

Abstract
Human arylamine N-acetyltransferases NAT1 and NAT2 are highly polymorphic genes that modify individual susceptibility to cancers caused by exposure to arylamine pro-carcinogens. Strong similarities exist between rat Nats and human NATs, and rat Nat2 polymorphisms result in slow acetylator phenotype. Recently, a third rat Nat, rNat3*1, was reported. Although in vivo toxicological and carcinogenic studies are often conducted in rats, relatively little is known about Nat sequences among available inbred rat strains. We report here that rNat1 and rNat2 open reading frames (ORFs) in twelve inbred rat strains (ACI, BN, BUF, CDF, COP, DA, LEW, LOU/M, MW, PVG, SHR, WF) corresponded to reference rNat1*13 and rNat2*20. While ten of the twelve strains had reference rNat3*1 ORFs, strains ACI and COP had a variant rNat3*2 ORF characterized by a G619>T transversion (A207S). The rNat3*2 SNP reduced Nat3 protein levels and N- and O-acetyltransferase activity when recombinantly expressed in bacteria. Recombinant expression of rNat3 1 and rNat3 2 in COS-1 cells yielded equivalent protein levels but undetectable catalytic activities. Relative tissue expression of rNat1, rNat2, and rNat3 mRNAs were assessed in liver and twelve extrahepatic tissues (lung, spleen, kidney, heart, esophagus, stomach, urinary bladder, prostate, colon, duodenum, jejunum, ileum) from male F344 rats exsanguinated prior to sacrifice. Semi-quantitative RT-PCR experiments demonstrate that the relative expression of the rNat transcripts in liver and twelve extrahepatic tissues was rNat1 > rNat2, while rNat3 transcripts were not detected. This study concludes that rNat1 and rNat2 are primarily responsible for acetylation phenotype in rats.
Keywords: Rat, N-acetyltransferase, tissue-specific, expression, rNat3
INTRODUCTION
Human arylamine N-acetyltransferase 1 (NAT1) and 2 (NAT2) (EC 2.3.1.5) exhibit genetic polymorphisms that modify pharmaceutical drug toxicities and influence individual susceptibility to cancers caused by exposure to environmental arylamine pro-carcinogens (Hein et al., 2000). NAT enzymes catalyze acetyl group transfer from acetyl-CoA to an aromatic amine’s exocyclic amine (N-acetylation) or the oxygen of its oxidized exocyclic amine (O-acetylation) (Hein, 1988). Arylamine N-acetylation often results in xenobiotic elimination and subsequent excretion. If, however, the exocyclic nitrogen of an arylamine pro-carcinogen is first oxidized by cytochrome P450, NATs may further activate the carcinogen through O-acetylation, resulting in reactive arylnitrenium intermediates that can form mutagenic DNA adducts (Hanna, 1996).
Mice, hamsters, rabbits, and rats have been used to model the human NAT polymorphism (Boukouvala and Fakis, 2005). The recent increase in rat genetic and genomic studies due to the sequencing of the rat genome promises to enhance the usefulness of the rat model for in vivo studies (Jacob, 1999; Lazar et al., 2005; Rat Genome Sequence Project Consortium, 2004). The rat will play a key role in advancing our understanding of tissue-specific NAT gene and protein expression, in vivo enzyme activity, functional gene interactions, and the influence of environmental exposures on NAT activity and expression in vivo.
The rat genome contains three loci encoding Nat isozymes that are unique in substrate selectivity, structural stability, and kinetic activity (Walraven et al., 2006). Previous studies have reported similarities between rat Nat1 (rNat1) and human NAT2, and between rat Nat2 (rNat2) and human NAT1, with respect to nucleotide and amino acid sequence, structural stability, and substrate selectivity (Walraven et al., 2006). The rat genome also includes a third active N-acetyltransferase locus, rat Nat3 (rNat3), for which no known human homolog exists (Walraven et al., 2006). When recombinantly expressed in E. coli, rNat3 produces a functional enzyme with relatively weak activity toward few known substrates, and demonstrates intermediate structural stability relative to rNat1 and rNat2 (Walraven et al., 2006). Although rNat3 has low activity in vitro, the in vivo expression of rNat3 relative to rNat1 and rNat2 must be considered before the relative contribution of rNat3 to the rat N-acetylation phenotype can be determined. Recent mouse Nat knockout studies demonstrated that Nat3 N-acetylation activity was not detected in mice lacking functional Nat1 and Nat2, suggesting that Nat3 does not significantly contribute to N-acetylation in mice under normal physiological conditions (Sugamori et al., 2003). Studies of rat Nat expression are needed to provide important insights into the relative contribution of rNat1, rNat2, and rNat3 to the N-acetylation phenotype in rats.
The only rat Nat polymorphisms reported to date are in the rNat2 slow alleles from inbred rat strains WKY (rNat2*21A) and NSD (rNat2*21B), which share the single nucleotide polymorphism (SNP) transitions G361>A (V121I), G399>A (silent), G796>A (V266I), and differ from each other only by silent transitions G522>A in *21A and G672>A in *21B (Doll and Hein, 1995). These variant alleles code for slow acetylator Nat2 proteins and make the rat a useful in vivo model for the rapid and slow acetylator phenotype. Variants for the rNat1 and rNat3 genes have not been previously reported for the available inbred rat strains. Defining the Nat genomic sequences of available inbred strains is important for designing toxicological studies using inbred rat models.
Here we report the relative expression of rNat1, rNat2, and rNat3 genes in thirteen different rat tissues, their complete sequencing in twelve different inbred rat strains, and the discovery and recombinant characterization of a novel polymorphic rNat3*2 allele in bacterial and mammalian cells.
METHODS
Rat Genomic DNA Isolation & Sequencing
Rat whole blood samples were obtained from Harlan Biosciences (Indianapolis, IN) for inbred rat strains ACI, DA, LOU/M, MW, PVG, SHR, and WF. Rat ear clippings were obtained from Charles River Laboratories, Inc. (Wilmington, MA) for inbred rat strains BN, LEW, COP, BUF, and CDF.
White blood cell nuclei were isolated from whole blood samples with three cycles of suspension in 0.32 M sucrose, 10 mM Tris pH 7.5, 5 mM MgCl2, 1% Triton X-100 followed by centrifugation. Nuclei were resuspended in phosphate-buffered saline (138 mM NaCl, 2.7 mM KCl, 10 mM phosphate buffer, pH 7.4) and genomic DNA was isolated using the QIAamp DNA Mini Kit (Qiagen, Valencia, CA).
