1 function-blocking antibody (HEC7) (Muller, 2003). To our surprise, we found that although HEC7 inhibited diapedesis, it affected both para- and transcellular routes comparably (Figure 1C). Immunofluorescence imaging of PECAM-1 (Figure 1D) and JAM-1 (Figure S1 in the Supplemental Data available with this article online) showed that these molecules were similarly enriched around both trans- and paracellular diapedesis events in previously described “transmigratory cups” (Barreiro et al., 2002; Carman and Springer, 2004). Though the role of PECAM-1 in lymphocyte diapedesis in vivo remains unclear, these data suggest that PECAM-1 and JAM-1 may not be specific for paracellular transmigration.
Clusters of Dynamic, Micron-Scale Invaginations into the Endothelial Surface Precede Transcellular Diapedesis
Live-cell imaging experiments, combining fluorescence and DIC (differential interference contrast) modalities, were used to dissect mechanisms for transcellular diapedesis. To monitor the endothelial cell-surface dynamics, we imaged lymphocyte migration on microvascular endothelial cells transfected with intercellular adhesion molecule (ICAM)-1 fused to GFP (ICAM-1-GFP). Lymphocytes migrated laterally over the endothelium and underwent both trans- and paracellular diapedesis. Strikingly, clusters of micron-scale fluorescent rings (~0.2–1 μm in diameter) formed intermittently under most spread and laterally migrating lymphocytes (Figure 2 and Movie S1). The initial appearance of a single or several rings was typically followed by the rapid (within ~30 s) appearance of clusters containing 10–30 rings (covering an area of ~100 μm2) as lymphocytes spread (Figure 2). For laterally migrating lymphocytes, rings formed predominantly just behind the leading edge (see Movie S3) but also formed under the trailing edges and various lateral extensions (see below and Movie S9). Individual rings were highly dynamic and appeared, disappeared, and changed size and shape on the time scale of tens of seconds (Figure 2).
Figure 2. Transcellular Diapedesis Is Preceded by Clusters of Micron-Scale ICAM-1-Rich Rings on the Endothelium.
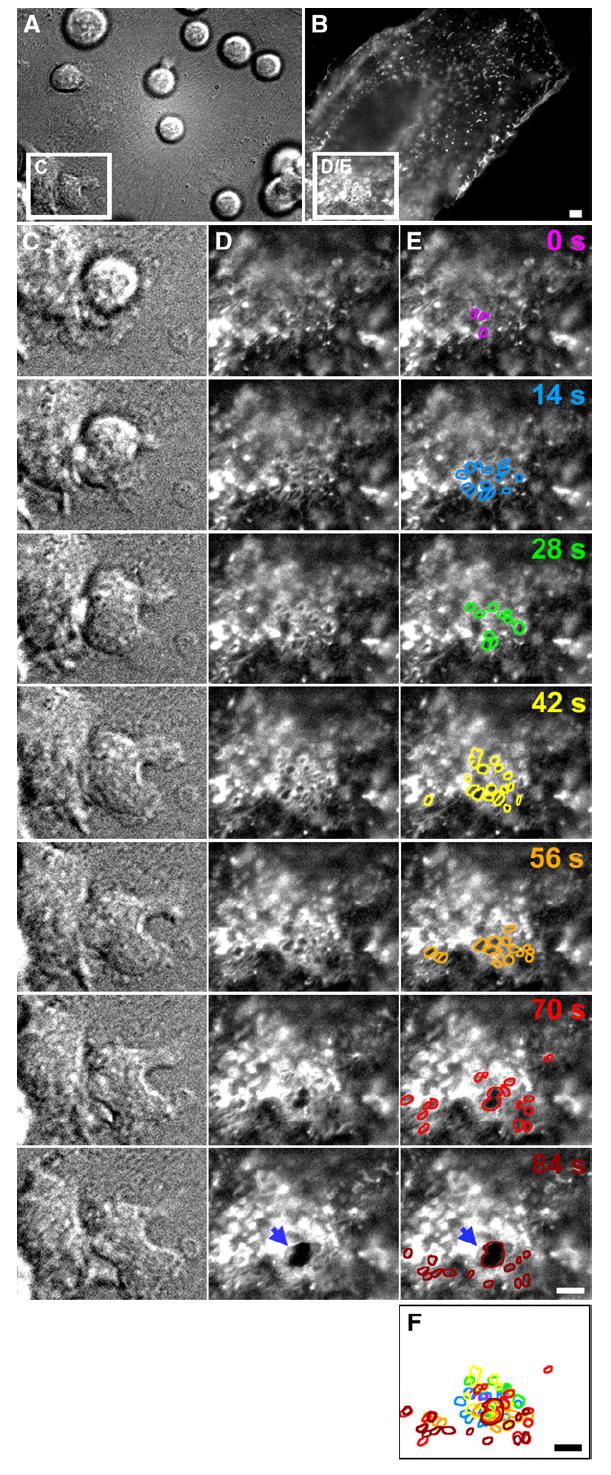
TNF-α-activated HDMVEC transiently transfected with ICAM-1-GFP were subjected to live-cell dynamic imaging upon addition of lymphocytes as described in Experimental Procedures. (A) and (C) are DIC, and (B), (D), and (E) are fluorescence images of ICAM-1-GFP. (A) and (B) depict a wide field of view in which most of a single GFP-positive endothelial cell (in the context of a confluent endothelial monolayer) is present. Boxed regions in (A) and (B) depict a location distant from endothelial intercellular junctions. Columns (C), (D), and (E) depict expanded sequential views (at 14 s intervals) of these boxed regions, in which the right-most lymphocyte initiates transcellular diapedesis. The micrographs shown in column (E) are identical to those shown in column (D), but individual ICAM-1-GFP-rich ring-shaped structures have been highlighted, with distinct colors correlating to each time point. (F) represents the overlay of these lines. Note that formation of a transcellular pore (see arrows in [D] and [E] at the 84 s time point), through which the lymphocyte subsequently completes transcellular diapedesis (shown in Movie S1), emerges from the center of two of these rings, which appear to fuse. Scale bars represent 5 μm.
Most importantly, formation of transcellular pores at the initiation of transcellular diapedesis was essentially always preceded by the appearance of rings (Figure 2 and Movie S1). An analysis of 58 transcellular diapedesis events from representative videos demonstrated that as few as 4 and as many as 216 rings (average ± SEM = 62.1 ± 7.1) were seen prior to the formation of each transcellular pore. In most cases, the initial pores widened rapidly (expanding their diameter an average rate ± SEM = 2.6 ± 0.2 μm/min; n = 28) to a maximum of ~6 μm, as lymphocyte diapedesis proceeded, and then constricted and resealed (Movie S1). These observations suggested a potential functional relationship between the ring-shaped structures and the initiation of transcellular diapedesis.
To determine whether the observed structures represented planar rings of ICAM-1-GFP enrichment or involved topological perturbations in the endothelial membrane, we conducted analyses similar to those above by using, instead of ICAM-1-GFP, palmitoylated YFP (memb-YFP) as a general membrane marker. This membrane reporter revealed essentially identical ring-shaped structures, with dynamics and association with transcellular pores similar to those seen with ICAM-1-GFP (Movies S2 and S3), demonstrating that the “rings” represented topological changes in the endothelial membrane surface.
We reasoned that the fluorescent rings could arise from either apical exvaginations or invaginations of the endothelial plasma membrane. To distinguish between these possibilities, endothelial cells were cotransfected with palmitoylated RFP (memb-RFP) as a membrane marker and unmodified GFP as a marker for the cytoplasmic volume. Live-cell imaging demonstrated a striking inverse correlation between memb-RFP rings and GFP distribution: within each memb-RFP ring, the GFP signal was diminished (Figure 3A, Movies S4 and S8; see below). This suggests that the ring-shaped membrane structures represent invaginations of the apical endothelial membrane.
Figure 3. “Ring Structures” Represent Endothelial Cell-Surface Invaginations.
