


Abstract
We have used 2-aminopurine (2AP) as a fluorescent probe in the template strand of a 13/20mer primer/template (D) to detect deoxynucleoside triphosphates (N)-dependent conformational changes exhibited by RB69 DNA polymerase (ED) complexes. The rates and amplitudes of fluorescence quenching depend hyperbolically on the [dTTP] when a dideoxy-primer/template (ddP/T) with 2AP as the templating base (n position) is used. No detectable fluorescence changes occur when a ddP/T with 2AP positioned 5′ to the templating base (n + 1 position) is used. With a deoxy-primer/template (dP/T) with 2AP in the n position, a rapid fluorescence quenching occurs within 2 ms, followed by a second, slower fluorescence quenching with a rate constant similar to base incorporation as determined by chemical quench. With a dP/T having 2AP in the n + 1 position, there is a [dNTP]-dependent fluorescence enhancement that occurs at a rate comparable to dNMP incorporation. Collectively, the results favor a minimal kinetic scheme in which population of two distinct biochemical states of the ternary EDN complex precedes the nucleotidyl transfer reaction. Observed differences between dP/T and ddP/T ternary complexes indicate that the 3′ hydroxyl group of the primer plays a critical role in determining the rate constants of transitions that lead to strong deoxynucleoside triphosphate binding prior to chemistry.
INTRODUCTION
A general feature of replicative DNA polymerases, such as RB69 pol, is their ability to select a dNTP complementary to the templating base with great accuracy during primer extension. The frequency of misincorporation errors rarely exceed 10−6 per replicated base pair because incorporated mispaired bases are usually excised by the polymerase itself or by a separate replicase subunit having 3′–5′ exonuclease activity (1–3). DNA polymerases also have to move rapidly and processively along the template strand as they catalyze the formation of phosphodiester bonds (4–6). This movement during leading strand synthesis is facilitated by DNA helicases that catalyze unwinding of the duplex DNA (7–9); single-strand DNA-binding proteins that keep the template-strand accessible to the polymerase and a doughnut-shaped trimeric sliding clamp with a central cavity diameter that can accommodate dsDNA and a groove on the outer surface which provides an interface for interaction with the C-terminal tail of the polymerase (10–14). The arrangement of these proteins in relation to each other and to the template ensures that the polymerase and the sliding clamp are tethered to the DNA until an appropriate signal triggers their disengagement (15–17).
Although enormous strides have been made during the last decade in understanding how DNA polymerases function in DNA replication and repair, there are still questions that remain about certain aspects of the mechanisms used by DNA polymerases to achieve fidelity. Among them is the relationship between base discrimination and the rate-limiting step in the nucleotidyl transfer reaction (18–20). Since most DNA polymerases share common structural motifs as well as basic mechanisms for primer extension and nucleotide excision, it has been a general belief that a kinetic scheme determined for a well characterized DNA pol would be universally applicable to all DNA polymerases (21–23). More recent data suggests that subtle differences in the rate and equilibrium constants of the enzymatic cycles among DNA polymerases confer specific catalytic activities that are important for their specialized tasks in the cell (21).
To gain a complete understanding of the role that nucleotide-linked conformational changes play in base discrimination and in the mechanism of nucleotidyl transfer, it is necessary to distinguish biochemical intermediates that are populated before nucleotidyl transfer from states that follow primer extension. If the prechemical transition acts as a check point for base discrimination as has been proposed (18), the conformational changes that accompany the dNTP binding-induced transition from the open to the closed state is predicted to be rate limiting for nucleoside triphosphate incorporation. This expectation appears to have been fulfilled for T7 DNA pol and the Klenow fragment (20), which are members of the A family polymerases, but Klentaq1, also an A family polymerase, is an exception since it has been shown that the transition from the open to the closed state is much faster than chemistry (24). Chemistry appears to be rate limiting for pol β (19), however, it is not clear which mechanism applies to members of the B family which includes T4 and RB69 pol (20). To address this issue we have used 2-aminopurine (2AP) as a fluorescent probe together with a nonextendable primer/template (ddP/T) which has allowed us to estimate both the rates of domain closing and opening in the presence of an incoming dNTP.
The nucleotide analog, 2AP, does not change the standard B-type conformation when it is part of a DNA duplex and is therefore a good adenosine analog (25). 2AP has been widely used as a fluorescent probe because its spectral properties respond to changes in its immediate environment that occur during nucleotidyl transfer. 2AP fluorescence is quenched when it stacks against adjacent bases or aromatic residues in proteins (25,26). Fluorescence enhancement occurs when stacking of 2AP with aromatic residues is perturbed (27,28). The fluorescence of 2AP is insensitive to base-pairing or other hydrogen bonding interactions (29). Furthermore, fluctuations in stacking of 2AP with an adjacent base does not interfere with the standard Watson–Crick hydrogen bonds between 2AP and thymidine (28). Thus, 2AP can be used as the templating base (n position) and changes in its fluorescence, when deoxynucleoside triphosphate (dTTP) is added to a DNA pol:ddP/T complex, allow monitoring of conformational alterations in the complex independent of fluorescence changes that arise as a consequence of chemistry. 2AP can also be placed just 5′ to the templating base (n + 1 position) so that conformational changes in the environment around the 2AP can potentially be monitored by fluorescence change in RB69:dP/T complexes with any dNTP as long as it is complementary to the templating base (26–28). This extends the range of this approach since the incoming dNTP does not have to be restricted to dTTP as is the case when 2AP is in the n position.
In this report, we have determined the kinetic parameters for nucleotide binding to the RB69 DNA polymerase primer/template complex and subsequent base incorporation by monitoring the nucleotide-dependent fluorescence changes of 2AP that has been incorporated into the n or n + 1 position of a primer/template. To distinguish conformational changes preceding chemistry from rates of phosphoryl transfer and subsequent steps, we have employed a ddP/T (with 2AP in the n position) that is chemically inert with respect to nucleotidyl transfer reaction (26,30–35).
We selected RB69 pol, a member of B family DNA polymerases, because crystal structures have been determined for the apo enzyme (36) as well as for complexes of the enzyme with ddP/Ts and complementary dNTPs (37–40). Our results provide evidence for the existence of a transient conformational state of a RB69 pol:dP/T:dTTP complex (2AP in the n position) that is populated after dTTP binding but before chemistry. We have evaluated the relative contribution of the rate of this conformational change to the rate-limiting step for incorporation of a correct base. We find no evidence for the existence of three distinct intermediates populated before the chemical step as proposed for T4 DNA polymerase (35), a close relative of RB69 pol. Rather, the experimental data is reliably accounted for by a mechanism in which two distinct biochemical states of the ternary complex, one that binds incoming nucleotide weakly and one that binds strongly, precedes nucleotidyl transfer. This information is crucial for understanding RB69 fidelity at the base selection stage.