Rat ear clippings were homogenized in buffer (10 mM Tris-HCl, 10 mM KCl, 2 mM EDTA, 4 mM MgCl2, pH 7.6) using a rotor-stator homogenizer, followed by centrifugation, resuspension in buffer, and cell disruption by adding 1/10th volume 10% SDS. After incubation at 37°C, protein was precipitated by adding 6 M ammonium acetate, followed by centrifugation. Isopropyl alcohol was added to the supernatant to precipitate DNA. The DNA pellet was washed twice with 70% ethanol prior to resuspension in water overnight.
The rNat1, rNat2, and rNat3 open reading frames (ORF) were amplified by polymerase chain reaction (PCR) from rat genomic DNA with high-fidelity Phusion DNA Polymerase (New England Biolabs, Ipswich, MA) using gene-specific primers (Figure 1A). To reduce the chance of detecting PCR artifacts as variants, independent duplicate reactions were performed for each gDNA/primer set and combined prior to sequencing. PCR products were purified using the QIAquick PCR Purification Kit (Qiagen) and sequenced with gene specific rNat1, rNat2, or rNat3 primers (Table 1) as previously described (Walraven et al., 2006). Results were compared to published ORF sequences from Sprague Dawley rNat1 (U17260), rNat2 (U17261), and rNat3 (AY253757) using SeqMan II v.5.03 (DNAStar Inc., Madison, WI). The rNat3 polymorphism in strains ACI and COP was verified using independent sources of ACI and COP genomic DNA.
Figure 1.
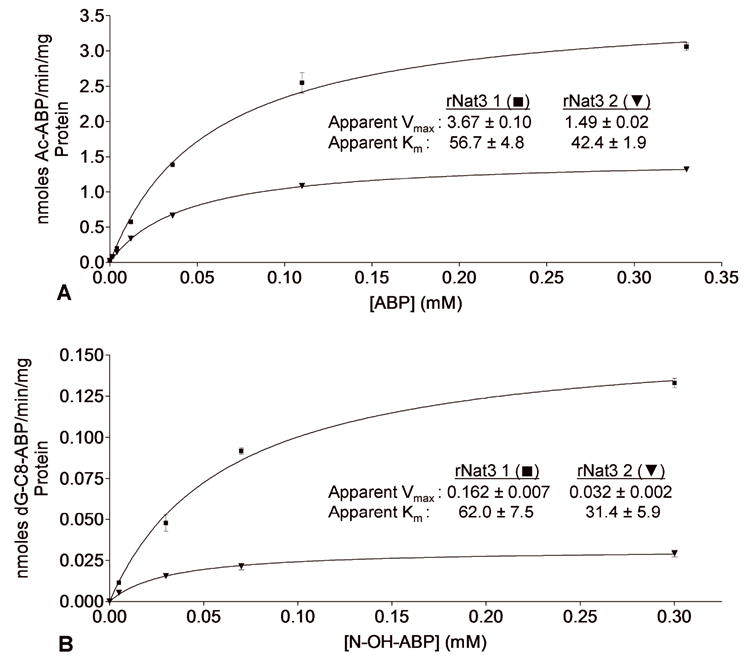
A N-acetylation of 4-aminobiphenyl (ABP) catalyzed by rNat3 1 and rNat3 2. Activity is reported as nanomoles of acetylated ABP per minute reaction time per milligram of total protein. Apparent Km (μM) and Vmax (nmoles/min/mg) for ABP N-acetylation are displayed on the graph. Error bars indicate standard error (n = 3). B Metabolic activation (O-acetylation) of N-hydroxy-ABP (N-OH-ABP) catalyzed by rNat3 1 and rNat3 2. Activity is reported as nanomoles of dG-C8-ABP formed per minute reaction time per milligram of total protein. Apparent Km (μM) and Vmax (nmoles/min/mg) for N-OH-ABP O-acetylation are displayed on the graph. Error bars indicate standard error (n = 3).
Table 1.
rNat1, rNat2, and rNat3 PCR and Sequencing (“Seq”) Primers
| Primer | Sequence | Features |
|---|---|---|
| rNat1-pf1 | 5′-CTGGAATTCATGGACATCGAAGCATACTTCGAAAGGAT-3′a,b | EcoRI, PCR/Seq, forward |
| rNat1-pr1 | 5′-AGAGCATGCATGGAGAACCAATTTACCCTAAATAGT-3′ a,b | SphI, PCR/Seq, reverse |
| rNat1-pf2 | 5′-ATGCGAGCTAGCccaccATGGACATCGAAGCATACTTCGAA-3′ b,d | NheI, PCR, Kozak, forward |
| rNat1-pr2 | 5′-ATGCGACTCGAGCTAAATAGTAAAAACGAGTTCGCCATGT-3′ b | XhoI, PCR, reverse |
| rNat1-f1 | 5′-TCTGCTGTACTGGGCTCTGA-3′ | Seq, forward |
| rNat1&2-f2 | 5′-AAGAGAATGGAACCTGGTAC-3′ | Seq, forward |
| rNat1-f3 | 5′-GCCTCTGTGTTTGTAAGCAC-3′ | Seq, forward |
| rNat1&2-r1 | 5′-TCTTCTGTCAAACGAAAGAT-3′ | Seq, reverse |
| rNat1-r2 | 5′-TGGACCATTTCACTGCTATA-3′ | Seq, reverse |
| rNat2-pf1 | 5′-TCTGAATTCATGGACATTGAAGCATACTTTGAAAGAATTGGTTAT-3′ a,b | EcoRI, PCR/Seq, forward |
| rNat2-pr1 | 5′-CATGCATGCACACCAAAACTGCATATTCTAAATGGT-3′ a,b | SphI, PCR/Seq, reverse |
| rNat2-pf2 | 5′-ATGCGAGCTAGCccaccATGGACATTGAAGCATACTTTGAAAGA-3′ b,d | NheI, PCR, Kozak, forward |
| rNat2-pr2 | 5′-ATGCGACTCGAGCTAAATGGTTAAAAAATCGATCACCATGT-3′ b | XhoI, PCR, reverse |
| rNat2-f1 | 5′-TAGAAGTCATCTTTGATCAA-3′ | Seq, forward |
| rNat2-f3 | 5′-CAGCATCCTTGTTTACAAGT-3′ | Seq, forward |
| rNat2-r2 | 5′-ATGGACTCAAAATCCTCAAT-3′ | Seq, reverse |
| rNat2-r3 | 5′-TGAATCATGCCACTGCTGTA-3′ | Seq, reverse |
| rNat3-pf1 | 5′-GTAGCACCCGGGATGGACATTGAAGCGTACTT-3′b,c | XmaI, PCR/Seq forward |
| rNat3-pr1 | 5′-ATGCGACTGCAGCTAAATAGTAAAAAAGCCAAT-3′b,c | PstI, PCR/Seq reverse |
| rNat3-pf2 | 5′-ATGCGAGCTAGCccaccATGGACATTGAAGCGTACTTTGAAAG-3′ b,d | NheI, PCR, Kozak, forward |
| rNat3-pr2 | 5′-ATGCGACTCGAGCTAAATAGTAAAAAGCCAATTACCACATTTG-3′b | XhoI, PCR, reverse |
| rNat3-f1 | 5′-TCTTCTTTACTGGGCTCTGA-3′c | Seq, forward |
| rNat3-f2 | 5′-TGCCATCTTCCACTTGACAGAA-3′c | Seq, forward |
| rNat3-f3 | 5′-TGCATACTACCAGAAATCTCCAA-3′c | Seq, forward |
| rNat3-r1 | 5′-CAGTAAAGAAGATGATTGACTTGAA-3′c | Seq, reverse |
| rNat3-r2 | 5′-GTCAATTCCAGAGGCTCCCACAT-3′c | Seq, reverse |