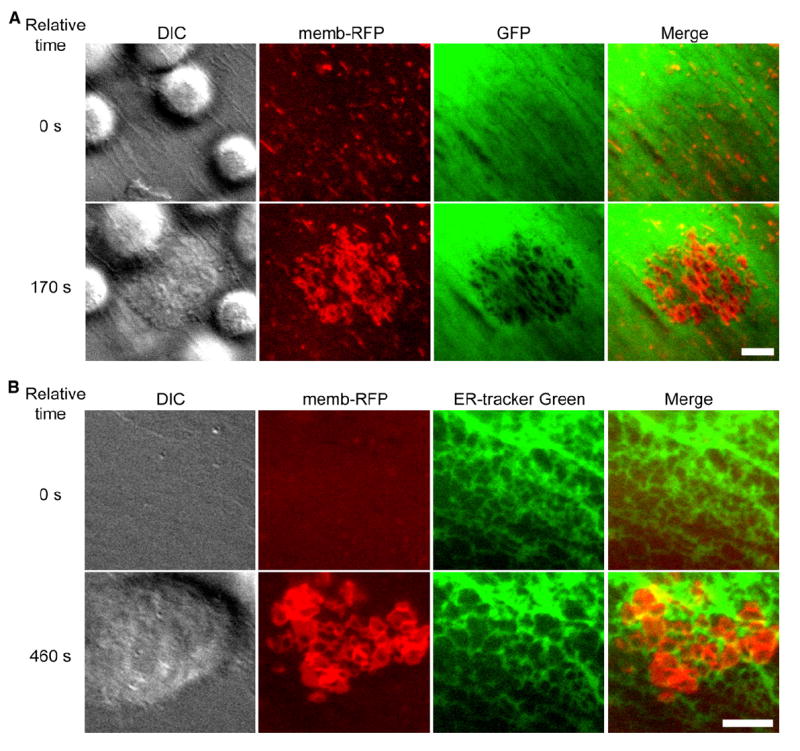
TNF-α-activated HLMVEC were subjected to live-cell dynamic imaging upon addition of lymphocytes.
(A) HLMVEC were transiently cotransfected with memb-RFP and unmodified GFP. Panels depict DIC, memb-RFP, GFP, and merge of memb-RFP and GFP. Upper panels show a time point shortly after lymphocytes have settled on the endothelium, but before formation of memb-RFP ring structures (relative time = 0 s). Lower panels show a time point after lymphocyte spreading and formation of rings (relative time 170 s). Note that for each memb-RFP ring, a circular region of diminished GFP signal is formed. See corresponding Movie S4.
(B) HDMVEC were transiently transfected with memb-RFP and prestained with ER-tracker green. Panels depict DIC, memb-RFP, ER-tracker green, and Merge of memb-RFP and ER-tracker green. Upper panels show a time point before (relative time = 0 s) and lower panels show a time point after (relative time 460 s) the formation of memb-RFP ring structures. Note that memb-RFP ring structures seem to form within the individual reticula of the ER often distorting and expanding these structures. These features can be more readily appreciated in the corresponding Movie S5. Scale bars represent 5 μm.
To probe this idea further, we investigated the distribution of other intracellular components in endothelium. Endoplasmic reticulum (Endotracker-Green [Figure 3B and Movie S5]), GFP-actin (Movie S6), and GFP-tubulin (Movie S7) were also displaced and distorted at sites of ring formation. Rings also formed directly over the nucleus; however, there was little apparent distortion of this organelle and never transcellular pore formation at this site (Movies S3 and S8).
Endothelial Invaginations Result from Podosome-like Leukocyte Protrusions
To determine the role of the leukocyte and endothelium in the formation of the invaginations, we employed K562 cells, which lack protrusive activity (Carman et al., 2003). K562 LFA-1 transfectants, which were activated with a mAb to lymphocyte function-associated antigen (LFA)-1, elicited formation of ICAM-1-enriched cup-like structures on HDMVEC, but not ring-shaped invaginations or transcellular pores (data not shown). In contrast, SDF-1-stimulated lymphocytes readily induced ring-shaped invaginations, and occasionally transcellular pores, on CHO-K1 epithelial cells stably expressing ICAM-1-GFP (Figure S2A). These data suggest a critical proactive role for the lymphocyte in formation of the endothelial invaginations.
Given the size, dynamics, and clustered patterns of the endothelial cell invaginations, we hypothesized that they might be formed in response to podosome protrusions generated by leukocytes. After a brief incubation of lymphocytes with endothelium, samples were fixed and stained for ICAM-1 and F-actin, along with lymphocyte β2-integrin (αLβ2; LFA-1) or talin-1. High-resolution confocal imaging revealed the existence of clusters of ventral leukocyte protrusions, each ~0.2–1 μm in diameter, with LFA-1 (Figures 4A and 4B)- and talin-1 (Figure 4C)-rich outer rings and F-actin-rich inner cores (Figures 4A-4C). These features support the designation of the lymphocyte protrusions as podosomes. We term the corresponding ICAM-1-rich invaginations on the endothelial surface “podoprints.” Podoprints were of slightly greater diameter than their cognate podosomes (Figures 4A and 4B).
Figure 4. Endothelial Invaginations Result from Podosome-like Protrusive Structures.
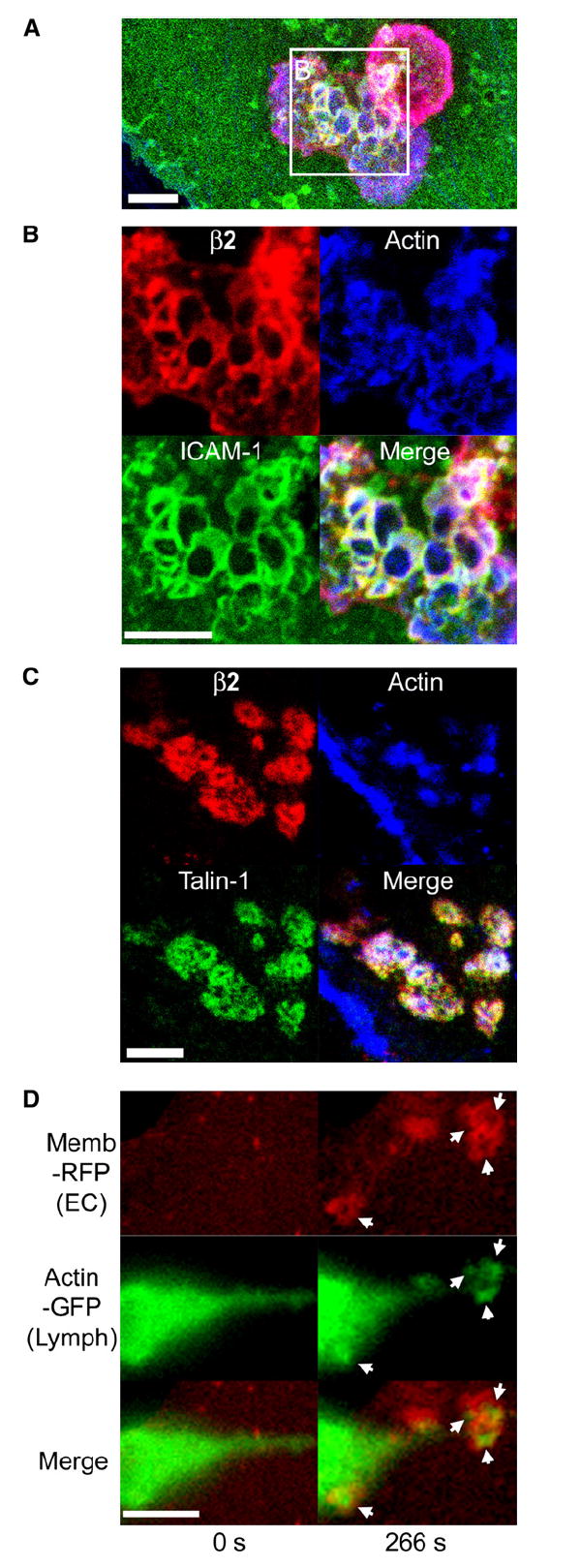
(A–C) Confocal imaging of lymphocyte-endothelial interactions. Lymphocytes were incubated with TNF-α-activated HDMVEC for 5 min followed by fixation, staining for F-actin (blue), leukocyte β2 integrin (red), endothelial ICAM-1 (green in [A] and [B]), or talin-1 (green in [C]), and confocal microscopy described in Experimental Procedures. Images are representative Z-stack projections of confocal sections near the plane of the leukocyte-endothelial interaction interface.