EXPERIMENTAL PROCEDURES
Reagents
Escherichia coli strain BL21(DE3) was obtained from Stratagene Corp. DH5α cells were from Invitrogen. dNTPs, and T4 polynucleotide kinase were purchased from New England Biolabs; γ-[32P]-ATP was from Perkin Elmer Life Sciences Inc; and Ni-NTA resin was obtained from Qiagen. Other chemicals were analytical grade. RB69 pol cDNA was a generous gift from J. Karam (Tulane University). Oligonucleotides were provided by W. M. Keck Foundation Biotechnology Resource Laboratory (Yale University). The sequences of the DNA primer/template substrates (P/Ts) used in this study are shown in Figure 1. The fluorescent nucleotide analog 2AP is in the n or n + 1 position of the template.
Figure 1.

Primer/template sequences used in the fluorescence studies. The templating base is underlined. P stands for 2-aminopurine (2AP). (A) When 2AP is the templating base it is in the n position. (B) When 2AP is located 5′ to the templating base, it is in the n + 1 position. The ddP/Ts are identical to the dP/Ts except that for ddP/Ts the last nucleotide at the 3′ end of the primer is 2′, 3′-dideoxy-C.
Protein expression and purification
Site-directed mutagenesis was used to substitute Ala for Asp at residues 222 and 327 to create the exonuclease-deficient (exo−) RB69 pol which we refer to throughout the article as RB69 pol. Expression of the (exo−) RB69 pol was carried out as previously described (41). For ease in subsequent isolation, the cDNA for RB69 pol was subcloned into the pSP72 vector (Promega) so that the expressed protein would have six histidine residues appended to its C-terminus. This permitted facile purification of the (exo−) RB69 pol on a Ni-NTA column (42). For the final purification RB69 pol was loaded on a Source Q column (Pharmacia) and eluted with a linear gradient of increasing sodium chloride. Purified RB69 pol migrated as a single band on SDS–PAGE, was free of imidazole as judged by a 280/260 nm ratio of 1.91 and had pre-steady-state kinetic parameters for the pol reaction that corresponded closely to those obtained by Capson et al. (43) for T4 pol. After elution from the Source Q column, RB69 pol was dialyzed against pol storage buffer (20 mM Tris–HCl, pH 7.5; 0.1 mM EDTA; 5 mM 2-mercaptoethanol; 25% glycerol) and stored in small aliquots at –80°C. Protein concentrations were determined by the Bradford assay and by absorbance at 280 nm using an ε280 = 1.14 mg−1 cm−1).
Equilibrium binding titrations
Fluorescence emission spectra of the ddP/T (2AP in the n position) (200 nM) together with RB69 pol (1 or 2 μM), 10 mM MgCl2, 66 mM Tris–HCl, pH 7.5, 0.5 mM EDTA and varying [dTTP] were acquired at 25°C with a Photon Technology International Alphascan scanning spectrofluorometer. Samples were excited at 310 nm to minimize inner filter effects. Fluorescence emission spectra were collected from 320 to 460 nm and the intensities were corrected for the intrinsic fluorescence of RB69 pol. Peak intensities at 365 nm were plotted as a function of [dTTP] and fit to Equation (1) to obtain the overall dissociation equilibrium constants  for dNTP binding:
for dNTP binding:
| 1 |
where FI is the observed fluorescence intensity, FIo is the fluorescence in the absence of dTTP, FI∞ is the fluorescence at saturating dTTP and  is the overall dissociation constant for dTTP binding. This expression assumes binding to a single site. The same procedure was used for the ddP/T with 2AP in the n + 1 position.
is the overall dissociation constant for dTTP binding. This expression assumes binding to a single site. The same procedure was used for the ddP/T with 2AP in the n + 1 position.
Kinetic measurements and analysis
Transient kinetic fluorescence experiments were performed using an Applied Photophysics SX18MV-R stopped-flow apparatus (Leatherhead, UK) thermostatted at the indicated temperatures ±0.1°C. The excitation wavelength for 2AP was 313 nm. Fluorescence emission was monitored at 90° through a 345 nm long pass colored glass filter. The final concentrations of the reaction components after mixing were 66 mM Tris–HCl (pH 7.5), 200 nM dP/T or ddP/T, 1 μM RB69 pol, 10 mM MgCl2 and varying concentrations of dNTPs. For some experiments the final concentrations of RB69 pol and the P/T were 2 μM and 400 nM, respectively.
Time courses of fluorescence changes were fitted to a sum of exponentials [Equation. (2)] using Pro-K software provided with the instrument or with Kaleidagraph (Synergy Software, Reading, PA, USA):
| 2 |
where F(t) is the fluorescence at time t, F∞ is the final fluorescence intensity at equilibrium, Ai is the amplitude and ki is the observed pseudo-first order rate constant characterizing the ith relaxation process and n is the total number of observed relaxations. The value of n was one (single exponential) or two (double exponential). Most time courses shown are the average of five to seven traces.
Chemical quench experiments
All chemical quench reactions were performed using a KinTek RQF-3 quench flow instrument (KinTek Corp., Austin, TX, USA). Single turnover experiments were carried out at 22 or 4°C. The complex of 5′-[32P] labeled primer/template (dP/T) and RB69 pol was mixed with various concentrations of dNTP. The final concentrations after mixing were 200 nM dP/T, 1 μM RB69 pol, 10 mM MgCl2 and the indicated [dNTP]. Reactions were quenched with 0.5 M EDTA. The disappearance of substrate and the formation of product were monitored after gel electrophoresis (20% polyacrylamide/50% urea) by phosphorimager and quantified using NIH imaging software. Time courses for product formation were fitted to single exponentials. The [dNTP] dependence of the observed rate constant (kobs) was fitted to a rectangular hyperbola [Equation. (3)] where the maximum observed rate constant is the rate constant for dNMP incorporation (kpol) and  represents the apparent overall dissociation equilibrium constant for dNTP binding to the RB69 pol:dP/T complex prior to dNMP incorporation.
represents the apparent overall dissociation equilibrium constant for dNTP binding to the RB69 pol:dP/T complex prior to dNMP incorporation.
| 3 |
Kinetic simulations
Kinetic simulations were performed with Tenua (provided by Dr D. Wachsstock; available free online at: http://bililite.com/tenua/), which is based on the kinetic simulation program KINSIM (44).
RESULTS
Our goal in this study was to use the fluorescent nucleotide analog (2AP) at either the templating base (n position) or just 5′ to the templating base (n + 1 position) to detect transition intermediates during the RB69 polymerase primer extension reaction, and to determine the rate constants for their formation and loss both prior to and after phosphoryl transfer. We used a ddP/T to monitor fluorescence changes associated with ternary complex formation in the absence of base incorporation. A dP/T was used to monitor changes in 2AP fluorescence when nucleotidyl transfer reaction takes place.