| rNat3-r3 | 5′-GATAAGAATGGTGCCTACTAAA-3′c | Seq, reverse |
| rNat1-f-int | 5′-CGAGCAGTTCCTTTTGAGAATCTTAG-3′ | Internal forward |
| RNat1-r-int | 5′-CAAGGAACAGAACGATGTGCTTAC-3′ | Internal reverse |
| Rnat2-f-int | 5′-CCATGGAGCTGAATTTAGAAGTCAT-3′ | Internal forward |
| Rnat2-r-int | 5′-ATGCGACTCGAGCTAAATGGTAAAAAATCGATCACCATGT-3′ | Internal reverse |
| Rnat3-f-int | 5′-CTCCAACCTGCAAATATAGCAACA-3′ | Internal forward |
| Rnat3-r-int | 5′-GTCTTTTAGTTTGTCCCAGATACCAAA-3′ | Internal reverse |
Restriction site underlined.
Kozak sequence in lower case
Cloning, Sequencing, and Expression
Cloning into pKK223-3, sequencing, bacterial transformation, and expression were carried out as previously described (Chung et al., 1989; Doll and Hein, 1995; Walraven et al., 2006). Briefly, the reference rNat3*1 and variant rNat3*2 ORFs were directionally cloned into bacterial expression vector pKK223-3 (Pharmacia-LKB Biotechnology, Piscataway, NJ). Plasmid insert sequences were verified by DNA sequencing of both DNA strands prior to transformation and expression in JM105 E. coli.
rNat1*13, rNat2*20, rNat3*1 and rNat3*2 ORFs were cloned into vector pcDNA5/FRT (Invitrogen, Carlsbad, CA) and expressed in COS-1 cells (American Type Culture Collection, Rockville, MD) in parallel with a negative control plasmid (no insert). rNat ORFs were amplified by independent duplicate PCRs using rNat-specific primers (Table 1) and directionally cloned into pcDNA5/FRT using restriction enzymes NheI (forward primers) and XhoI (reverse primers). A Kozak translation initiation sequence was included in the forward primers. Sequence was confirmed by sequencing of both DNA strands using rNat-specific primers and plasmid-specific T7 and BGH primers (Table 1). COS-1 cells were cultured, transfected, and cell lysates generated and stored as previously described (Zang et al., 2004).
Total protein concentration for all lysates was measured by the Bradford method (Bradford, 1976). As controls for pKK223-3 and pcDNA5/FRT cloning, independent duplicates of each clone were generated in parallel and carried through initial sequencing, transformation/transfection, and catalytic activity assays to verify representative cloning.
Enzyme Activity Assays
Bacterial lysates containing recombinantly expressed rNat3 1, rNat3 2, or empty plasmid (negative control) were assayed for N-acetylation of 4-aminobiphenyl (ABP), or 2-aminofluorine (AF) and the reactions were analyzed for production of N-acetyl-ABP or N-acetyl-AF by high performance liquid chromatography (HPLC) as previously described (Walraven et al., 2006).
Bacterial lysates were diluted to 3 mg/ml in homogenization buffer and assayed for O-acetylation of N-hydroxy-ABP followed by HPLC analysis for formation of ABP-deoxyguanosine adduct as previously described (Hein et al., 2006b).
Michaelis-Menten kinetic parameters Km (μM) and Vmax (nanomoles per minute per milligram) were determined by measuring initial velocities for a range of substrate concentrations, and fitting to the data a one-phase exponential association nonlinear regression curve using GraphPad Prism software (GraphPad Software Inc., San Diego, CA).
COS-1 cell lysates containing recombinantly expressed rNat1, rNat2, rNat3 1, rNat3 2, or empty plasmid (negative control) were assayed for N-acetylation of ABP as described above. Results were normalized to β-galactosidase activity as a transfection control as previously described (Zang et al., 2004).
Thermostability
Bacterial lysates (1 mg/ml) were incubated at 50°C for 0-80 min, or at temperatures ranging from 30°C to 50°C for 10 min, and then assayed for N-acetylation of AF as described above. Results were normalized to the activity of untreated lysate. The heat inactivation half-life (T½) and first order rate constant (k) were determined by fitting a first-order decay curve to the data using GraphPad Prism.
Western Blotting
Multiple independent recombinant expressions were tested for rNat3 protein by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and Western blotting using rNat3-selective antiserum prepared with a peptide corresponding to amino acid residues 269 to 287 as previously described (Walraven et al., 2006).