(A) A merged image of β2 integrin, F-actin, and ICAM-1 is shown in which two adjacent lymphocytes, one spread and somewhat dumbbell-shaped (lower left) and one rounded (upper right), adhere to the surface of the endothelium.
(B) An expanded view of the boxed region in (A) is shown as both separate and merged fluorescent channels.
(C) In a separate experiment, samples were stained for β2 integrin, F-actin, and talin-1.
(D) Live-cell imaging of lymphocyte actin. Lymphocytes transiently expressing GFP-actin were incubated with activated HDMVEC expressing memb-RFP and subjected to live-cell imaging. Left and right panels depict a lymphocyte at time points before (relative time point = 0 s) and after (relative time point = 266 s), respectively, endothelial podoprint formation. Memb-RFP, GFP-actin, and merged images are as indicated. Arrows indicate the appearance of podosome-like (Evans et al., 2003) actin puncta in lymphocytes that are centered within the memb-RFP rings of the endothelial podoprints. Note the distinctly green areas (actin) evident at the center of each ring. See corresponding Movie S8. Scale bars represent 5 μm.
We further examined lymphocyte actin dynamics during podosome and podoprint formation in live cells (Figure 4D and Movie S9). GFP-actin expressed in transiently transfected primary human lymphocytes formed dynamic puncta similar to those typically observed for podosomes (Evans et al., 2003) that were spatially and temporally correlated with the formation of podoprints on the endothelium (Figure 4D and Movie S9).
In addition to the IL-2-cultured effector type lymphocytes used throughout this study, we also examined IL-15-cultured memory type (Weninger et al., 2001) lymphocytes. These formed podoprints, podosomes, and transcellular pores through TNF-α-activated endothelium with roughly similar efficiency as IL-2-cultured lymphocytes (data not shown). Alternatively, freshly isolated peripheral blood lymphocytes (PBLs) underwent little adhesion, migration, or diapedesis in this inflamed endothelial model, which correlated with infrequent initiation of podosomes (<5% of PBLs) or transcellular pores (<1% of PBLs) (data not shown) and the predominance of naive lymphocytes, which fail to emigrate at sites of inflammation in vivo (Weninger et al., 2001).
Podosomes have not previously been observed in lymphocytes but have been widely observed in leukocytes of myeloid lineage (Buccione et al., 2004; Linder and Aepfelbacher, 2003). Primary human monocytes, a representative myeloid lineage cell, induced podoprints that were essentially identical to those induced by lymphocytes (Figure S2B).
Lymphocyte Podosomes Extend and Invade into the Endothelial Cytoplasm
Transmission electron microscopy (TEM) was used to further investigate the lymphocyte-protrusive structures and their relation to transcellular diapedesis. Consistent with fluorescence imaging data, TEM revealed lymphocytes inserting numerous ventral protrusions into opposed endothelial cells (Figure 5). Endothelial invaginations were not observed in the absence of lymphocyte protrusions. In many cases, the lymphocyte protrusions and endothelial cell invaginations extended deeply into the endothelial cytoplasm (Figure 5A, see “I”). These often extended to a depth in which the apical endothelial plasma membrane was brought into close proximity to the basal aspect of the endothelium (Figure 5B, see “I1–I3”). We also observed lymphocytes that had formally breached the endothelium and were at various stages of transcellular migration at sites distant from intact interendothelial cell junctions (Figure S3). In addition, podosomes were seen that pressed against the endothelial cell nucleus; however, these were invariably shallow and never provided a pathway for lymphocyte transmigration (Figures 5C and 5D).
Figure 5. In Vitro and In Vivo Ultrastructure of Lymphocyte Podosomes and Invasive Podosomes.
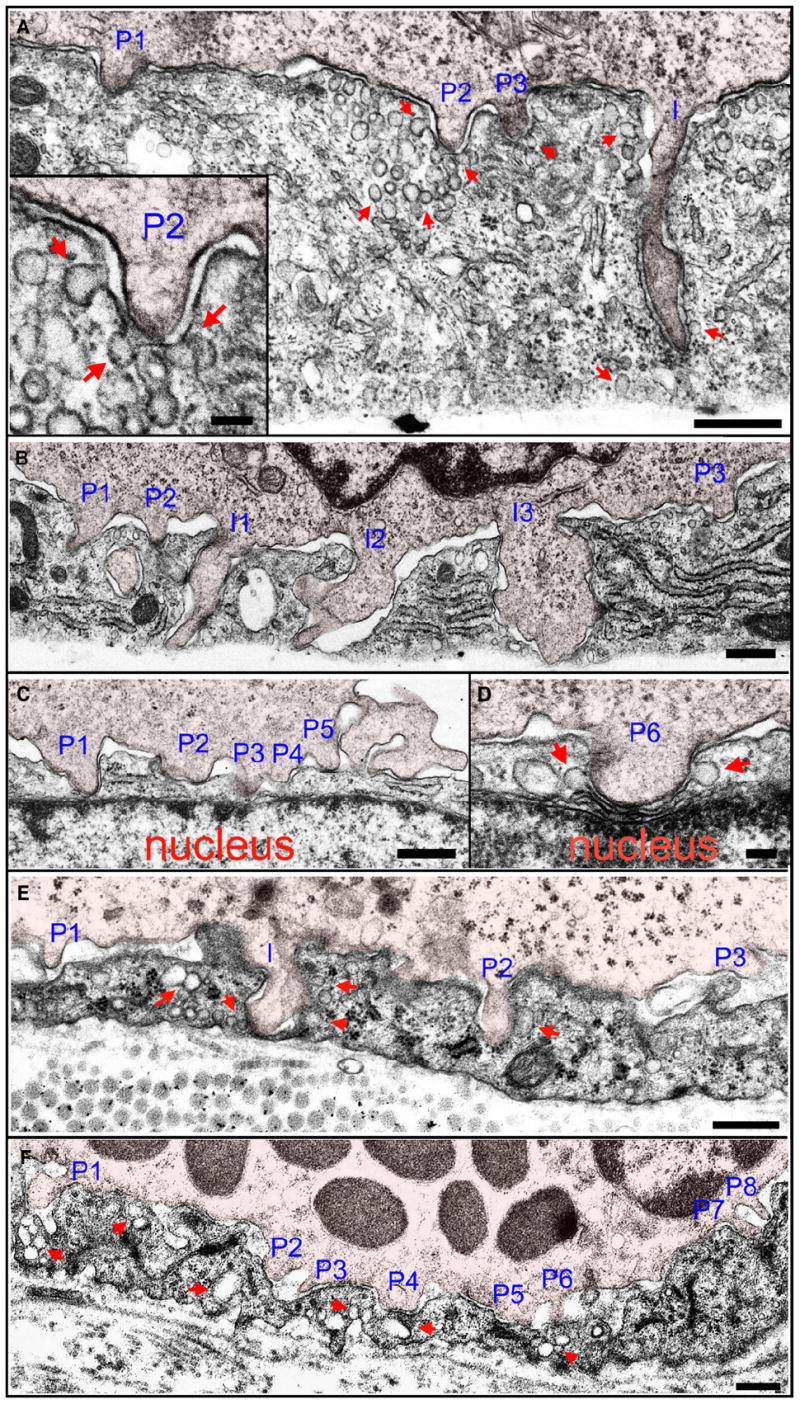
(A–D) Podosomes in vitro. Lymphocytes migrating on TNF-α-activated HDMVEC were fixed after a 5 min coincubation and processed for transmission EM as described in Experimental Procedures.
(A) A lymphocyte extends three shallow (~0.3 μm) podosomes (“P1–P3”) and one deep (~1.5 μm) invasive podosome (“I”) into endothelial invaginations. Inset is an expanded view of podosome “P2.”
(B) A lymphocyte extends three shallow (~0.2–0.5 μm) podosomes (“P1–P3”) and three deep (~1.5 μm) invasive podosomes (“I1–I3”) that span to nearly the basal surface.
(C and D) Shallow lymphocyte podosomes “P1–P6” that have formed directly over the endothelial cell nucleus. “P1” shows the apical plasma membrane nearly in contact with the nuclear envelope and “P6” shows it mildly indenting, but not invaginating, the nucleus.