Equilibrium fluorescence measurements
RB69 pol (1 μM) binding to the ddP/T (200 nM) with 2AP in the n position (Figure 1) increased the 2AP fluorescence by ∼6-fold (Figure 2A). Doubling the [RB69 pol] to 2 μM yielded a comparable ∼6-fold enhancement, consistent with saturation being achieved at 1 μM RB69 pol and an RB69 pol:ddP/T affinity of <100 nM, as reported for T4 pol with the same sized P/T (13/20-mer) (43).
Figure 2.

Equilibrium binding of dTTP to the RB69 DNA pol:ddP/T complex. (A) Fluorescence emission spectra of wild-type RB69 pol (exo−) enzyme in the presence of 13/20-mer ddP/T, with 2AP in the n position (shown by the solid lines). The concentration of ddP/T was 200 nM and RB69 pol was 1 μM, respectively. The [dTTP] (from top to bottom) are 0, 5, 10, 20, 40 and 80 μM. The dotted line is the fluorescence trace for the ddP/T alone. (B) The [dTTP]-dependence of fluorescence signal intensity at 365 nm. The solid line is the best fit to a single-site binding isotherm [Equation (1)] yielding a  of 9 (±1) μM for dTTP binding to the RB69 pol:ddP/T complex.
of 9 (±1) μM for dTTP binding to the RB69 pol:ddP/T complex.
dTTP binding to a RB69 pol:ddP/T complex (2AP in the n position) quenches the 2AP fluorescence (Figure 2A). We interpret the fluorescence quenching with dTTP as a consequence of a conformational rearrangement during the pol reaction cycle that affects 2AP stacking (30,32–35). The best fit of the [dTTP]-dependence of the equilibrium fluorescence intensity at 365 nm to a single-site binding isotherm [Equation (1)] yields an overall dissociation equilibrium constant of 9 (±1) μM for dTTP binding (Figure 2B), a value about seven times tighter than the observed for dTTP utilization estimated by chemical quench (63 μM, Table 1)]. The from chemical quench measurements is the product of all dissociation equilibrium constants preceding chemistry. The equilibrium titrations of dTTP binding to RB69 pol:ddP/T (Figure 2B) should report the same overall affinity for dTTP binding prior to base incorporation. The apparent discrepancy will be addressed in the Discussion section.
Table 1.
Comparison of kinetic parameters for RB69 pol:P/T:dNTP complexes from stopped-flow, equilibrium fluorescence and chemical quench assays
| Stopped flow fluorescence | Chemical quenchc | |||||
|---|---|---|---|---|---|---|
| Primer/Template | 2AP Positiona | kobs,max (s−1) |
b (μM) |
(μM) |
kpol (s−1) |
(μM) |
| ddP/T | n | 849 ± 45 | 34 ± 11 | ∼2 14 ± 7d | ||
| dP/T | n | Super fast >1000 fast 220 ± 33 slow 24 ± 6 | 78 ± 32 ND | 83 ± 10e | 293 ± 20 | 63 ± 11 |
| dP/T | n+1 | 200 ± 18 | 28 ± 3 | 256 ± 7 | 16 ± 2 | |
| Equilibrium titration | ||||||
| ddP/T | n | 9 ± 1f | ||||
aSee Figure 1 for P/T sequences. When 2AP is in the n position, the incoming nucleotide is dTTP. When 2AP is in the n + 1 position, the incoming nucleotide is dCTP.
b values for dP/T were calculated by fitting to Equation (3), except that the 1/K1 value for ddP/T was derived from Equation (4).
cPre-steady-state kinetic parameters for the incorporation of dTMP versus 2AP (2AP in the n position) and dCMP versus dG (2AP in the n + 1 position). dFrom the [dTTP]-dependence of the amplitude change (Figure 3C). eFrom the [dTTP]-dependence of the start point change (Figure 4C). fCalculated from equilibrium fluorescence titrations (Figure 2). All reactions were carried out at 22°C.
With a ddP/T (2AP in the n + 1 position, Figure 1B), addition of the complementary dCTP, even at concentrations 4-fold higher than the value of determined by chemical quench, did not change the 2AP fluorescence (data not shown). Thus, dCTP binding to the RB69 pol:ddP/T complex does not alter the base-stacking interactions of 2AP when it is in the n + 1 position with the sequence we have used (33).
Kinetic analysis using a ddP/T with 2AP in the n position
Time courses after mixing RB69 pol:ddP/T complex (2AP at the n position) with a range of [dTTP] under pseudo-first order conditions ([dTTP] » [RB69 pol:ddP/T complex]) follow single exponentials (Figure 3A) with observed rate constants (Figure 3B) and amplitudes (Figure 3C) that depend hyperbolically on the [dTTP]. The nucleotide concentration dependence of the observed pseudo-first order rate constant (kobs) was fitted to a rectangular hyperbola in the form of Equation (4):
| 4 |
where K1 is the association equilibrium constant for the formation of collision complex (EDN)** (Scheme 1) that is in rapid equilibrium with free enzyme:P/T complex (ED**) and nucleotide (N). The forward and reverse isomerization rate constants for formation of (EDN)* are k+2 and k−2, respectively, as defined by the following two-step reaction scheme:
Figure 3.

Kinetics of dTTP binding to the ddP/T (2AP in the n position) in complex with RB69 pol at 22°C. (A) Time course of fluorescence changes after mixing (top to bottom): 0, 10, 20, 40, 80, 100 and 160 μM dTTP with 300 nM RB69 pol:ddP/T complex. The solid lines through the data are the best fits to single exponentials. (B) The [dTTP]-dependence of kobs. The solid line through the data is the best fit to a hyperbola, yielding the maximum rate of 849 (±45) s−1 and 1/K1 of 34 (±11) μM. (C) Amplitude of fluorescence change as a function of [dTTP]. The value obtained is the best fit to Equation (1) and is 14 (±7) μM.
Scheme 1.

in which the ** superscript denotes a higher fluorescence than *. The best fit of the [dTTP] dependence of the observed rate constants to Equation (4) yields a 1/K1 of ∼34 μM, k+2 ∼ 849 s−1 (Table 1), and k−2 ∼ 44 s−1 (Figure 3B). The overall affinity calculated from the rate and equilibrium constants  is ∼2 μM, comparable, given experimental uncertainty, to the value obtained by equilibrium titration (9 ± 1 μM; Figure 2B) and from the best fit of the [dTTP]-dependence of the fluorescence quenching amplitudes of the kinetic time courses to Equation (1) (14 ± 7 μM; Figure 3C; Table 1). The rate and equilibrium constants of dTTP binding to RB69 pol:ddP/T are weakly dependent on temperature (Figure S1, Supplementary Data).
is ∼2 μM, comparable, given experimental uncertainty, to the value obtained by equilibrium titration (9 ± 1 μM; Figure 2B) and from the best fit of the [dTTP]-dependence of the fluorescence quenching amplitudes of the kinetic time courses to Equation (1) (14 ± 7 μM; Figure 3C; Table 1). The rate and equilibrium constants of dTTP binding to RB69 pol:ddP/T are weakly dependent on temperature (Figure S1, Supplementary Data).
