

Summary
The objective of this study was to evaluate whether an increased hazard of developing ischemic heart disease (IHD) is associated with any of the three genotypes A560T832/A560T832, A560T832/A560G832 and A560T832/T560T832, defined by variations in two non-coding SNPs in the 5′ promoter region of the apolipoprotein E (APOE) gene. These genotypes were selected because they distinguished between high and low levels of HDL-C, TG and/or T-C in our earlier study of multiple samples defined by gender and population. We found a significant increase (p<0.05) in the hazard of IHD in females with the A560T832/T560T832 genotype that remained significant after fitting the effects of dyslipidemia, other established risk factors, and the structural isoform variations of the ApoE molecule. We discuss why this statistically significant genetic predictor may not be an appropriate screening test for IHD in the Danish population at large.
Keywords: APOE gene, regulatory variation, prediction of IHD
Introduction
Cholesterol accumulation in arterial walls contributes to the development of atherosclerosis, an important progenitor of ischemic heart disease (IHD) and other manifestations of cardiovascular disease (Steinberg, 2004). A plethora of single nucleotide polymorphisms (SNPs) have been characterized in genes coding for proteins which are involved in lipid metabolism (Crawford et al. 2004; Nickerson, 2005; Fullerton et al. 2000, 2004; Nickerson et al. 2000; Knoblauch et al. 2004; Nabel, 2003). Interindividual differences in blood concentrations of total cholesterol (T-C), triglycerides (TG) and/or high-density lipoprotein cholesterol (HDL-C) are associated with genotypic variations defined by some of these SNPs (Knoblauch et al. 2004; Stengård et al. 2002, 2006; Viiri et al. 2005; Davignon et al. 1999; Frikke-Schmidt et al. 2000a, 2004, 2007; Lussier-Cacan et al. 2002; Nelson et al. 2001). Variations in the risk of atherosclerotic disease endpoints have been documented for a subsample of these genotypic variations (Nabel, 2003; Sposito et al. 2004; Stengård et al.1995; Song et al. 2004; Frikke-Schmidt et al. 2000b; Lambert et al. 2000a; Viitanen et al. 2001; Ye et al. 2003). Most of the observed phenotype-genotype associations are, however, specific to a particular population or context, genetic or environmental, within a population (Stengård et al. 2002; Frikke-Schmidt et al. 2000a, 2000b, 2004; Lussier-Cacan et al. 2002; Nelson et al. 2001). The association of variation in measures of lipid metabolism and risk of IHD with particular genotypic variations of the apolipoprotein E (APOE) gene stands apart as being observed in multiple populations. In this study we focus on those phenotypic effects of APOE genotypes that are less sensitive to variation in genetic and environmental context.
Apolipoprotein E (ApoE) is a structural constituent of many atherogenic lipoprotein particles, such as TG-rich chylomicrons, chylomicron remnants, very-low-density lipoproteins (VLDLs) and high-density lipoproteins (HDLs), and is involved in their transport from one tissue or cell type to another (Davignon et al.1988; Mahley, 1988; Mahley & Rall, 2000). The ApoE molecule has three Common isoforms – E2, E3 and E4 (Davignon et al.1988; Utermann et al. 1977). In a study that resequenced 5.5 kb of the APOE gene, including related 5′ and 3′ flanking regions, we identified ten biallelic SNPs that segregated in multiple populations (Fullerton et al. 2000, Nickerson et al. 2000). These “public” SNPs include the two coding SNPs in the fourth exon at positions 3937 and 4075 (Fig. 1) that are responsible for the structural differences between the E2, E3 and E4 isoforms. Inter-individual differences in blood measures of lipid metabolism are repeatedly associated with SNP genotypes that determine this structural variation. On average, blood T-C concentration in individuals with the E4 isoform is higher than in individuals who do not have this isoform (Stengård et al. 2002; Davignon et al. 1988, 1999; Frikke-Schmidt et al. 2000a, 2004). The E4 isoform also has pleiotropic effects on blood HDL-C and TG concentrations in some populations (Frikke-Schmidt et al. 2000a, 2004; Lussier-Cacan et al. 2002; Nelson et al. 2001). Animal experiments (Davignon et al. 1999) and studies in humans (Davignon et al. 1999; Stengård et al. 1995; Song et al. 2004) have repeatedly demonstrated that the E4 isoform tends to predict the development of atherosclerosis. In humans it has been suggested that the atherogenic potential of the E4 isoform is not solely mediated through the associated hypercholesterolinemia (Davignon et al. 1999).
Figure 1.
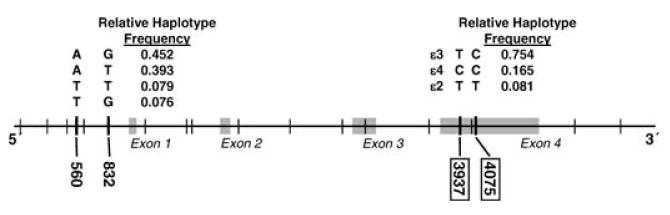
Genomic structure and locations of the two non-coding SNPs in the 5′ promoter region, and the two coding-SNPs and the exon four region of the APOE gene.