(E and F) Podosomes in vivo. A guinea pig model of dermatitis was prepared and processed for TEM as described in Experimental Procedures. The representative lymphocyte (E) and basophil (F) are from an extensive analysis, including four different experiments and examination of at least 100 distinct lymphocytes and at least 100 distinct basophils. More than half of all both lymphocytes and basophils exhibited at least one podosome in any given section.
(E) Lymphocyte podosomes (“P1–P3;” ~0.2–0.5 μm) and an invasive podosome (“I;” ~0.8 μm) project into the endothelial surface of an inflamed vessel. Note that “I” has traversed the entire depth of the endothelial cell and placed the luminal and abluminal membranes into extremely close apposition.
(F) Multiple basophil podosomes (“P1–P8;” ~0.2–0.5 μm) protrude into the endothelial surface.
In all panels, leukocytes are indicated by a 5% opacity red overlay. In all panels, arrows indicate endothelial vesicles and VVOs, both apparently free in the cytoplasm and fused or docked to the plasma membrane, enriched near leukocyte protrusions. Scale bars represent 500 nm in (A)–(C), (E), and (F) and 100 nm in insert in (A) and (D).
For 143 lymphocyte protrusions from randomly selected sections, dimensions were determined in the y axis normal to the endothelial monolayer and in the xz plane of the monolayer. The average width along the x axis was 344 ± 13 nm. For several podosomes, we also estimated the width in the z axis by determining the minimal number of 90 nm-thick serial sections that contained the entire podosome. The average z:x ratio was 1.2 ± 0.06 (n = 8), demonstrating that these structures were basically symmetrical in agreement with the confocal analysis above. The depth of protrusions into the endothelium in the y axis was 482 ± 29 nm (n = 143). Thus, these structures were consistent with podosomes (width and depth both ~200–500 nm [Buccione et al., 2004; Linder and Aepfelbacher, 2003]).
However, diameters ranged from 94 to 1046 nm and depths ranged from 81 to 2145 nm. Thus, many of the protrusions exceeded typical size criteria for podosomes and exhibited widths and, particularly, depths that were more consistent with invadopodia (Figures 5A and 5B, see “I” and “I1–I3,” respectively; Buccione et al., 2004; Linder and Aepfelbacher, 2003). We term these extended protrusions “invasive podosomes.” Importantly, the sizes were continuously distributed between the extremes, rather than bimodally (Figure S4), consistent with a continuum of related protrusive structures rather than the coexistence of categorically distinct ones.
To assess the physiologic relevance of these structures, we examined leukocyte-endothelial interactions in two distinct settings in vivo. TEM of inflamed vessels in a guinea pig model of dermatitis demonstrated both lymphocytes (Figure 5E) and basophils (Figure 5F) bearing multiple ventral podosomes and invasive podosomes (essentially identical to those seen in vitro) that protruded into endothelial invaginations. Similar observations were made for murine mononuclear cells bound to tumor-associated vessels (Figure S5). These results demonstrate that the podosomes and invasive podosomes form in vivo and in the context of shear flow. We further confirmed this latter point by assessing lymphocyte podoprint formation on HLMVECs in the presence of 2 dyne/cm2 laminar shear flow in vitro. In this setting, the overall efficiency and dynamics of podosome, podoprint, and transcellular pore formation were similar to those seen under static conditions (data not shown).
Podosomes Are Required for Efficient Transcellular Pore Formation
The regulatory protein WASP has been shown to be critical for podosome formation (Calle et al., 2004; Linder and Aepfelbacher, 2003). To investigate the role of WASP in our system, we obtained lymphocytes from WAS and XLT (X-linked thrombocytopenia) patients bearing the WASP-destabilizing mutations W64R or P58A, respectively. Immunoblotting demonstrated that WASP expression was either undetectable (W64R) or ~10% (P58A) the amount of control lymphocytes (Figure 6A). Live-cell imaging with these lymphocytes demonstrated that spreading and lateral migration on endothelium was generally normal (Movie S10). Indeed, lateral migration velocities for W64R (4.9 ± 0.3 μm/min, n = 69) and P58A (5.1 ± 0.5 μm/min, n = 21) lymphocytes were indistinguishable from control (4.6 ± 0.4 μm/min, n = 28). In contrast, however, podoprint induction (a readout for podosome formation) was reduced to 25% of control (Figure 6B and Movie S10). In addition, dynamic imaging suggested a selective reduction in transcellular diapedesis (Movie S10). This phenotype was quantified through fixed-cell imaging experiments (Figures 6C and 6D). Whereas paracellular diapedesis was similar to control (Figure 6C), transcellular diapedesis was reduced by more than 80% in WASP-deficient lymphocytes (Figure 6D).
Figure 6. Podosomes and Transcellular Diapedesis Are Dependent on Lymphocyte Src and WASP Activity.
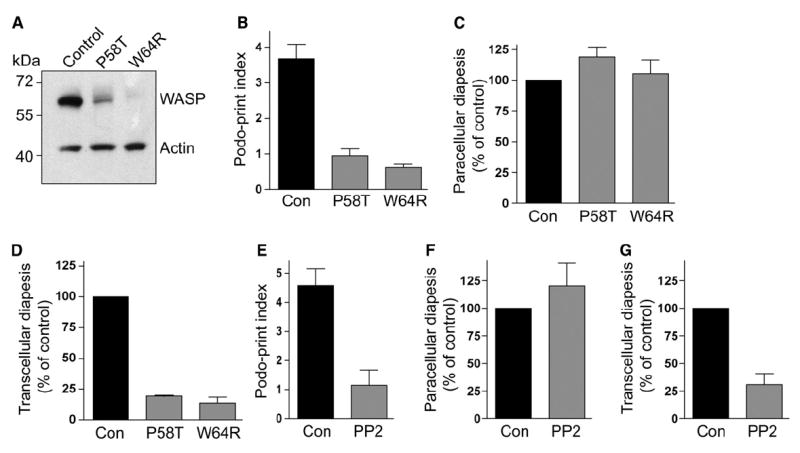
(A) Lysates from IL-2-cultured control and WASP-deficient (P58A and W64R) lymphocytes were immunoblotted for WASP and actin as described (Jin et al., 2004).
(B) Podosome formation by WASP-deficient lymphocytes. Control, P58A, and W64R lymphocytes were incubated with TNF-α-activated HLMVEC transiently transfected with memb-YFP and subjected to live-cell dynamic imaging. As a readout for podosome formation, podoprint indices were quantified as described in Experimental Procedures. Values are mean ± SEM of measurements obtained from at least 40 lymphocytes. See corresponding Movie S10.
(C and D) Para- and transcellular diapedesis in WAS lymphocytes. Control, P58A, and W64R lymphocytes were incubated with activated HLMVEC for 5 min and then fixed, stained, and scored for para- and transcellular diapedesis.
(C) Paracellular diapedesis by P58A and W64R lymphocytes were expressed as a percentage of the control value (36.5% ± 1.7% of total cells).
(D) Transcellular diapedesis by P58A and W64R lymphocytes were expressed as a percentage of the control value (22.5% ± 1.6% of total cells). Values are mean of normalized values ± SEM for at least three separate experiments.
(E) Podoprint indices for lymphocytes pretreated with the Src inhibitor PP2 (1 μM, 60 min) or DMSO (control) were assessed as in (B).
(F and G) Para- and transcellular diapedesis in lymphocytes pretreated with PP2 (1 μM, 60 min) or DMSO (control) was as described in (C) and (D), respectively. Control values for para- and transcellular diapedesis were 21.5% ± 2.6% (F) and 12.9% ± 1.6% (G) of total cells, respectively.
Podosomes are reported to depend strongly on Src kinase activity (Buccione et al., 2004; Linder and Aepfelbacher, 2003). Thus, we pretreated lymphocytes with Src inhibitor PP2 or vehicle (DMSO, control) and conducted dynamic and fixed-cell analysis similar to those described above for WASP-deficient cells. Live-cell imaging demonstrated that the PP2-treated lymphocytes spread, polarized, and migrated laterally on endothelium with velocities (4.6 ± 0.5 μm/min, n = 14) similar to control (5.2 ± 0.5 μm/min, n = 24). However, the Src-inhibited lymphocytes displayed significantly reduced podoprint formation (p = 0.0005) (Figure 6E) that correlated with a selective decrease in transcellular diapedesis (Figures 6F and 6G). Together, the results with WASP-deficient and Src-inhibited lymphocytes demonstrate the importance of leukocyte podosomes in transcellular pore formation.