Kinetic analysis using dP/T with 2AP in the n position
Time courses of fluorescence change after mixing dTTP with the RB69 pol:dP/T complex (2AP in the n position) reveal a very fast quenching phase that was completed in the stopped-flow instrument dead time (∼2 ms), as indicated by the decreasing start points of the transients (Figure 4A). The magnitude of the start point changes depends on the [dTTP] with an apparent affinity of 83 μM (Figure 4C, Table 1), comparable to the nucleotide binding affinity (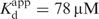 , Table 1) determined for the best fit of the observed rate constant of the next phase following the start point change (Figure 4B). The further fluorescence decay after the starting point change follows a double exponential at 22°C (Figure 4A) and 4°C (Figure S2, Supplementary Data) with both observed rate constants that depend hyperbolically on the [dTTP] (Figure 4B and Figure S2). At 22°C, the maximum rate of the fast phase that follows the change in start points is 220 s−1 and the apparent overall equilibrium constant
, Table 1) determined for the best fit of the observed rate constant of the next phase following the start point change (Figure 4B). The further fluorescence decay after the starting point change follows a double exponential at 22°C (Figure 4A) and 4°C (Figure S2, Supplementary Data) with both observed rate constants that depend hyperbolically on the [dTTP] (Figure 4B and Figure S2). At 22°C, the maximum rate of the fast phase that follows the change in start points is 220 s−1 and the apparent overall equilibrium constant  is 78 μM, comparable to the dTMP incorporation rate constant (kpol) and nucleotide affinity determined from chemical quench experiments (kpol = 293 s−1;
is 78 μM, comparable to the dTMP incorporation rate constant (kpol) and nucleotide affinity determined from chemical quench experiments (kpol = 293 s−1; 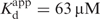 ) (Figure 5), suggesting that these two transitions are monitoring the same process or that the biochemical transition that coincides with the fast phase of fluorescence change precedes and limits rapid base incorporation. The Y-axis intercept of the observed fast rate constant after the starting point change versus [dTTP] (Figure 4B) is indistinguishable from the origin, indicating that the process is essentially irreversible.
) (Figure 5), suggesting that these two transitions are monitoring the same process or that the biochemical transition that coincides with the fast phase of fluorescence change precedes and limits rapid base incorporation. The Y-axis intercept of the observed fast rate constant after the starting point change versus [dTTP] (Figure 4B) is indistinguishable from the origin, indicating that the process is essentially irreversible.
Figure 4.

Kinetics of dTTP binding to the dP/T (2AP in the n position) in complex with RB69 pol at 22°C. (A) Time course of fluorescence changes after mixing (top to bottom) 10, 20, 40, 80 and 160 μM dTTP with 200 nM RB69 pol:dP/T complex. The thick solid lines through the data are the best fits to double exponentials. The thin lines between time 0 and 2 ms are the extrapolations to the Y-axis from each of the scans to give the start points for each [dTTP]. (B) Dependence of kobs on [dTTP]. The solid lines through the data best fit to a hyperbola for both the fast phase (shown by filled circle) and the slow phase (shown by filled triangle). The maximum rate of the fast phase is 220 (±33) s−1 with a 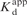 of 78 (±32) μM. The maximum rate for slow phase is 24 (±6) s−1. (C) The missing phase, as reflected by the change in start points of the fluorescence scans, was plotted as a function of [dTTP]. The data best fits to a hyperbolic equation, yielding a of 83 (±10) μM for dTTP binding, in agreement with the estimated in (B).
of 78 (±32) μM. The maximum rate for slow phase is 24 (±6) s−1. (C) The missing phase, as reflected by the change in start points of the fluorescence scans, was plotted as a function of [dTTP]. The data best fits to a hyperbolic equation, yielding a of 83 (±10) μM for dTTP binding, in agreement with the estimated in (B).
Figure 5.

Kinetics of dNMP incorporation determined by rapid chemical quench. (A) The dependence of kobs on [dTTP] using a dP/T (2AP in the n position). The solid line is the best fit to a hyperbola yielding a kpol of 293 (±20) s−1 and of 63 (±11) μM. (B) The dependence of kobs on [dCTP] using a dP/T (2AP in the n + 1 position). The solid line is the best fit to a hyperbola yielding a kpol of 256 (±7) s−1 and a of 16 (±2) μM.
These results indicate that there are at least two biochemical transitions coupled to dTTP binding and incorporation with a dP/T substrate; one is very rapid (>>1000 s−1) and completed within the 2 ms instrument dead time, and the second occurs at ∼220 s−1 when [dTTP] is saturating. This overall behavior, including the comparable values of the rate and equilibrium constants measured by fluorescence and chemical quench (Table 1) were similar at 4°C except that the fluorescence and the chemical quench were 4- to 6-fold slower. The magnitude of the rapid ‘missing’ phase (i.e. reduction in starting point) was smaller at 4°C than at 22°C (Figure S2, Supplementary Data) suggesting that we were following a process that occurred after formation of the collision complex, since formation of the collision complex should be only weakly affected by temperature. Finally, there is also a slow phase which saturates at 24 s−1 at (22°C) and must reflect a biochemical transition after base incorporation, which occurs at ∼293 s−1 (Figure 5A, Table 1). This slow phase was reduced to 10 s−1 at 4°C (Figure S2, Supplementary Data).
Chemical quench experiments using dTTP versus templating 2AP with the dP/T sequence shown in Figure 1A gave a kpol of 293 s−1 and a of 63 μM (Figure 5A), which differs from previous measurements yielding values of 165 s−1 for kpol and 367 μM for for dTTP binding and incorporation to 2AP at template n position by T4 pol (32). However, the degree of discrimination between dTTP versus dA and dTTP versus 2AP with the dP/T we have used in this study (Figure 1A, Table 1) was considerably lower than what has been reported in T4 pol (35), indicating the importance of sequence context on the kinetic parameters in this system.
Kinetic analysis using a dP/T with 2AP at the n + 1 position
With a dP/T where 2AP is in the n position we were restricted in our choice of substrates to dTTP, as it is the only nucleoside triphosphate that can form a Watson–Crick (W–C) base pair with 2AP. To determine if the multiple phases in the time courses of fluorescence change could be observed with base pairs other than dTTP:2AP, we used 2AP in the n + 1 position with dG as the templating base (Figure 1B). Time courses of fluorescence change after mixing RB69 pol:dP/T (2AP at the n + 1 position) with increasing [dCTP] were best fit to single exponentials (Figure 6A) with the kobs depending hyberbolically on the [dCTP] (Figure 6B). Curve fitting yielded for dCTP of 10–12 μM (Table 1), comparable to the value derived from chemical quench experiments (16 μM; Table 1). The maximum rate constant is ∼200 s−1 (Table 1) and is in the same range as the kpol value obtained from chemical quench experiments (∼256 s−1; Figure 5B, Table 1).