Rat Tissue Harvest and RNA Extraction
F344 male rats (n = 4) were anesthetized by intramuscular injection of ketamine/xylazine (80/12 mg/kg) (Sigma) (Flecknell P, 1996). Rats were exsanguinated and sacrificed by cardiac puncture using Krebs-Henseleit bicarbonate buffer (Krebs et al., 1932) saturated with a mixture of 95% O2 and 5% CO2. Liver, lung, spleen, kidney, heart, esophagus, stomach, bladder, prostate, colon, duodenum, jejunum, and ileum tissues were harvested immediately following exsanguination, snap-frozen in liquid nitrogen, and stored at -80°C. Frozen tissues were thawed in RNAlater-ICE (Ambion, Foster City, CA) according to the manufacturer’s instructions, then disrupted and homogenized using the Mini-BeadBeater-1 with 1 mm zirconia beads (BioSpec Products, Inc., Bartlesville, OK). RNA was isolated using the RNeasy Mini Kit with on-column RNase-free DNase (Qiagen). RNA quality was confirmed by electrophoresis on a 1% agarose gel.
Reverse Transcription and PCR
Reverse transcription (RT) reactions were performed using SuperScript III reverse transcriptase (Invitrogen, Carlsbad, CA) with random hexamers and 1 μg RNA for every rat tissue. RT-PCR reactions were performed using AmpliTaq DNA polymerase (Applied Biosystems, Foster City, CA) and gene specific primers (Table1) to amplify an internal region of each Nat ORF. PCR products were amplified by 30 cycles of 95°C for 30 sec, 64°C for 1 min, and 72°C for 1.5 min. Reactions prepared with RNA and no reverse transcriptase, and reactions containing water instead of cDNA were used as controls for DNA contamination in the RNA and reagents, respectively. The cDNA PCR products were evaluated on 1% agarose gels. The Nat primers used for these PCR reactions were tested for specificity with full-length PCR-amplified rNat1, rNat2, and rNat3 ORFs. Genomic DNA control reactions validated the specificity and effectiveness of the PCR conditions, and demonstrated the relative amplification efficiency of each set of rNat primers. Genomic DNA control reactions were performed over a range of genomic DNA concentrations (3 pg/μl to 2 ng/μl reaction concentration) to ensure that the RT-PCR test reactions were in the linear range of amplification. In addition, genomic DNA PCR reactions were loaded in each gel to control for equal exposure during image capture. Representative RT-PCR results for each tissue were chosen from among four replicates and analyzed by densitometry using Quantity One 1-D Analysis Software (Bio-Rad Laboratories, Hercules, CA).
Computational Protein Structure Modeling
The human NAT1 F125S mutant crystal structure (Protein Data Bank ID# 2IJA) was used as a template for rNat3 homology modeling by satisfaction of spatial restraints using Modeller 8v2 (Sali and Blundell, 1993). Both the rNat3 1 and rNat3 2 homology models were tested for quality using PROCHECK (Laskowski et al., 1993) and ProSa 2003 (Sippl MJ, 1993). Molecular graphics images were generated using the UCSF Chimera package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (Pettersen et al., 2004).
RESULTS
Rat Nat Sequencing
rNat1, rNat2, and rNat3 ORFs were completely sequenced from genomic DNA PCR products of twelve different inbred rat strains [GenBank accession numbers DQ133344-DQ133355 (rNat1), DQ133356-DQ133367 (rNat2), and DQ099537-DQ099526 (rNat3)]. All twelve strains had the reference rNat1 and rNat2 ORF sequences. While ten of the strains had reference rNat3 ORFs, strains ACI and COP had the same variant rNat3 ORF (Table 2). This variant was named rNat3*2 (http://louisville.edu/medschool/pharmacology/NAT.html) and is characterized by a G619>T transversion that results in the amino acid coding change A207S. This variant was verified in strains ACI and COP by a second round of complete sequencing using genomic DNA from different animals. A summary of rNat genotypes is listed in Table 2, including the previously determined rNat genotypes for F344, SPRD, NSD, and WKY rat strains (Doll and Hein, 1995; Land et al., 1996).
Table 2.
Summary of Arylamine N-acetyltransferase Alleles in 16 Inbred Rat Strains
| Rat Strain | rNat1 | rNat2 | rNat3 |
|---|---|---|---|
| ACI | WT | WT | G619T |
| BN | WT | WT | WT |
| BUF | WT | WT | WT |
| CDF | WT | WT | WT |
| COP | WT | WT | G619T |
| DA | WT | WT | WT |
| LEW | WT | WT | WT |
| LOU/M | WT | WT | WT |
| MW | WT | WT | WT |
| PVG | WT | WT | WT |
| SHR | WT | WT | WT |
| WF | WT | WT | WT |
| F344 | WT a | WT a | WT c |
| SPRD | WT b | WT b | WT c |
| WKY | WT a | G361A, G399A, G522A, G796A a | WT c |
| NSD | WT | G361A, G399 A, G672A, G796A a | No data |
Recombinant Enzyme Activity and Stability
Sequencing results demonstrated successful cloning of rNat3*1 and rNat3*2 ORFs into bacterial expression vector pKK223-3. Lysates expressing rNat3 2 displayed significantly lower activity than lysates expressing rNat3 1 when assayed for N-acetylation of ABP (Figure 1A) and O-acetylation of N-hydroxy-ABP (Figure 1B). Previous reports of recombinant rNat1, rNat2, and rNat3 expression in the same bacterial system demonstrated that rNat3 1 activity is lower than rNat1 and rNat2 (Walraven et al., 2006).
Clones of rNat1*13, rNat2*20, rNat3*1, and rNat3*2 ORFs in vector pcDNA5/FRT were transfected into COS-1 cells. Lysates expressing rNat1 and rNat2 demonstrated ABP N-acetylation activities of 3.12 ± 0.26 and 7.289 ± 0.39 nmoles/min/mg, respectively, while rNat3 1 and rNat3 2 activities were not significantly above background (< 1.5 nmoles/min/mg).
Thermostability
When incubated at 50°C, the rNat3 1 and rNat3 2 enzymes demonstrated T½ values of 8.59 min and 7.25 min, respectively, and inactivation rate constants of 4.84 ± 0.19 hr-1 and 5.73 ± 0.15 hr-1, respectively (Figure 2A). When incubated for 10 min at temperatures ranging from 30°C to 50°C, no significant difference in stability was observed between rNat3 1 and rNat3 2 over the entire range of temperatures (Figure 2B). Together these results indicate that the rNat3 2 residue change does not affect the stability of the rNat3 protein.
Figure 2.