Regulated Membrane Fusion in Endothelial Cells Is Required for Efficient Transcellular Pore Formation
During both in vitro and in vivo ultrastructure analysis, we noted an enrichment of endothelial vesicles and VVOs (vesiculo-vacuolar organelles) (Feng et al., 2002) immediately adjacent to sites of podosome protrusion (Figure 5 and Figure S5). Quantitative analysis revealed a ~4-fold increase in total vesicle density in cultured endothelium within 500 nm of podosomes, compared to endothelium without attached lymphocytes or with attached lymphocytes but lacking podosomes (Figure 7A). Moreover, the density of vesicles and VVOs that were fused or docked to the endothelial cell plasma membrane was also increased by ~4-fold at these locations (Figure 7A). These data suggest that lymphocyte podosomes trigger local recruitment and fusion of endothelial vesicles with the plasma membrane.
Figure 7. Endothelial Calcium- and SNARE-Mediated Membrane Fusion Is Required for Efficient Transcellular Diapedesis.
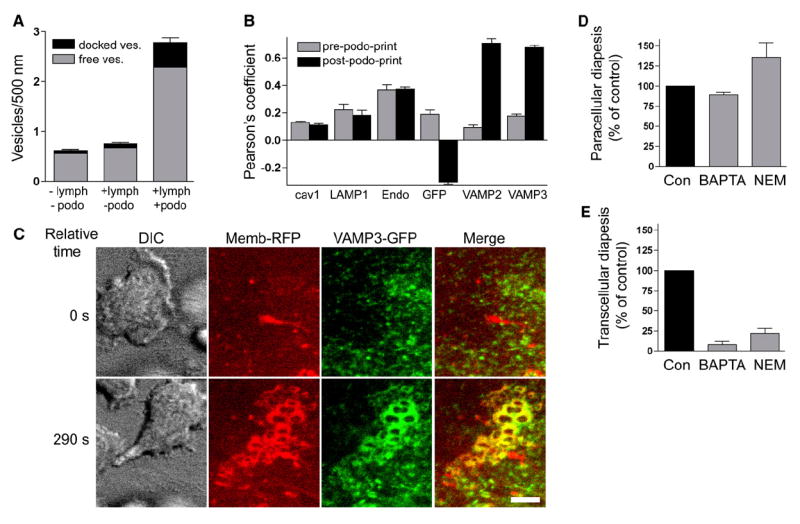
(A) The density of both free (gray) and plasma-membrane-fused or docked (black) vesicles in HDMVEC was quantified in samples without (−) or with (+) lymphocyte incubation and without (−) or with (+) podosomes, as described in Experimental Procedures. Values are mean ± SEM for at least 74 linear segments, each 500 nm long.
(B) Pearson’s correlation for the distribution of caveolin-1-GFP (cav1), LAMP1-GFP (LAMP1), pACGFP-Endo (Endo), GFP, VAMP2-GFP (VAMP2), and VAMP3-GFP (VAMP3) relative to memb-RFP, both prior to (gray bars) and during (black bars) podoprint formation, was quantified from live-cell imaging experiments as described in Experimental Procedures. Values are mean ± SEM for at least five measurements.
(C) DIC, memb-RFP, VAMP3-GFP, and merged memb-RFP and VAMP3-GFP images are as indicated. Upper panels show a time point before (relative time = 0 s) and lower panels show a time point after (relative time 290 s) the formation of memb-RFP podoprints. See corresponding Movie S12. Scale bars represent 5 μm.
(D and E) Activated HLMVEC were pretreated with BAPTA-AM (20 μM, 60 min), NEM (300 μM, 5 min), or DMSO (control) and then incubated with lymphocytes for 5 min followed by fixation, staining, and scoring. Control values for para- and transcellular diapedesis were 29% ± 1.2% (D) and 14.9% ± 1.8% (E) of total cells, respectively.
We sought to identify and monitor these vesicles via live-cell fluorescence imaging during podoprint formation. However, markers for caveolae (cav1-GFP) (Movie S11), endosomes (pACGFP-Endo) (not shown), and lysosomes (LAMP1-GFP; Lyso-Tracker Green) (not shown) were not enriched adjacent to podoprints, as quantified by Pearson’s correlation analysis of the distribution of these markers relative to memb-RFP (Figure 7B). In contrast, we found that two important fusogenic proteins in endothelium, the SNAREs VAMP2 (p < 0.0001) and VAMP3 (p < 0.0001), became significantly enriched in podoprints and early-stage transcellular pores, exhibiting large increases in Pearson’s coefficients (Figures 7B and 7C and Movie S12).
To investigate whether fusion activity is important for transcellular pore formation, we blocked function of the NSF-, SNAP-, and SNARE-complex in endothelium either by chelating intracellular calcium with BAPTA-AM or by inactivating NSF (NEM-sensitive factor) with N-ethyl maleimide (NEM). Through live-cell imaging, we observed that whereas podoprints were essentially normal, transcellular pore formation was reduced in each of these settings (not shown). This was quantified by fixed-cell imaging experiments; these showed significant reductions in transcellular pore formation of 87% (p < 0.0001) and 72% (p = 0.0004) by BAPTA and NEM treatments, respectively (Figure 7E). Paracellular diapedesis was only modestly reduced (by ~10%) by BAPTA in this setting and was actually increased by NEM (Figure 7D), correlating with partial junction disruption effect by NEM (not shown).
DISCUSSION
Crossing endothelial barriers is a critical requirement for leukocyte trafficking and immune system function. Our results here, coupled with recent in vitro (Carman and Springer, 2004; Cinamon et al., 2004; Millan et al., 2006; Nieminen et al., 2006; Yang et al., 2005) and previous in vivo (Feng et al., 1998; Greenwood et al., 1994; Lossinsky and Shivers, 2004; Marchesi and Gowans, 1963; Wolburg et al., 2005) investigations, demonstrate that transcellular diapedesis is a quantitatively important contributor to overall physiologic leukocyte trafficking. Lossinsky and Shivers (2004) have proposed that diapedesis occurs through the “path of least resistance,” an idea that was recently supported by in vivo studies (Wang et al., 2006). For transcellular diapedesis in particular, the means by which leukocytes identify sites permissive to transcellular pore formation have remained mysterious. The dynamic podosome- and invasive podosome-mediated protrusive behavior uncovered in the current study provides a mechanism for leukocytes to both discover and exploit such pathways.
Through combined fluorescence and ultrastructure imaging studies, we discovered highly dynamic leukocyte protrusions that form against the endothelial surface. Based on their architecture, dynamics, and WASP and Src dependence, these protrusions were determined to represent podosomes and extended “invasive podosomes.” These were formed by lymphocytes, monocytes, and basophils and observed both in vitro and several settings in vivo. Most importantly, transcellular diapedesis, but not paracellular diapedesis or lateral migration, was found to be highly dependent on podosomes and invasive podosomes.
The dozens of podosome protrusions that form rapidly during lateral migration seem to provide an efficient means for leukocytes to probe or palpate the local surface resistance of the endothelium (Figure S6). In response to podosomes, endothelial cell-surface invaginations, i.e., podoprints, were formed, which displaced and distorted the cytoplasm, actin filaments, microtubules, and endoplasmic reticulum to varying degrees. Shallow podosomes and podoprints were also frequently observed to encounter the endothelial nucleus, but with modest distortion and never transcellular pore formation at these sites (see model, Figure S6). We presume that the nuclear envelope and its underlying lamina provide too great a resistance for deeper penetration.
The finding of podoprints over nuclei suggests that leukocytes do not know a priori that they cannot diapedese directly through the nucleus, but must discover a route through trial and error. Even in nonnuclear areas, the vast majority of podosomes were inserted less than 600 nm into the endothelium (~half of the total average endothelial cell thickness) and were rapidly retracted after ~10–30 s without transcellular pore formation (Figure S6). This suggests that endothelial locations permissive for deeper protrusion and transcellular diapedesis are limiting. We anticipate that the efficiency of transcellular diapedesis is governed by the sum local resistances provided by endothelial organelles including the plasma membrane, nucleus, cytoskeleton, and endoplasmic reticulum. We propose that podosome-mediated palpation during lateral migration provides leukocytes with a stochastic mechanism to efficiently identify locations of relatively low total endothelial resistance (Figure S6). When such locations are encountered, podosomes are able to progressively extend (becoming invasive podosomes) all the way to the basal endothelial surface, resulting in transcellular pore formation (Figure S6).