Figure 6.

Kinetics of dCTP binding to the dP/T (2AP in the n + 1 position) in complex with RB69 pol. (A) Time course of fluorescence changes after mixing (bottom to top) 0, 10, 20, 40, 80 and 160 μM dCTP with 200 nM RB69 pol:dP/T complex. The solid lines through the data best fits a single exponential. (B) The dependence of kobs on [dCTP]. The solid lines through the data (as shown by filled circle) best fits a hyperbola, yielding the maximum rate for fluorescence enhancement of 200 (±18) s−1 and of 28 (±3) μM.
Kinetic simulations
Kinetic simulations were carried out based on Scheme 2 (see Discussion section) and our experimentally determined rates and equilibrium constants for dP/T's (with 2AP in the n position). The simulations are in excellent agreement with the observed time courses of 2AP fluorescence decay (Figure 7), indicating that our Scheme 2 is consistent with the kinetic mechanism of nucleotidyl transfer catalyzed by RB69 pol.
Scheme 2.

Figure 7.

Comparison between stopped-flow data (dP/T with 2AP in the n position) and the result from kinetic simulation. As described in Experimental Procedures, simulations were carried out using the kinetic mechanism shown in Scheme 2. The simulated curve is represented by the solid line in red and is superimposed onto the data from stopped-flow fluorescence experiments shown in black. The three dTTP concentrations included for comparison are 40, 100 and 160 μM, (top to bottom) respectively.
DISCUSSION
The minimal kinetic scheme for the nucleotidyl transfer reaction catalyzed by T4 pol, a close homolog of RB69 pol, was determined first by Capson et al. (43) using rapid chemical quench methods. Subsequent work using stopped-flow fluorescence provided evidence for a conformational change prior to chemistry induced by binding of the noncomplementary dATPαS to the binary complex (45). This work was extended further with T4 pol using 2AP at different positions in the template strand by Reha-Krantz and colleagues (32–35), who showed that the pre-chemistry conformational change was partially rate limiting. In our study, only one detectable conformational change preceding phosphoryl transfer induced by correct nucleotide binding to RB69 pol was observed (discussed subsequently), which differs from the results obtained with T4 pol.
Evidence for the existence of a conformational change when these B family polymerases form a catalytically competent complex with DNA and a dNTP was based on a comparison of the crystal structures of the RB69 pol apo-enzyme (36) and the corresponding ternary complex (38), which show that fingers closing is the most prominent structural rearrangement involved in the formation of the ternary complex. The available crystal structures of binary and ternary RB69 pol complexes indicate that changes in base stacking occur and these could contribute to changes in 2AP fluorescence (38–40). It has been suggested that this conformational change provides a major checkpoint that is partly responsible for the base selectivity exhibited by many DNA polymerases (18).
Kinetic mechanism of nucleotide-dependent conformational changes in the RB69 pol complexes
Fluorescence quenching is observed both by equilibrium fluorescence titrations (Figure 2) and by stopped-flow fluorescence experiments (Figure 3), when a correct dTTP is added to an RB69 pol:ddP/T binary complex. We have assumed that the RB69 pol:ddP/T:dNTP collision complex formation does not perturb the 2AP environment, thus, no alteration in 2AP fluorescence would be expected at this point. The rapid change in fluorescence is interpreted to be a consequence of the subsequent isomerization of the open state to a closed state with lower fluorescence (Scheme 1).
The hyperbolic [dTTP]-dependence of the observed rate constant and amplitude change with an RB69 pol:ddP/T complex indicates that correct nucleotide binding is a two-step process in which a collision complex isomerizes to a partially quenched conformational state ‘preceding’ and ‘independent’ of base incorporation (Figure 3B and C and Scheme 1). The isomerization likely represents closing of the fingers, which favors 2AP stacking with the adjacent 3′ base in the primer/template duplex.
With a dP/T, the conformational change induced by correct dNTP binding was too fast to measure (>1000 s−1), as it occurs within the 2 ms dead time of the instrument (Figure 4A). Evidence for this transition comes from the [dTTP]-dependent decrease in start points. In contrast, when [dTTP] is added to the RB69 pol:ddP/T complex the start points do not change and the time course of the conformational change can be determined. The amplitude of quench observed with the ddP/T (Figure 3C) is approximately equal to the magnitude of decrease in start points (i.e. missing amplitudes) observed with the dP/T substrate (Figure 4C), suggesting these changes are monitoring the same process, albeit with different rates (i.e. the process is somewhat slower and observable with ddP/T).
Time courses of fluorescence change with a dP/T after the initial rapid quench follow double exponentials. The rate of the first phase is comparable to base incorporation measured by chemical quench. The second phase is much slower than incorporation and must reflect a step after chemistry, such as switching of the EDn+1 from the pol to the exo mode, which may alter 2AP stacking interactions. This second phase represents a process distinct from the pathway involving repeated steps in primer extension since incorporation of the next base is faster (200s−1, data not shown) than the 24 s−1 slow phase (Table 1 and Scheme 2).
The minimum kinetic scheme for the ddP/T and the dP/T before base incorporation are identical and consistent with Scheme 2, in which three fluorescence levels exist (as indicated by the number of superscript stars).
According to Scheme 2, the for nucleotide binding (i.e. all states preceding chemistry) and the apparent values measured by stopped-flow fluorescence and by chemical quench, represent the product of all equilibria preceding chemistry (1/K1K2), and is supported by the agreement among: (i) the of the decrease in start points which represent the missing phase (83 μM, Figure 4C); (ii) the from the chemical quench (63 μM, Figure 5A) and (iii) the value from the plot of kobs versus [dTTP] (78 μM, Figure 4B and Table 1) when dTMP incorporation by RB69 pol is assayed by stopped-flow fluorescence using a dP/T (2AP in the n position). Scheme 2 is consistent with either the chemical step or another conformation transition, not detectable by changes in 2AP fluorescence that precedes and limits the rate of base incorporation (data not shown in Scheme 2), as being the rate-determining step in the nucleotidyl transfer reaction catalyzed by RB69 pol. The inclusion of [EDn+ 1·PPi]* in Scheme 2 is based on the assumption that the fluorescent state of the complex just prior to chemistry is identical to that of the complex immediately after the formation of the phosphodiester bond.
It should be noted that the maximum forward rate constant (k+2) for isomerization of the collision complex, determined with a ddP/T pseudo-substrate is slower than the rate of the very rapid fluorescence quench seen with the dP/T (>1000 s−1), indicating that the ddP/T does not precisely mimic the dP/T substrate. This could be due to the absence of a 3′ terminal hydroxyl group on the ddP/T which serves as one of the ligands for the metal ion in the A site (46).
The slow rate of fluorescence quenching k+5 (24 s−1) subsequent to chemistry leads to a state with slightly lower fluorescence that we have not defined, but have represented as [EDn+ 1·PPi] in Scheme 2. This step competes with the pathway that leads to the (EDn+1) complex that can be extended by the next correct incoming dNTP with a rate far exceeding 24 s−1.