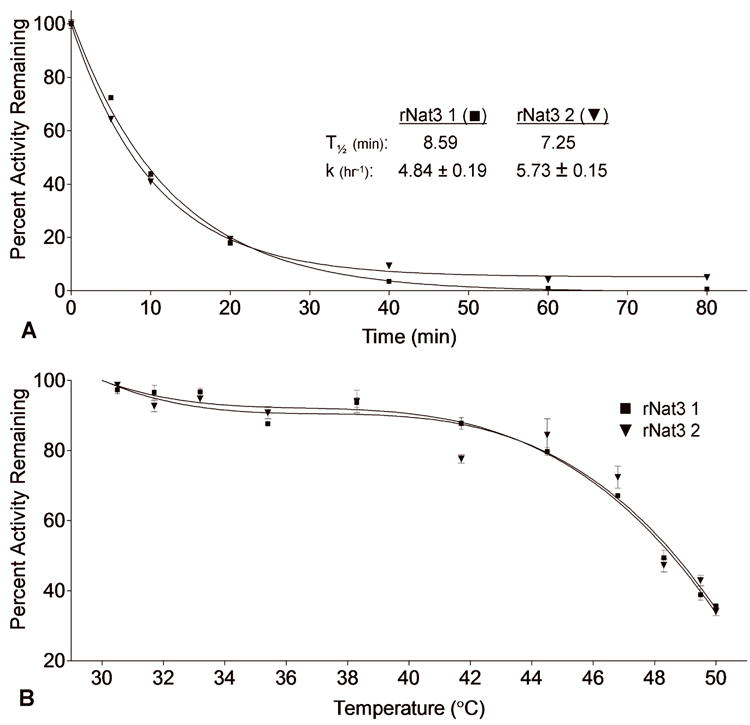
A Thermostability of rNat3 1 and rNat3 2 at 50°C. Following incubation, ABP N-acetylation was measured and results were normalized based on activity at time zero, and reported as percent activity remaining. Heat inactivation rate constant (k) and half-life (T½) are shown on the graph. Error bars indicate standard error (n = 3). B Thermostability of rNat3 1 and rNat3 2 incubated at 30-50°C for 10 min. Following incubation, ABP N-acetylation was measured, results were normalized based on activity at time zero, and reported as percent activity remaining. Error bars indicate standard error (n = 3).
Western Blotting
Variant rNat3 2 protein expression was significantly reduced relative to reference rNat3 1 protein in the recombinant bacterial system (Figure 3). Recombinantly expressed rNat3 1 and rNat3 2 protein was detectable in COS-1 cells by Western blot, but their protein levels did not differ (data not shown). These E. coli and COS-1 protein expression results were consistently replicated in three independent experiments. Since the rNat3 antiserum was prepared with a peptide corresponding to amino acid residues 269 to 287 (Walraven et al., 2006), A207S should not interfere with epitope recognition in the denatured rNat3 protein.
Figure 3.
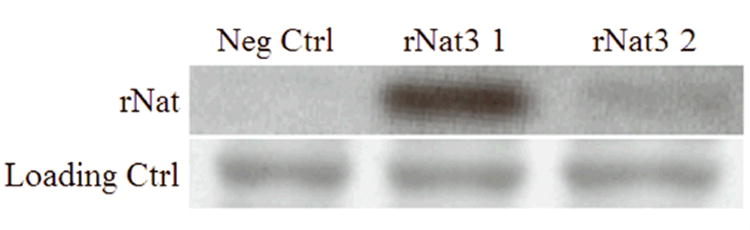
Bacterial lysates from negative control, rNat3 1 and rNat3 2 expressing cells were separated by SDS-PAGE and analyzed by Western blot using anti-Nat3 antiserum. Ponceau S red staining of the membrane was used as a loading control to verify equal protein loading in each lane.
Rat Tissue Nat Expression
Four F344 male inbred rats were exsanguinated and thirteen tissues harvested and processed from each. Relative levels of rNat1, rNat2, and rNat3 were assessed by semi-quantitative RT-PCR using primers that specifically amplified a segment within the single coding exon of each gene. For each gene, the same amplicon was generated from a genomic DNA template. Thus, parallel genomic PCRs with these primers controlled for the relative efficiency of each amplification from a DNA template with equal numbers of rNat1, rNat2, and rNat3 copies. Loading of the genomic DNA PCR controls in every gel also allowed normalization of photographic exposures for agarose gels run at different times. The cDNA PCR product intensities fell within in the linear range of the PCR amplification for each gene as established by amplification of serially diluted genomic DNA (data not shown).
Representative results from four different animals are shown in Figure 4. This analysis demonstrated rNat1 > rNat2 ≫ rNat3 expression in every tissue tested. Following densitometric analysis, the average ratio of rNat1:rNat2 intensities for each tissue was 4.1 ± 0.8, while the average ratio of rNat1:rNat2 for the genomic DNA standards was 1.0 ± 0.1. Under these PCR conditions no rNat3 amplification was detected for any of the tissues tested, even though rNat3 was readily amplified in the genomic DNA controls.
Figure 4.
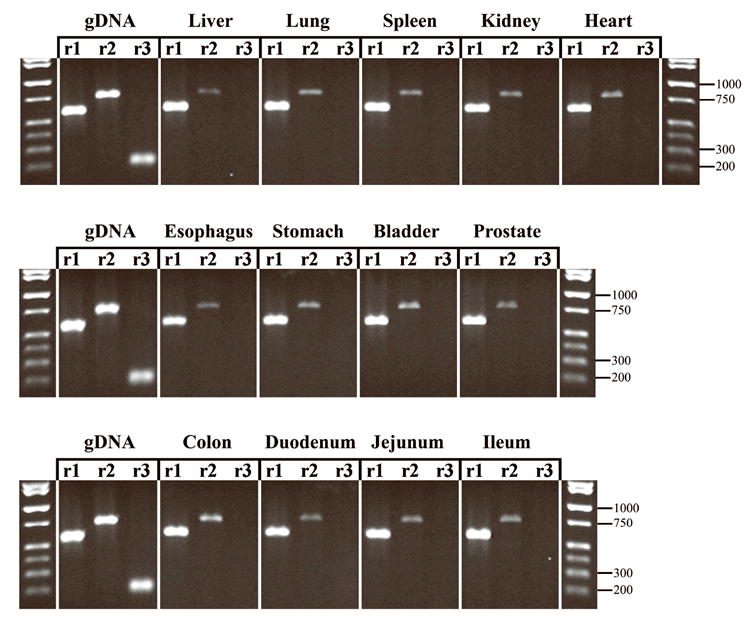
Semi-quantitative RT-PCR of rat tissue cDNA. rNat1, rNat2, and rNat3 PCR products are shown side-by-side for each tissue tested. The genomic DNA (gDNA) control set demonstrates the expected PCR product size and the relative band intensities expected of a rNat1:rNat2 :rNat3 template ratio of 1:1:1 in the linear range of amplification. Key molecular weight markers are shown on the right side of each lane.