Our studies here demonstrate a distinct proactive role for the endothelium in locally modulating permissiveness to leukocyte protrusions. We found that both plasma membrane-fused vesicles and VVOs and fusogenic proteins (i.e., the SNAREs VAMP2 and 3) were substantially enriched at endothelial sites of podosome protrusion. Furthermore, the efficiency of transcellular pore formation was attenuated by endothelial treatments (i.e., BAPTA and NEM) that inhibit SNARE fusogenic activity. We envision that podosomes induce endothelial signals (such as calcium fluxes initiated through endothelial adhesion receptors or stretch-activated ion channels) that trigger site-specific SNARE complex-mediated vesicle and VVO fusion. Such vesicle fusion could serve to lower local surface tension, by providing additional plasma-membrane surface area and thereby allow invasive podosomes to probe progressively deeper.
In vivo ultrastructure studies here in two distinct settings demonstrated physiologic relevance for both the leukocyte palpation behavior and the resulting endothelial vesicle enrichment. A re-examination of the existing literature provides further support for this. Indeed, though referred to variously as “processes,” “filopodia-like structures,” and “pseudopods,” ultrastructurally defined protrusions remarkably similar to the podosomes and invasive podosomes characterized in the current study have been demonstrated in vivo to be formed by lymphocytes (Fujita et al., 1991; Lossinsky and Shivers, 2004; Olah and Glick, 1985; Raine et al., 1990; Wolburg et al., 2005; Wolosewick, 1984), monocytes (Greenwood et al., 1994), and tumor cells (De Bruyn et al., 1989) during transcellular diapedesis across a variety of vascular and lymphatic endothelia, often in association with visible endothelial vesicle enrichment.
Beyond vascular and lymphatic endothelium, the act of palpating in order to identify the “path of least resistance” could be important for other substrates encountered by leukocytes during trafficking. For example, it was recently demonstrated that neutrophil extravasation in vivo occurs preferentially near pre-existing attenuations in the basement membrane, suggesting that leukocytes possess mechanisms to identify weak points in the matrix (Wang et al., 2006). It has also been demonstrated that neutrophils are capable of transcellular diapedesis through the pericytes underlying the vascular endothelium (Feng et al., 1998). Moreover, leukocytes are known to cross the intestinal epithelium by yet incompletely understood mechanisms (Zen and Parkos, 2003). Interestingly, we found here that lymphocytes readily formed podosomes and transcellular pores across epithelial cells, i.e., CHO-K1 expressing ICAM-1-GFP. Thus, podosome-dependent leukocyte-protrusive behavior might indeed have broad roles in leukocyte trafficking.
Our studies point to important new ideas regarding the function of podosomes and their relation to invadopodia, a recent subject of intense investigation. The structures formally defined as “invadopodia” have been demonstrated only in transformed cells where they function to confer invasiveness, i.e., the ability to cross tissue barriers. Functions for podosomes have remained unclear. To our knowledge, the functional role of leukocyte podosomes has not previously been studied in the context of the endothelium, a physiologic and “soft” substrate. Instead, the vast majority of studies have employed solid glass and plastic substrates, which would likely frustrate the formation of extended invasive podosomes and mask the protrusive and invasive functions (Yamaguchi et al., 2005b) that we have described here. Moreover, assays directly implicating podosomes (and related signaling activities including WASP and Src) in leukocyte locomotion and lateral migration have been performed on glass and plastic substrates with inclusion of only a single chemoattractant. This contrasts with the robust and physiologic substrate provided by the activated endothelium on which lymphocyte lateral migration was independent of podosomes. We found, in the physiologic setting of leukocyte-endothelial interaction, the dominant function of podosomes was in protrusion and invasion, functions akin to those ascribed to tumor-cell invadopodia.
A precursor role of podosomes relative to invadopodia has been hypothesized (Buccione et al., 2004; Linder and Aepfelbacher, 2003; Yamaguchi et al., 2005a) but remains controversial. On the “soft” endothelial substrate, lymphocyte podosomes were free, at permissive locations, to progressively extend to invadopodia-like depths (several microns) as they transition to “invasive podosomes.” These structures served an unambiguously invasive function; indeed, the act of passing directly through another cell would seem to be the ultimate act of invasiveness. Leukocyte podosomes have also recently been shown to possess invadopodia-like matrix-degrading capabilities (Buccione et al., 2004; Linder and Aepfelbacher, 2003). These observations suggest that leukocytes are capable of expressing a continuum of related podosomes and invadopodia-like structures.
Thus, we propose that (with the exception of osteoclasts, which have the distinction of interacting with a physiologic solid substrate, i.e., bone [Buccione et al., 2004; Linder and Aepfelbacher, 2003]) podosomes are probing or palpating organelles, uniquely designed for “path finding,” rather than locomotion per se, during the process of tissue-barrier crossing and invasion. Tumor-cell invadopodia likely represent the pathologic subversion of the physiologic invasive podosome.
These findings add to our understanding of basic mechanisms for leukocyte trafficking. Future investigation of how leukocyte podosomes are influenced by diverse in vivo settings may provide even greater understanding of both normal and pathologic immune cell trafficking.
EXPERIMENTAL PROCEDURES
Antibodies and Reagents
Cells and Cell Culture
Studies of human blood were approved by the institutional review board at the CBRI, Harvard Medical School. Written informed consent was obtained from all the subjects. Animal studies were conducted under protocols approved by the Committee on Use and Care of Animals at BIDMC, Harvard Medical School. Preparation of mitogen-activated, IL-2-cultured primary human lymphocytes (model effector type lymphocytes [Weninger et al., 2001]) was as described (Carman and Springer, 2004). Resulting cells were ~97% CD3+, CD56−, ~50% CD4+, and ~50% CD8+. Freshly isolated human PBL and monocytes were prepared as described (Carman et al., 2003; Carman and Springer, 2004). IL-2-cultured lymphocytes, from patients with WASP-destabilizing mutations (W64R or P58T) and healthy volunteers (control), were prepared and assessed by immunoblotting as described (Jin et al., 2004). The W64R patient presented a classic WAS phenotype (clinical score of 4), and the P58A patient presented a milder phenotype (clinical score of 2) with features of XLT (Jin et al., 2004). Stably transfected K562 cells expressing LFA-1 and CHO-K1 cells expressing ICAM-1-GFP were described (Carman et al., 2003). HUVEC, HDMVEC, and HLMVEC were purchased from Cambrex (East Rutherford, NJ) and cultured on fibronectin (20 mg/ml)-coated substrates in either EGM-2 (for HUVEC) or EGM-2MV (HDMVEC and HLMVEC) media (Cambrex). In addition, HDMVEC (HDMVEC*) and lymphatic ECs (Lymph*) were isolated from fresh neonatal foreskins (Richard et al., 1998). EC and lymphocyte transient transfection was by Amaxa Nucleofection according to the manufacturer’s instructions (Amaxa Inc., Gaithersburg, MD). pAcGFP1-Endo, membrane-YFP, monomeric-DSRed (RFP), pEGFP-tubulin, and pEGFP-actin plasmids were purchased from Clontech (Mountain View, CA). Memb-RFP was generated by overlap-extension PCR to add an N-terminal palmitoylation sequence (from membrane-YFP) to monomeric-DSRed. Other DNA constructs were kind gifts from T. Kirchhausen (LAMP1-GFP, CBR Institute for Biomedical Research), M. Lisanti (Caveolin-1-GFP, Thomas Jefferson University), E. Masuda (VAMP2-GFP, Rigel Pharmaceuticals, San Francisco, CA), and W.S. Trimble (VAMP3-GFP, University of Toronto).
Lymphocyte Diapedesis
ECs were plated at 90% confluency on fibronectin (20 μg/ml)-coated glass surfaces and cultured for 48–72 hr and then activated for 12 hr with TNF-α (50 ng/ml) before use. Where indicated, ECs were also preincubated with BAPTA-AM (20 μM, 1 hr), NEM (300 μM, 5 min), or equal dilutions of vehicle (DMSO). In addition, for specified experiments, ECs were prelabeled with either Endo- or Lyso-Tracker Green (100 nM for 30 min at 37°C). ECs were washed five times prior to addition of lymphocytes with Hank’s balanced salt solution supplemented with 20 mM HEPES (pH 7.2) and 1% human serum albumin (Buffer A). Lymphocytes were washed and resuspended in Buffer A and then added to EC monolayers and incubated at 37°C for the indicated times. Where indicated, lymphocytes were preincubated with the Src-inhibitor PP2 (1 μM, for 60 min) or an equal dilution of vehicle (DMSO).