When 2AP is in the n + 1 position, fluorescence enhancement occurs after base incorporation
In contrast to the 6-fold fluorescence enhancement observed when RB69 pol forms a binary complex with a P/T (2AP in the n position), there is only a small enhancement observed for the binary complex when 2AP is in the n + 1 position, indicating that binding of the enzyme has a minor effect on 2AP stacking in this case. Addition of saturating [dCTP] to a RB69 pol:ddP/T binary complex (2AP in the n + 1 position), where G is the templating base, did not produce any change in 2AP fluorescence (data not shown). Since binding of a correct dNTP to the templating base induces a conformational change in the polymerase ternary complex, it was surprising that dCTP binding did not affect 2AP stacking (when it is in the n + 1 position) sufficiently to give a change in the fluorescence signal. The same observation with T4 pol was reported by another group (33). Only with the corresponding dP/T, and after incorporation of the correct dNMP, did fluorescence enhancement occur. The maximum rate (kmax) for fluorescence enhancement was comparable to kpol for dCMP incorporation (Table 1), suggesting that the enhancement occurs concurrently, or subsequent to, nucleotidyl transfer when the 2AP translocates from the n + 1 to the n position where it would be unstacked. We conclude that when 2AP is in the n + 1 position of a dP/T with the sequence flanking the 2AP, shown in Figure 1B, it is not sensitive to conformational changes associated with the closed ternary complex formation and cannot distinguish conformational changes from base incorporation. We found that the behavior of the RB69 pol:dP/T complex (2AP in the n + 1 position) differs from that of T4 DNA polymerase (35), a close relative of RB69 pol. Hariharan et al. (35) observed a rapid 2AP fluorescence enhancement rate (314 s−1) that preceded a slow rate (164 s−1) of dAMP incorporation opposite a templating T. In contrast, we found that, with RB69 pol, the rates of enhancement and dCMP incorporation were nearly equivalent. In addition, the mechanism proposed by these authors, in which the conformational change associated with base unstacking (2AP in the n + 1) position occurs before base incorporation, predicts a fluorescence enhancement with both T4 and RB69 pols when a non-extendable primer/template with 2AP in the n + 1 position is employed. This was not observed with a non-extendable primer either with T4 pol (33) or RB69 pol.
Comparison of the stopped-flow fluorescence behavior of RB69 pol complexes with other DNA pol complexes
A very rapid initial phase of fluorescence quenching with the dP/T substrate (2AP in the n position) that is nearly complete within the instrument dead time has also been observed with the Klenow fragment (KF) (30). However significant differences in the time courses for fluorescence quenching exist between KF and RB69 pol. With KF, the change in start points observed with a ddP/T, upon addition of increasing [dTTP] [Figure 3B in (30)], was not seen with the RB69 pol:ddPT complex (Figure 3A). Another difference between KF and RB69 pol is the behavior of the enzyme:ddP/T complex (2AP in the n + 1 position) where, upon addition of the correct dNTP, fluorescence enhancement was observed with KF [Figure 4B in (30)] but not with RB69 pol. Also the behavior of the KF:dP/T complex (2AP in the n + 1 position) [Figure 4A in (30)] differed from the behavior of the corresponding RB69 pol:dP/T complex (Figure 6A). In the case of KF there was fluorescence enhancement followed by quenching upon incorporation of the correct dNMP. For RB69 pol, only fluorescence enhancement was observed (Figure 6A). The explanation for these distinctions rests on the structural differences among the various biochemical intermediates with respect to the stacking environment of 2AP during the nucleotide addition cycle.
In experiments with pol β, using a ddP/T with 2AP in either the n or n + 1 positions, a fast phase for fluorescence change was observed (26). It was suggested that this rapid phase corresponds to finger closing and that chemistry, rather than a conformational change, is rate limiting for the pol reaction (19). Recent FRET experiments with Klentaq1 also support the idea that fingers closing are faster than the kinetically determined rate-limiting step. The authors propose that the rate-limiting step, which presumably involves rearrangement of residues in the nucleotide binding pocket, occurs after the fingers close (24).
Changes in 2AP fluorescence correlate with crystal structures of RB69 pol complexes
In an attempt to understand the structural basis for the change in 2AP fluorescence, we have relied first on the commonly accepted idea that stacking of 2AP with adjacent bases or aromatic residues in the protein results in quenching of 2AP fluorescence and that enhancement of 2AP fluorescence occurs when the stacking constraints are relaxed (25). We then examined the published crystal structures of RB69 pol complexed with different P/Ts and complementary incoming dNTPs to estimate the extent that the templating base and its 5′ neighbor stacked against adjacent bases. The ternary RB69 pol complex (38) with a ddP/T and a complementary dNTP has the templating base (n position) stacked against its 3′ neighbor in the P/T duplex even though the nucleotidyl transfer reaction has not yet occurred (Figure 8). The base in the n + 1 position is 11.5 Å away from the templating base and is displaced by ∼180° from the P/T duplex (38). It appears to be partially stacked against the adjacent base 5′ to it in the single-stranded template overhang (Figure 8). The position of the templating base before binding of the incoming dNTP, in terms of stacking, can only be inferred from the fluorescence behavior of 2AP and by analogy with other DNA pol:P/T binary complexes (47), as there is no crystal structure of binary RB69 pol:P/T complex in the pol mode, except one that has a furan moiety in the n position instead of a templating base (40). When a P/T (2AP in the n position) forms a binary complex with RB69 pol, 2AP fluorescence is also enhanced as observed with T4 pol (33). The observed bending of the template strand at the P/T junction, as seen in the crystal structure of the binary RB69 pol complex with an abasic site in place of the templating base, suggests that 2AP in the n position is less stacked in the binary complex than in the P/T alone. Our studies suggest that binding of the correct dNTP causes only the templating base (n position) to stack against its 3′ neighbor, however, unstacking of the base in the n + 1 position from its 5′ neighbor occurs either concurrently or subsequent to nucleotidyl transfer.
Figure 8.

The primer/template junction in the active site of RB69 pol ternary complex [PDB entry 1IG9, (38)]. The primer strand and the template strand are shown in light green; the templating base (in the n position) is shown in magenta, the base 5′ to it (in the n + 1 position) is shown in blue. The α-helix present at the active site is shown in gray. The triphosphate tail of the incoming dTTP is shown in orange. This figure was made using PyMOL (DeLano Scientific).