Computational Protein Structure Modeling
The rNat3 1 and rNat3 2 homology models were of high stereochemical quality according to PROCHECK analyses, and high three-dimensional folding quality according to ProSa 2003 analyses. The rNat3 1 and rNat3 2 homology structures compared favorably to the template human NAT1 structure (2IJA) in all of these parameters. PROCHECK analysis of the human NAT1 structure (2IJA) yielded an average G-factor of 0.04, with 90.0 percent of the residues in the most favored regions, and zero in the disallowed regions, of the Ramachandran plot. PROCHECK analyses of the rNat3 1 and rNat3 2 models yielded average G-factors of -0.05 and -0.04, respectively, with 92.7 and 93.1 percent of the residues in the most favored regions, respectively, and zero in the disallowed regions for both. ProSa 2003 analysis yielded very consistent combined energy z-scores of -9.55, -9.64, and -9.94 for the human NAT1 template, the rNat3 1 model, and the rNat3 2 model, respectively.
According to the rNat3 models, residue 207 is located on the surface of the protein in the inter-domain region between domains II and III, adjacent to the active site pocket (Figure 5) (Payton et al., 2001). Since residue 207 is on the surface and has no apparent intramolecular side chain interactions, changing residue 207 from alanine to a slightly larger serine should not introduce steric interference in that region of the protein. Changing the small hydrophobic methyl side chain of alanine to the small hydrophilic side chain of serine would be expected to stabilize the protein surface-solvent interface.
Figure 5.
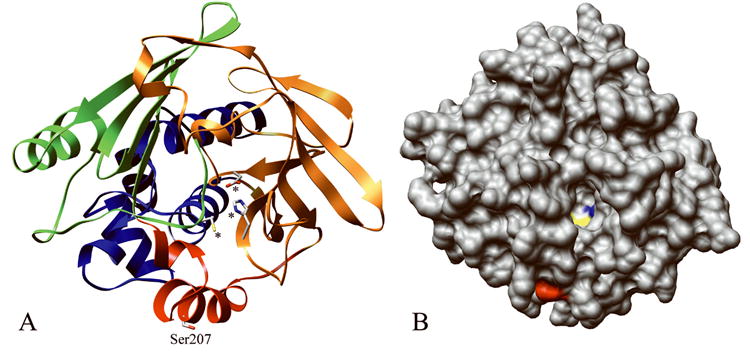
Computational structures of rNat3 1 and rNat3 2 modeled after human NAT1 crystal structure (PDB ID# 2IJA). A The location of residue Ser207 (labeled) in the inter-domain region (red), and catalytic triad residues Cys68, His107, Asp122 are demonstrated (marked by asterisks). Protein domains I, II, and III are indicated by ribbon colors blue, orange, and green, respectively. B The rNat3 2 homology model with protein surface shown, demonstrates the location of Ser207 on the protein surface (red), near the active site opening in which the catalytic Cys68 (yellow) and His107 (blue) are visible.
DISCUSSION
Prior to this study, the complete set of rNat1, rNat2, and rNat3 gene sequences were known for only three inbred rat strains (Table 2). In addition, nothing was known about the relative contribution of these three Nat genes to the overall Nat expression in rat tissues. This study facilitates future research in the rat by addressing these issues and introducing the novel rNat3*2 allele.
N-acetyltransferase expression has been detected in blood for a number of organisms, including rat (Gollamudi et al., 1980), mouse (Mattano and Weber, 1987), rabbit (Drummond et al., 1980), Syrian hamster (Hein et al., 1986) and human (Lindsay and Baty, 1988). Because the degree of blood perfusion varies greatly among tissues, our evaluation of tissue-specific Nat expression took into account the contribution of Nat expressed in blood. For this study, the rats were exsanguinated prior to tissue harvest to eliminate the influence of blood Nat expression on the evaluation of tissue-specific Nat expression.
Specific rNat1 and rNat2 transcripts were detected in all thirteen rat tissues analyzed, which is consistent with similar findings in hamster (Hein et al., 2006a), mouse (Loehle et al., 2006; Sugamori et al, 2003), and human (Barker et al., 2006; Husain et al., 2007). The results of this study demonstrate that rNat1 mRNA is more highly expressed than rNat2 mRNA in every rat tissue tested. Since rNat1 and rNat2 probably share a common promoter and non-coding exon (Boukouvala and Fakis, 2005; Ebisawa et al., 1995), the rNat1 and rNat2 expression differences are likely due to mRNA stability differences, differential mRNA splicing, and/or transcription attenuation between the rNat1 and rNat2 ORFs, though more studies would be necessary to identify the exact mechanism. Combined with the observation that rNat1 protein is much more stable than rNat2 protein (Walraven et al., 2006), this study suggests that rNat1 is the most prevalent rNat protein in rat tissues, though any unknown differences in translational efficiency might also affect this balance. These results represent the first direct comparison of the expression of two different Nat isoforms in the same tissue, providing insight into the relative expression of different Nat genes. Further comparative analyses of rNat protein levels are needed to investigate possible differences in tissue-specific translational efficiencies. Further analyses of rNat mRNA sequences are necessary to identify potential alternatively spliced rNat mRNA species, which may contribute to the complexity of rNat transcription and translational rates.
Rat Nat3 transcripts were not detectable in tissues with the PCR conditions used to detect rNat1 and rNat2 (Figure 4), indicating that constitutive rNat3 expression is much lower than for rNat1 and rNat2. Very low expression was also observed for mouse Nat3 relative to Nat1 and Nat2 (Sugamori et al., 2007). The similar low expression of rat and mouse Nat3 may be suggestive of similarities in the promoter region of these genes. A comparison of rat genomic sequence near the rNat3 ORF with a spliced mouse Nat3 cDNA of experimental origin that was recently added to GenBank (AK077364) provides support for this idea. BLAT alignment (Kent, 2002) of the mouse genome with this mouse Nat3 cDNA identifies two upstream exons that are separated from each other by only 89 base pairs, but lie 23.3 kb upstream from the Nat3 coding exon. A highly similar region in the rat genomic sequence lies approximately 26 kb upstream of the rat Nat3 coding exon, and includes similar splice recognition sequences and a generally very high similarity that extends 240 base pairs upstream of the exons. This 240 base pair region likely includes a large part of the promoter region, suggesting potential homology between the mouse and rat Nat3 promoter regions. Alignment of this region with other Nat promoters detects only a very faint similarity.