Fixed-Cell Imaging
Samples were fixed and stained for β2 (LFA1/7-488, -Cy3, or -Cy5), ICAM-1 (IC1/11-488 or IC1/11-Cy3), VE-cadherin (55-7H1-488, -Cy3, or -Cy5), PECAM-1 (9G11-Cy3), JAM-1 (polyclonal anti-JAM-1-488), talin-1 (polyclonal anti-talin-1 followed by goat anti-rabbit-488), and F-actin (phalloidin-488, -546, or -647) as described (Carman and Springer, 2004). The stages and route of leukocyte diapedesis were determined from the relative distribution (assessed by confocal microscopy) of ICAM-1 and LFA-1 and VE-cadherin, PECAM-1, JAM-1 or actin fluorescence in both the x-y and z dimensions (Carman and Springer, 2004). Diapedesis adjacent to endothelial cell-cell junctions was scored as paracellular; diapedesis where no part of the transmigration passage was within 1 μm of a junction was scored as transcellular.
Live-Cell Imaging
For live-cell imaging, ECs growth on Bioptechs delta-T chambers were maintained at 37°C in Buffer-A before and during addition of leukocytes. DIC and epifluorescence images were acquired at intervals of 10–15 s as indicated for durations of ~20 min. Migrating lymphocytes were determined to be above (bright edges) or below (dark edges) the endothelium based on their diffractive properties in DIC. Initiation of diapedesis was determined by the appearance, under bound lymphocytes, of an endothelial region in which the fluorescent membrane reporter was completely displaced, forming a “black spot.” This readout was validated by confocal microscopy as described (Carman and Springer, 2004). Endothelial junctions could readily be seen by DIC. However, determination of transcellular diapedesis in most cases was simplified by imaging individual reporter-positive EC transfectants surrounded by reporter-negative ECs. Unambiguous transcellular pores could be seen as dark spots formed >3 μm away from the edge of these individual cells (the criteria of 3 μm was used here, rather than 1 μm used above for confocal analysis, to account for the relative decrease in resolution with this technique). Details for analysis of live-cell imaging are provided in Supplemental Experimental Procedures.
Light-Microscopy Image Acquisition and Processing
Wide-field DIC and epifluorescence imaging were on a Zeiss Axiovert S200 epifluorescence microscope (Germany), with a 63× oil objective and a Hamamatsu Orca CCD (Japan). Confocal imaging was performed with a Biorad Radiance 2000 laser-scanning confocal system on an Olympus BX50BWI microscope with a 100× water-dipping objective. For serial Z-stacks, the Z-step was between 0.1 and 0.3 μm. Pearson’s analysis was conducted with Volocity 3.6.1 software (Improvision). Other analysis was with Openlab software (Improvision). Images were exported to Adobe Photoshop or Apple Quicktime software for preparation of final images or videos, respectively.
Transmission Electron Microscopy
TNF-α-activated HDMVEC grown on fibronectin-coated coverglass, with or without lymphocyte coincubation (as indicated) were fixed with 2.5% glutaraldehyde and 2% paraformaldehyde in 1.0 M sodium cacodylate buffer (pH 7.4), for 2 hr, postfixed in 1.5% sym-collidine-buffered OsO4 for 1 hr, stained en bloc with uranyl acetate, dehydrated in a series are graded alcohols, and embedded in Eponate. The coverglass was then separated from the basal aspect of the endothelium by brief treatment with liquid nitrogen (a step that causes mild disruption to the basal plasma membrane, lowering its contrast in EM), and the samples were then re-embedded with the monolayer parallel to the long axis of the embedding block. Thin Eponate sections of 90 nm, cut perpendicular to the long axis of the block (and to the plane of the monolayer), were visualized with a Philips CM-10 electron microscope at 80 kV. Preparation of Hartley guinea pigs sensitized for cutaneous basophil hypersensitivity reactions (Dvorak and Dvorak, 1993) and solid MOT tumors grown in C3Heb/FeJ mice (Nagy et al., 1995) were described. Tissue was processed for TEM as described (Feng et al., 2002). Details for the quantitation of ultrastructure are provided in Supplemental Experimental Procedures.
Supplementary Material
Six figures, twelve movies, and Experimental Procedures are available at http://www.immunity.com/cgi/content/full/26/6/■■■/DC1/.
Acknowledgments
We thank C. Peruzzi for providing isolated HDMVEC and lymphatic endothelium. This work was supported by grants from the Arthritis Foundation (C.V.C.) and the National Institutes of Health (T.A.S., H.F.D., H.D.O., and R.S.G.).
References
- Adamson P, Etienne S, Couraud PO, Calder V, Greenwood J. Lymphocyte migration through brain endothelial cell monolayers involves signaling through endothelial ICAM-1 via a rhodependent pathway. J Immunol. 1999;162:2964–2973. [PubMed] [Google Scholar]
- Barreiro O, Yanez-Mo M, Serrador JM, Montoya MC, Vicente-Manzanares M, Tejedor R, Furthmayr H, Sanchez-Madrid F. Dynamic interaction of VCAM-1 and ICAM-1 with moesin and ezrin in a novel endothelial docking structure for adherent leukocytes. J Cell Biol. 2002;157:1233–1245. doi: 10.1083/jcb.200112126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buccione R, Orth JD, McNiven MA. Foot and mouth: podosomes, invadopodia and circular dorsal ruffles. Nat Rev Mol Cell Biol. 2004;5:647–657. doi: 10.1038/nrm1436. [DOI] [PubMed] [Google Scholar]
- Calle Y, Chou HC, Thrasher AJ, Jones GE. Wiskott-Aldrich syndrome protein and the cytoskeletal dynamics of dendritic cells. J Pathol. 2004;204:460–469. doi: 10.1002/path.1651. [DOI] [PubMed] [Google Scholar]
- Carman CV, Springer TA. A transmigratory cup in leukocyte diapedesis both through individual vascular endothelial cells and between them. J Cell Biol. 2004;167:377–388. doi: 10.1083/jcb.200404129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carman CV, Jun C-D, Salas A, Springer TA. Endothelial cells proactively form microvilli-like membrane projections upon ICAM-1 engagement of leukocyte LFA-11. J Immunol. 2003;171:6135–6144. doi: 10.4049/jimmunol.171.11.6135. [DOI] [PubMed] [Google Scholar]
- Cinamon G, Shinder V, Shamri R, Alon R. Chemoattractant signals and beta 2 integrin occupancy at apical endothelial contacts combine with shear stress signals to promote transendothelial neutrophil migration. J Immunol. 2004;173:7282–7291. doi: 10.4049/jimmunol.173.12.7282. [DOI] [PubMed] [Google Scholar]
- De Bruyn PP, Cho Y, Michelson S. Endothelial attachment and plasmalemmal apposition in the transcellular movement of intravascular leukemic cells entering the myeloid parenchyma. Am J Anat. 1989;186:115–126. doi: 10.1002/aja.1001860202. [DOI] [PubMed] [Google Scholar]
- Dvorak AM, Dvorak HF. Cutaneous basophil hypersensitivity: a 20-year perspective, 1970-1990. In: Foreman JC, editor. Immunopharmacology of Mast Cells and Basophils. London: Academic Press; 1993. pp. 153–180. [Google Scholar]
- Etienne-Manneville S, Manneville JB, Adamson P, Wilbourn B, Greenwood J, Couraud PO. ICAM-1-coupled cytoskeletal rearrangements and transendothelial lymphocyte migration involve intracellular calcium signaling in brain endothelial cell lines. J Immunol. 2000;165:3375–3383. doi: 10.4049/jimmunol.165.6.3375. [DOI] [PubMed] [Google Scholar]