Several of our experimental observations suggest that the change in 2AP fluorescence is monitoring a conformational change, most likely fingers domain closing, because the amplitudes of the decrease in start point with increasing [dTTP] using a dP/T is comparable to the amplitudes of quenching observed with a ddP/T (Figures 3A and 4A). In addition, the amplitude changes seen from fluorescence equilibrium titrations indicate that a structural rearrangement affecting 2AP stacking occurs after addition of a correct dNTP (dTTP) to the highly fluorescent binary complex. This results from isomerization of an open collision complex to a closed ternary complex with lower fluorescence in the absence of chemistry.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
We thank Dr Cathy Joyce and Dr Harold Lee for helpful discussions and critical reading of the manuscript. We thank Liz Vellali for skillful preparation of the manuscript. This work was supported by United States Public Health Service Grant GM63276 to W.K. and grants from the National Science Foundation (MCB-0546353), American Heart Association (0655849T) and NIH (GM071688) to E.M.D.L.C. W.C. is supported by an American Heart Association postdoctoral fellowship award (0625997T). Funding to pay the Open Access publication charges for this article was provided by United States Public Health Service Grant GM63276 to W. Konigsberg.
Conflict of interest statement. None declared.
REFERENCES
- 1.Loeb LA, Kunkel TA. Fidelity of DNA synthesis. Annu. Rev. Biochem. 1982;51:429–457. doi: 10.1146/annurev.bi.51.070182.002241. [DOI] [PubMed] [Google Scholar]
- 2.Kunkel TA. DNA replication fidelity. J. Biol. Chem. 1992;267:18251–18254. [PubMed] [Google Scholar]
- 3.Kunkel TA, Bebenek K. DNA replication fidelity. Annu. Rev. Biochem. 2000;69:497–529. doi: 10.1146/annurev.biochem.69.1.497. [DOI] [PubMed] [Google Scholar]
- 4.Baker TA, Bell SP. Polymerases and the replisome: machines within machines. Cell. 1998;92:295–305. doi: 10.1016/s0092-8674(00)80923-x. [DOI] [PubMed] [Google Scholar]
- 5.Kornberg A, Baker T. DNA Replication. New York: Freeman & Company Publishing; 1992. [Google Scholar]
- 6.Benkovic SJ, Valentine AM, Salinas F. Replisome-mediated DNA replication. Annu. Rev. Biochem. 2001;70:181–208. doi: 10.1146/annurev.biochem.70.1.181. [DOI] [PubMed] [Google Scholar]
- 7.Nossal NG. Molecular Biology of Bacterial T4. Washington D.C: ASM; 1994. [Google Scholar]
- 8.Nossal NG, Alberts B. Molecular Biology of Bacterial T4. Washington D.C: ASM; 1983. [Google Scholar]
- 9.Schrock RD, Alberts B. Processivity of the gene 41 DNA helicase at the bacteriophage T4 DNA replication fork. J. Biol. Chem. 1996;271:16678–16682. doi: 10.1074/jbc.271.28.16678. [DOI] [PubMed] [Google Scholar]
- 10.Shamoo Y, Williams K, Konigsberg W. Molecular Biology of Bacterial T4. Washington D.C: ASM; 1994. [Google Scholar]
- 11.Williams K, Shammo Y, Spicer EK, Coleman JE, Konigsberg W. Molecular Biology of Bacterial T4. Washington D.C: ASM; 1994. [Google Scholar]
- 12.Hingorani MM, O’Donnell M. Sliding clamps: a (tail)ored fit. Curr. Biol. 2000;10:R25–R29. doi: 10.1016/s0960-9822(99)00252-3. [DOI] [PubMed] [Google Scholar]
- 13.Goodrich LD, Lin TC, Spicer EK, Jones C, Konigsberg WH. Residues at the carboxy terminus of T4 DNA polymerase are important determinants for interaction with the polymerase accessory proteins. Biochemistry. 1997;36:10474–10481. doi: 10.1021/bi9708949. [DOI] [PubMed] [Google Scholar]
- 14.Berdis AJ, Soumillion P, Benkovic SJ. The carboxyl terminus of the bacteriophage T4 DNA polymerase is required for holoenzyme complex formation. Proc. Natl Acad. Sci. USA. 1996;93:12822–12827. doi: 10.1073/pnas.93.23.12822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hacker KJ, Alberts BM. The rapid dissociation of the T4 DNA polymerase holoenzyme when stopped by a DNA hairpin helix. A model for polymerase release following the termination of each Okazaki fragment. J. Biol. Chem. 1994;269:24221–24228. [PubMed] [Google Scholar]
- 16.Soumillion P, Sexton DJ, Benkovic SJ. Clamp subunit dissociation dictates bacteriophage T4 DNA polymerase holoenzyme disassembly. Biochemistry. 1998;37:1819–1827. doi: 10.1021/bi972526a. [DOI] [PubMed] [Google Scholar]
- 17.Carver TE, Jr, Sexton DJ, Benkovic SJ. Dissociation of bacteriophage T4 DNA polymerase and its processivity clamp after completion of Okazaki fragment synthesis. Biochemistry. 1997;36:14409–14417. doi: 10.1021/bi971423p. [DOI] [PubMed] [Google Scholar]
- 18.Johnson KA. Conformational coupling in DNA polymerase fidelity. Annu. Rev. Biochem. 1993;62:685–713. doi: 10.1146/annurev.bi.62.070193.003345. [DOI] [PubMed] [Google Scholar]
- 19.Showalter AK, Tsai MD. A reexamination of the nucleotide incorporation fidelity of DNA polymerases. Biochemistry. 2002;41:10571–10576. doi: 10.1021/bi026021i. [DOI] [PubMed] [Google Scholar]
- 20.Joyce CM, Benkovic SJ. DNA polymerase fidelity: kinetics, structure, and checkpoints. Biochemistry. 2004;43:14317–14324. doi: 10.1021/bi048422z. [DOI] [PubMed] [Google Scholar]
- 21.Joyce CM, Steitz TA. Function and structure relationships in DNA polymerases. Annu. Rev. Biochem. 1994;63:777–822. doi: 10.1146/annurev.bi.63.070194.004021. [DOI] [PubMed] [Google Scholar]
- 22.Brautigam CA, Steitz TA. Structural and functional insights provided by crystal structures of DNA polymerases and their substrate complexes. Curr. Opin. Struct. Biol. 1998;8:54–63. doi: 10.1016/s0959-440x(98)80010-9. [DOI] [PubMed] [Google Scholar]
- 23.Steitz TA. DNA polymerases: structural diversity and common mechanisms. J. Biol. Chem. 1999;274:17395–17398. doi: 10.1074/jbc.274.25.17395. [DOI] [PubMed] [Google Scholar]
- 24.Rothwell PJ, Mitaksov V, Waksman G. Motions of the fingers subdomain of klentaq1 are fast and not rate limiting: implications for the molecular basis of fidelity in DNA polymerases. Mol. Cell. 2005;19:345–355. doi: 10.1016/j.molcel.2005.06.032. [DOI] [PubMed] [Google Scholar]