Our study of rNat sequences in twelve inbred rat strains (Table 2), combined with previously determined inbred rat strain rNat sequences, suggests that the reference rNat1*13, rNat2*20, and rNat3*1 alleles are the most common rNat alleles in inbred rat strains. Strains ACI and COP, both of which possess the rNat3*2 allele, are closely related genetically since ACI was developed by crossing the AUG and COP strains (Canzian, 1997). Other rare rNat variant alleles include rNat2*21A and rNat2*21B, both of which are slow acetylator alleles found in WKY and NSD inbred rat strains, respectively (Doll and Hein, 1995).
The low rNat3 expression and activity, and the absence of known rNat3 selective substrates precludes testing for differences between rNat3 1 and rNat3 2 in the tissues of inbred rats carrying these alleles. Reduced rNat3 2 catalytic activity in our recombinant bacterial system is consistent with reduced protein expression, and since no difference was observed in the enzymes’ thermostability in two different stability assays, a translational or pre-translational mechanism may be responsible. A difference in bacterial usage of codons GCA (rNat3*1) and UCA (rNat3*2) may alter the rNat3*2 translation. Codon usage tables for Escherichia coli K12 (www.kazusa.or.jp/codon/) estimate the usage of these codons at 20.2 (GCA) and 7.1 (UCA) per thousand codons, a nearly three-fold difference. In our high-expressing recombinant bacterial system this three-fold difference in codon usage may have altered translation efficiency and contributed to reduced protein expression for the rNat3*2 variant. The fact that protein expression differences between rNat3 1 and rNat3 2 were not observed when recombinantly expressed in mammalian COS-1 cells supports the codon usage explanation of the bacterial results, since differences in codon usage for GCA (rNat3*1) and UCA (rNat3*2) is less, at 14.6 and 11.8, respectively, in COS-1 cells. Thus, the differences between the recombinantly expressed rNat3 alleles may be absent in vivo, since the difference in Rattus norvegicus codon usage frequencies for GCA and UCA is also less, at 10.9 and 15.6 per thousand codons, respectively. Differences in translational efficiency due to codon usage are more likely to be observed in a high expression system where the tRNA pool is more depleted, than in a lower-expressing in vivo situation where codon usage differences are less likely to affect the translation rate due to tRNA supply depletion. The difference in bacterial recombinant protein expression between the two rNat3 alleles is not likely due to increased degradation or protein aggregation. Since the stability of the rNat3 2 variant protein is not compromised, and since the residue change on the surface of the protein is from a small hydrophobic (Ala) to small hydrophilic (Ser) side chain, there is no reason to suspect these mechanisms.
In comparison to rNat1 and rNat2, rNat3 has relatively low catalytic activity following recombinant expression in both bacterial and mammalian cells, with a limited number of known substrates (Walraven et al., 2006). If the rNat3 gene is actively expressed in vivo, its expression is far lower than that of rNat1 and rNat2. The low expression of the rNat3 gene and low catalytic activity of the rNat3 enzyme suggests that it has a negligible effect on rat N-acetylation phenotype for rats in standard laboratory conditions. These conclusions are consistent with similar recent findings using mouse Nat3 knockout models (Sugamori et al., 2007).
Acknowledgments
We thank Dr. Gavin Arteel for his expert guidance and assistance with whole-rat exsanguination techniques. This work represents partial fulfillment for the Ph.D. degree in Pharmacology and Toxicology to Jason Walraven at the University of Louisville School of Medicine.
FUNDING These studies were partially supported by National Institutes of Health grants CA34627 (DWH) and ES011564 (DWH to support JMW).
References
- Barker DF, Husain A, Neale JR, Martini BD, Zhang X, Doll MA, States JC, Hein DW. Functional properties of an alternative, tissue-specific promoter for human arylamine N-acetyltransferase 1. Pharmacogenet Genomics. 2006;16:515–525. doi: 10.1097/01.fpc.0000215066.29342.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Boukouvala S, Fakis G. Arylamine N-acetyltransferases: What we learn from genes and genomes. Drug Metab Rev. 2005;37:511–564. doi: 10.1080/03602530500251204. [DOI] [PubMed] [Google Scholar]
- Canzian F. Phylogenetics of the laboratory rat Rattus norvegicus. Genome Res. 1997;7:262–267. doi: 10.1101/gr.7.3.262. [DOI] [PubMed] [Google Scholar]
- Chung CT, Niemela SL, Miller RH. One-step preparation of competent Escherichia coli: transformation and storage of bacterial cells in the same solution. Proc Natl Sci USA. 1989;86:2172–2175. doi: 10.1073/pnas.86.7.2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doll MA, Hein DW. Cloning, sequencing, and expression of NAT1 and NAT2 encoding genes from rapid and slow acetylator inbred rats. Pharmacogenetics. 1995;5:247–251. doi: 10.1097/00008571-199508000-00009. [DOI] [PubMed] [Google Scholar]
- Drummond GS, Kelker HC, Weber WW. N-acetylation of drugs. Observations on the properties of partially purified N-acetyltransferase from peripheral blood of rabbit. Biochem J. 1980;187:157–162. doi: 10.1042/bj1870157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebisawa T, Sasaki Y, Deguchi T. Complementary DNAs for two arylamine N-acetyltransferases with identical 5’ non-coding regions from rat pineal gland. Eur J Biochem. 1995;228:129–137. doi: 10.1111/j.1432-1033.1995.tb20240.x. [DOI] [PubMed] [Google Scholar]