- Evans JG, Correia I, Krasavina O, Watson N, Matsudaira P. Macrophage podosomes assemble at the leading lamella by growth and fragmentation. J Cell Biol. 2003;161:697–705. doi: 10.1083/jcb.200212037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng D, Nagy JA, Pyne K, Dvorak HF, Dvorak AM. Neutrophils emigrate from venules by a transendothelial cell pathway in response to FMLP. J Exp Med. 1998;187:903–915. doi: 10.1084/jem.187.6.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng D, Nagy JA, Dvorak HF, Dvorak AM. Ultrastructural studies define soluble macromolecular, particulate, and cellular transendothelial cell pathways in venules, lymphatic vessels, and tumor-associated microvessels in man and animals. Microsc Res Tech. 2002;57:289–326. doi: 10.1002/jemt.10087. [DOI] [PubMed] [Google Scholar]
- Fujita S, Puri RK, Yu ZX, Travis WD, Ferrans VJ. An ultrastructural study of in vivo interactions between lymphocytes and endothelial cells in the pathogenesis of the vascular leak syndrome induced by interleukin-2. Cancer. 1991;68:2169–2174. doi: 10.1002/1097-0142(19911115)68:10<2169::aid-cncr2820681014>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- Greenwood J, Howes R, Lightman S. The blood-retinal barrier in experimental autoimmune uveoretinitis. Leukocyte interactions and functional damage. Lab Invest. 1994;70:39–52. [PubMed] [Google Scholar]
- Huang AJ, Manning JE, Bandak TM, Ratau MC, Hanser KR, Silverstein SC. Endothelial cell cytosolic free calcium regulates neutrophil migration across monolayters of endothelial cells. J Cell Biol. 1993;120:1371–1380. doi: 10.1083/jcb.120.6.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y, Mazza C, Christie JR, Giliani S, Fiorini M, Mella P, Gandellini F, Stewart DM, Zhu Q, Nelson DL, et al. Mutations of the Wiskott-Aldrich Syndrome Protein (WASP): hotspots, effect on transcription, and translation and phenotype/genotype correlation. Blood. 2004;104:4010–4019. doi: 10.1182/blood-2003-05-1592. [DOI] [PubMed] [Google Scholar]
- Linder S, Aepfelbacher M. Podosomes: adhesion hotspots of invasive cells. Trends Cell Biol. 2003;13:376–385. doi: 10.1016/s0962-8924(03)00128-4. [DOI] [PubMed] [Google Scholar]
- Lossinsky AS, Shivers RR. Structural pathways for macromolecular and cellular transport across the blood-brain barrier during inflammatory conditions. Histol Histopathol. 2004;19:535–564. doi: 10.14670/HH-19.535. [DOI] [PubMed] [Google Scholar]
- Marchesi VT. The site of leucocyte emigration during inflammation. Q J Exp Physiol. 1961;46:115–133. doi: 10.1113/expphysiol.1961.sp001522. [DOI] [PubMed] [Google Scholar]
- Marchesi VT, Gowans JL. The migration of lymphocytes through the endothelium of venules in lymph nodes: an electron microscope study. Proc R Soc Lond B Biol Sci. 1963;159:283–290. doi: 10.1098/rspb.1964.0002. [DOI] [PubMed] [Google Scholar]
- Millan J, Hewlett L, Glyn M, Toomre D, Clark P, Ridley AJ. Lymphocyte transcellular migration occurs through recruitment of endothelial ICAM-1 to caveola- and F-actin-rich domains. Nat Cell Biol. 2006;8:113–123. doi: 10.1038/ncb1356. [DOI] [PubMed] [Google Scholar]
- Muller WA. Leukocyte-endothelial-cell interactions in leukocyte transmigration and the inflammatory response. Trends Immunol. 2003;24:327–334. doi: 10.1016/s1471-4906(03)00117-0. [DOI] [PubMed] [Google Scholar]
- Nagy JA, Masse EM, Herzberg KT, Meyers MS, Yeo KT, Yeo TK, Sioussat TM, Dvorak HF. Pathogenesis of ascites tumor growth: vascular permeability factor, vascular hyperpermeability, and ascites fluid accumulation. Cancer Res. 1995;55:360–368. [PubMed] [Google Scholar]
- Nieminen M, Henttinen T, Merinen M, Marttila-Ichihara F, Eriksson JE, Jalkanen S. Vimentin function in lymphocyte adhesion and transcellular migration. Nat Cell Biol. 2006;8:156–162. doi: 10.1038/ncb1355. [DOI] [PubMed] [Google Scholar]
- Olah I, Glick B. Lymphocyte migration through the lymphatic sinuses of the chicken’s lymph node. Poult Sci. 1985;64:159–168. doi: 10.3382/ps.0640159. [DOI] [PubMed] [Google Scholar]
- Raine CS, Cannella B, Duijvestijn AM, Cross AH. Homing to central nervous system vasculature by antigen-specific lymphocytes. II. Lymphocyte/endothelial cell adhesion during the initial stages of autoimmune demyelination. Lab Invest. 1990;63:476–489. [PubMed] [Google Scholar]
- Richard L, Velasco P, Detmar M. A simple immunomagnetic protocol for the selective isolation and long-term culture of human dermal microvascular endothelial cells. Exp Cell Res. 1998;240:1–6. doi: 10.1006/excr.1998.3936. [DOI] [PubMed] [Google Scholar]
- Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, Parsons JT, Horwitz AR. Cell migration: integrating signals from front to back. Science. 2003;302:1704–1709. doi: 10.1126/science.1092053. [DOI] [PubMed] [Google Scholar]
- Schenkel AR, Mamdouh Z, Muller WA. Locomotion of monocytes on endothelium is a critical step during extravasation. Nat Immunol. 2004;5:393–400. doi: 10.1038/ni1051. [DOI] [PubMed] [Google Scholar]
- Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multi-step paradigm. Cell. 1994;76:301–314. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- von Andrian UH, Mackay CR. T-cell function and migration. Two sides of the same coin. N Engl J Med. 2000;343:1020–1034. doi: 10.1056/NEJM200010053431407. [DOI] [PubMed] [Google Scholar]
- Wang S, Voisin MB, Larbi KY, Dangerfield J, Scheiermann C, Tran M, Maxwell PH, Sorokin L, Nourshargh S. Venular basement membranes contain specific matrix protein low expression regions that act as exit points for emigrating neutrophils. J Exp Med. 2006;203:1519–1532. doi: 10.1084/jem.20051210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weninger W, Crowley MA, Manjunath N, von Andrian UH. Migratory properties of naive, effector, and memory CD8(+) T cells. J Exp Med. 2001;194:953–966. doi: 10.1084/jem.194.7.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolburg H, Wolburg-Buchholz K, Engelhardt B. Diapedesis of mononuclear cells across cerebral venules during experimental autoimmune encephalomyelitis leaves tight junctions intact. Acta Neuropathol (Berl) 2005;109:181–190. doi: 10.1007/s00401-004-0928-x. [DOI] [PubMed] [Google Scholar]
- Wolosewick JJ. Distribution of actin in migrating leukocytes in vivo. Cell Tissue Res. 1984;236:517–525. doi: 10.1007/BF00217218. [DOI] [PubMed] [Google Scholar]
- Yamaguchi H, Lorenz M, Kempiak S, Sarmiento C, Coniglio S, Symons M, Segall J, Eddy R, Miki H, Takenawa T, Condeelis J. Molecular mechanisms of invadopodium formation: the role of the N-WASP-Arp2/3 complex pathway and cofilin. J Cell Biol. 2005a;168:441–452. doi: 10.1083/jcb.200407076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi H, Wyckoff J, Condeelis J. Cell migration in tumors. Curr Opin Cell Biol. 2005b;17:559–564. doi: 10.1016/j.ceb.2005.08.002. [DOI] [PubMed] [Google Scholar]
- Yang L, Froio RM, Sciuto TE, Dvorak AM, Alon R, Luscinskas FW. ICAM-1 regulates neutrophil adhesion and transcellular migration of TNF-alpha-activated vascular endothelium under flow. Blood. 2005;106:584–592. doi: 10.1182/blood-2004-12-4942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zen K, Parkos CA. Leukocyte-epithelial interactions. Curr Opin Cell Biol. 2003;15:557–564. doi: 10.1016/s0955-0674(03)00103-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Six figures, twelve movies, and Experimental Procedures are available at http://www.immunity.com/cgi/content/full/26/6/■■■/DC1/.