- 25.Jean JM, Hall KB. 2-Aminopurine fluorescence quenching and lifetimes: role of base stacking. Proc. Natl Acad. Sci. USA. 2001;98:37–41. doi: 10.1073/pnas.011442198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dunlap CA, Tsai MD. Use of 2-aminopurine and tryptophan fluorescence as probes in kinetic analyses of DNA polymerase beta. Biochemistry. 2002;41:11226–11235. doi: 10.1021/bi025837g. [DOI] [PubMed] [Google Scholar]
- 27.Guest CR, Hochstrasser RA, Sowers LC, Millar DP. Dynamics of mismatched base pairs in DNA. Biochemistry. 1991;30:3271–3279. doi: 10.1021/bi00227a015. [DOI] [PubMed] [Google Scholar]
- 28.Nordlund TM, Andersson S, Nilsson L, Rigler R, Graslund A, McLaughlin LW. Structure and dynamics of a fluorescent DNA oligomer containing the EcoRI recognition sequence: fluorescence, molecular dynamics, and NMR studies. Biochemistry. 1989;28:9095–9103. doi: 10.1021/bi00449a021. [DOI] [PubMed] [Google Scholar]
- 29.Rachofsky EL, Osman R, Ross JB. Probing structure and dynamics of DNA with 2-aminopurine: effects of local environment on fluorescence. Biochemistry. 2001;40:946–956. doi: 10.1021/bi001664o. [DOI] [PubMed] [Google Scholar]
- 30.Purohit V, Grindley ND, Joyce CM. Use of 2-aminopurine fluorescence to examine conformational changes during nucleotide incorporation by DNA polymerase I (Klenow fragment) Biochemistry. 2003;42:10200–10211. doi: 10.1021/bi0341206. [DOI] [PubMed] [Google Scholar]
- 31.Zhong X, Patel SS, Werneburg BG, Tsai MD. DNA polymerase beta: multiple conformational changes in the mechanism of catalysis. Biochemistry. 1997;36:11891–11900. doi: 10.1021/bi963181j. [DOI] [PubMed] [Google Scholar]
- 32.Fidalgo da Silva E, Mandal SS, Reha-Krantz LJ. Using 2-aminopurine fluorescence to measure incorporation of incorrect nucleotides by wild type and mutant bacteriophage T4 DNA polymerases. J. Biol. Chem. 2002;277:40640–40649. doi: 10.1074/jbc.M203315200. [DOI] [PubMed] [Google Scholar]
- 33.Mandal SS, Fidalgo da Silva E, Reha-Krantz LJ. Using 2-aminopurine fluorescence to detect base unstacking in the template strand during nucleotide incorporation by the bacteriophage T4 DNA polymerase. Biochemistry. 2002;41:4399–4406. doi: 10.1021/bi015723p. [DOI] [PubMed] [Google Scholar]
- 34.Hariharan C, Reha-Krantz LJ. Using 2-aminopurine fluorescence to detect bacteriophage T4 DNA polymerase-DNA complexes that are important for primer extension and proofreading reactions. Biochemistry. 2005;44:15674–15684. doi: 10.1021/bi051462y. [DOI] [PubMed] [Google Scholar]
- 35.Hariharan C, Bloom LB, Helquist SA, Kool ET, Reha-Krantz LJ. Dynamics of nucleotide incorporation: snapshots revealed by 2-aminopurine fluorescence studies. Biochemistry. 2006;45:2836–2844. doi: 10.1021/bi051644s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang J, Sattar AK, Wang CC, Karam JD, Konigsberg WH, Steitz TA. Crystal structure of a pol alpha family replication DNA polymerase from bacteriophage RB69. Cell. 1997;89:1087–1099. doi: 10.1016/s0092-8674(00)80296-2. [DOI] [PubMed] [Google Scholar]
- 37.Shamoo Y, Steitz TA. Building a replisome from interacting pieces: sliding clamp complexed to a peptide from DNA polymerase and a polymerase editing complex. Cell. 1999;99:155–166. doi: 10.1016/s0092-8674(00)81647-5. [DOI] [PubMed] [Google Scholar]
- 38.Franklin MC, Wang J, Steitz TA. Structure of the replicating complex of a pol alpha family DNA polymerase. Cell. 2001;105:657–667. doi: 10.1016/s0092-8674(01)00367-1. [DOI] [PubMed] [Google Scholar]
- 39.Freisinger E, Grollman AP, Miller H, Kisker C. Lesion (in)tolerance reveals insights into DNA replication fidelity. EMBO J. 2004;23:1494–1505. doi: 10.1038/sj.emboj.7600158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hogg M, Wallace SS, Doublie S. Crystallographic snapshots of a replicative DNA polymerase encountering an abasic site. EMBO J. 2004;23:1483–1493. doi: 10.1038/sj.emboj.7600150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang G, Lin T, Karam J, Konigsberg WH. Steady-state kinetic characterization of RB69 DNA polymerase mutants that affect dNTP incorporation. Biochemistry. 1999;38:8094–8101. doi: 10.1021/bi990653w. [DOI] [PubMed] [Google Scholar]
- 42.Yang G, Franklin M, Li J, Lin TC, Konigsberg W. A conserved Tyr residue is required for sugar selectivity in a Pol alpha DNA polymerase. Biochemistry. 2002;41:10256–10261. doi: 10.1021/bi0202171. [DOI] [PubMed] [Google Scholar]
- 43.Capson TL, Peliska JA, Kaboord BF, Frey MW, Lively C, Dahlberg M, Benkovic SJ. Kinetic characterization of the polymerase and exonuclease activities of the gene 43 protein of bacteriophage T4. Biochemistry. 1992;31:10984–10994. doi: 10.1021/bi00160a007. [DOI] [PubMed] [Google Scholar]
- 44.Barshop BA, Wrenn RF, Frieden C. Analysis of numerical methods for computer simulation of kinetic processes: development of KINSIM – a flexible, portable system. Anal. Biochem. 1983;130:134–145. doi: 10.1016/0003-2697(83)90660-7. [DOI] [PubMed] [Google Scholar]
- 45.Frey MW, Sowers LC, Millar DP, Benkovic SJ. The nucleotide analog 2-aminopurine as a spectroscopic probe of nucleotide incorporation by the Klenow fragment of Escherichia coli polymerase I and bacteriophage T4 DNA polymerase. Biochemistry. 1995;34:9185–9192. doi: 10.1021/bi00028a031. [DOI] [PubMed] [Google Scholar]
- 46.Rittenhouse RC, Apostoluk WK, Miller JH, Straatsma TP. Characterization of the active site of DNA polymerase beta by molecular dynamics and quantum chemical calculation. Proteins. 2003;53:667–682. doi: 10.1002/prot.10451. [DOI] [PubMed] [Google Scholar]
- 47.Li Y, Korolev S, Waksman G. Crystal structures of open and closed forms of binary and ternary complexes of the large fragment of Thermus aquaticus DNA polymerase I: structural basis for nucleotide incorporation. EMBO J. 1998;17:7514–7525. doi: 10.1093/emboj/17.24.7514. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.