- Flecknell P. Anaesthesia and analgesia for rodents and rabbits. In: Laber-Laird K, Swindle MM, Flecknell P, editors. Handbook of Rodent and Rabbit Medicine. Elsevier Science; New York: 1996. pp. 225–227. [Google Scholar]
- Gollamudi R, Muniraju B, Schreiber EC. N-acetyltransferases of rat liver and blood: substrate specificities. Enzyme. 1980;25:309–315. doi: 10.1159/000459272. [DOI] [PubMed] [Google Scholar]
- Hanna PE. Metabolic activation and detoxification of arylamines. Curr Med Chem. 1996;3:195–210. [Google Scholar]
- Hein DW, Kirlin WG, Ferguson RJ, Thompson LK, Ogolla F. Identification and inheritance of inbred hamster N-acetyltransferase isozymes in peripheral blood. J Pharmacol Exp Ther. 1986;239:823–828. [PubMed] [Google Scholar]
- Hein DW. Acetylator genotype and arylamine-induced carcinogenesis. Biochim Biophys Acta. 1988;948:37–66. doi: 10.1016/0304-419x(88)90004-2. [DOI] [PubMed] [Google Scholar]
- Hein DW, Doll MA, Fretland AJ, Leff MA, Webb SJ, Xiao GH, Devanaboyina US, Nangju NA, Feng Y. Molecular genetics and epidemiology of the NAT1 and NAT2 acetylation polymorphisms. Cancer Epidemiol Biomarkers Prev. 2000;9:29–42. [PubMed] [Google Scholar]
- Hein DW, Doll MA, Nerland DE, Fretland AJ. Tissue distribution of N-acetyltransferase 1 and 2 catalyzing the N-acetyltation of 4-aminobiphenyl and O-acetylation of N-hydroxy-4-aminobiphenyl in the congenic rapid and slow acetylator Syrian hamster. Mol Carcinog. 2006a;45:230–238. doi: 10.1002/mc.20164. [DOI] [PubMed] [Google Scholar]
- Hein DW, Fretland AJ, Doll MA. Effects of single nucleotide polymorphisms in human N-acetyltransferase 2 on metabolic activation (O-acetylation) of hetereocyclic amine carcinogens. Int J Cancer. 2006b;119:1208–1211. doi: 10.1002/ijc.21957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husain A, Zhang X, Doll MA, States JC, Barker DF, Hein DW. Identification of N-acetyltransferase 2 (NAT2) transcription start sites and quantification of NAT2-specific mRNA in human tissues. Drug Metab Dispos. 2007;35:721–727. doi: 10.1124/dmd.106.014621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob HJ. Functional genomics and rat models. Genome Res. 1999;9:1013–1016. doi: 10.1101/gr.9.11.1013. [DOI] [PubMed] [Google Scholar]
- Kent WJ. BLAT – The BLAST-Like Alignment Tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs HA, Henseleit K. Untersuchungen über die harnstoffbildung im tierkorper. Hoppe-Seylers Z Physiol Chem. 1932;210:33–66. [Google Scholar]
- Land SJ, Jones RF, King CM. Genetic analysis of two rat acetyltransferases. Carcinogenesis. 1996;17:1121–1126. doi: 10.1093/carcin/17.5.1121. [DOI] [PubMed] [Google Scholar]
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Cryst. 1993;26:283–291. [Google Scholar]
- Lazar J, Moreno C, Jacob HJ, Kwitek AE. Impact of genomics on research in the rat. Genome Res. 2005;15:1717–1728. doi: 10.1101/gr.3744005. [DOI] [PubMed] [Google Scholar]
- Lindsay RM, Baty JD. Inter-individual variation of human blood N-acetyltransferase activity in vitro. Biochem Pharmacol. 1988;37:3915–3921. doi: 10.1016/0006-2952(88)90074-3. [DOI] [PubMed] [Google Scholar]
- Loehle JA, Cornish V, Wakefield L, Doll MA, Neale JR, Zang Y, Sim E, Hein DW. N-acetyltransferase (Nat) 1 and 2 expression in Nat2 knockout mice. J Pharmacol Exp Ther. 2006;319:724–728. doi: 10.1124/jpet.106.108662. [DOI] [PubMed] [Google Scholar]
- Mattano SS, Weber WW. Kinetics of arylamine N-acetyltransferase in tissues from rapid and slow acetylator mice. Carcinogenesis. 1987;8:133–137. doi: 10.1093/carcin/8.1.133. [DOI] [PubMed] [Google Scholar]
- Payton M, Mushtaq A, Yu T-W, Wu L-J, Sinclair J, Sim E. Eubacterial arylamine N-acetyltransferase identification and comparison of 18 members of the protein family with conserved active site cysteine, histidine, and aspartate residues. Microbiology. 2001;147:1137–1147. doi: 10.1099/00221287-147-5-1137. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera - A Visualization System for Exploratory Research and Analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Rat Genome Sequence Project Consortium . Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature. 2004;428:439–521. doi: 10.1038/nature02426. [DOI] [PubMed] [Google Scholar]
- Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- Sippl MJ. Recognition of errors in three-dimensional structures of proteins. Proteins. 1993;17:355–362. doi: 10.1002/prot.340170404. [DOI] [PubMed] [Google Scholar]
- Sugamori KS, Wong S, Gaedigk A, Yu V, Abramovici H, Rozmahel R, Grant DM. Generation and functional characterization of arylamine N-acetyltransferase Nat1/Nat2 double-knockout mice. Mol Pharmacol. 2003;64:170–179. doi: 10.1124/mol.64.1.170. [DOI] [PubMed] [Google Scholar]
- Sugamori KS, Brenneman D, Wong S, Gaedigk A, Yu V, Abramovici H, Rozmahel R, Grant DM. Effect of arylamine acetyltransferase Nat3 gene knockout on N-acetylation in the mouse. Drug Metab Dispos. 35:1064–1070. doi: 10.1124/dmd.107.015396. [DOI] [PubMed] [Google Scholar]
- Walraven JM, Doll MA, Hein DW. Identification and characterization of functional rat arylamine N-acetyltransferase 3: comparisons with rat arylamine N-acetyltransferases 1 and 2. J Pharmacol Exp Ther. 2006;319:369–375. doi: 10.1124/jpet.106.108399. [DOI] [PubMed] [Google Scholar]
- Zang Y, Zhao S, Doll MA, States JC, Hein DW. The T341C (Ile114Thr) polymorphism of N-acetyltransferase 2 yields slow acetylator phenotype by enhanced protein degradation. Pharmacogenetics. 2004;14:717–723. doi: 10.1097/00008571-200411000-00002. [DOI] [PubMed] [Google Scholar]