Two of the eight public non-coding SNPs of the APOE gene, located in the 5′ promoter region at positions 560 and 832, define three genotypes, A560T832/A560T832, A560T832/A560G832 and A560T832/T560T832, that distinguish between high and low levels of HDL-C, TG and/or T-C, separately for females and males, in independently ascertained samples from three populations (Stengård et al. 2006). This association was confirmed in a fourth independent sample from the Copenhagen City Heart Study (CCHS) (Stengård et al. 2006). These findings led us to postulate that individuals with 5′ genotypes at increased risk for dyslipidemia are at increased risk for developing atherosclerotic disease. The goal of the study reported here was to evaluate this hypothesis in a large sample of middle-aged and elderly females and males who have been evaluated for the onset of IHD in the CCHS (Frikke-Schmidt et al. 2000a, 2004; Schnohr et al. 2001). Our specific questions were: 1) do individuals with the proposed high risk 5′ genotypes of APOE identified in our previous studies of dyslipidemia have higher hazards of developing IHD than individuals who do not have these genotypes, separately for females and males? and 2) does an observed increase in the hazard persist when dyslipidemia, other established risk factors, and the structural isoform variations determined by the APOE gene, are considered in a risk assessment? We found a significant increase (p<0.05) in the hazard of IHD in females with the A560T832/T560T832 genotype. This increase remained statistically significant after fitting the effects of dyslipidemia, other established risk factors of IHD, and genotypes defined by variations in the two coding SNPs at positions 3937 and 4075 that define the ApoE isoforms. Although the positive predictive value of the A560T832/T560T832 genotype (0.099) was only slightly higher than the cumulative incidence of IHD in the population at large (0.070), it was 5 times higher in females with diabetes (0.375). Here we discuss why this statistically significant genetic predictor may not be an appropriate screening test for IHD in the Danish population at large.
Methods
Participants
The CCHS is a prospective study of the Danish population at large, aged 20 years or older (Frikke-Schmidt et al. 2000a, 2004; Schnohr et al. 2001). A baseline survey was carried out between 1976 and 1978. The first follow-up survey was performed between 1981 and 1983, and a second one between 1991 and 1994 (Frikke-Schmidt et al. 2000a, 2004; Schnohr et al. 2001). The second follow-up survey (1991-1994) served as a baseline survey for the study reported here. Altogether, 16,563 individuals were invited to the latter survey, 10,135 participated (response rate 61%), and 9,259 gave blood for DNA extraction. A subsample Comprising middle-aged and elderly individuals who were at least 45 years old when they were seen in this second follow-up survey, and who did not have IHD, was selected for our study. Clinical data and genotype information on the four SNPs in APOE (Fig. 1) considered in this study were available for 3686 females and 2,772 males who satisfied the selection criteria. Informed consent was obtained from all participants. More than 99% were Europeans of Danish descent. The study was approved by the Danish Ethics Committee for the City of Copenhagen and Frederiksberg (No. 100.2039/91).
Variable Definitions
IHD was evaluated for the 6458 participants of the CCHS during the period from the date they were seen in connection with the second follow-up survey until December 31, 1999. Information on diagnoses of IHD (World Health Organization International Classification of Diseases, 8th edition, codes 410 to 414; 10th edition, I20 to I25, defining myocardial infarction, angina pectoris, and acute and chronic atherosclerosis related heart disease) was gathered from the Danish National Hospital Discharge Register, from the Danish National Register of Causes of Death, and from medical records of general practitioners and hospitals. During follow-up 576 (257 females, 7.0% and 319 males, 11.5%) of the 6458 participants were diagnosed as having developed IHD. The potential length of the follow-up period ranged from 5.5 to 8.2 years. The observed median follow-up time was 6.5 years (range, 0.01 to 8.2 years). The total exposure to risk of IHD was 39,648 person-years.
Blood HDL-C, TG and T-C concentrations were measured by standard enzymatic assays (Boehringer Mannheim, GmbH Diagnostics, Mannheim, Germany) at the Department of Clinical Biochemistry, Rigshospitalet, Copenhagen University Hospital, Copenhagen (Frikke-Schmidt et al. 2004). Recommendations of the National Cholesterol Education Program Expert Panel, National Institute of Health, USA, were used to define dyslipemic subgroups (National Cholesterol Education Program National Heart, Lung, and Blood Institute, 2002). Dyslipidemia was diagnosed when an individual's blood T-C concentration exceeded 200mg/dl (5.18 mmol/L), TG exceeded 150mg/dl (1.60 mmol/L) or HDL-C was less than 40mg/dl (1.04 mmol/L). The definitions of other established risk factors for IHD, smoking, diabetes mellitus, and hypertension, are described in a previous report of the CCHS (Frikke-Schmidt et al. 2007). Briefly, each factor was dichotomized to define a high risk group, ever-smoked (current smoker at any of the three examinations), ever had diabetes (self reported disease, use of insulin, use of oral hypoglycemic drugs and/or non-fasting plasma glucose ≥11.1 mmol/L at any of the three examinations) or ever-hypertensive (systolic blood pressure ≥140 mHg and/or diastolic blood pressure ≥90 mmHg, or use of antihypertensive drugs at any of the three examinations), respectively. The four SNPs in APOE were genotyped using PCR and restriction enzyme digestion or allele specific amplification methods, as previously described (Frikke-Schmidt et al. 2000a, 2004; Hixson & Vernier, 1990). The locations of the SNPs are given in a feature map of the gene in Fig. 1.
Statistical Analyses
Fisher's F-ratio was used to test if there was a statistically significant difference in the phenotypic variance of a quantitative trait between females and males (Sokal & Rohlf, 1995). Student's t was used to test the statistical significance of the difference between gender means when the F-ratio was not significant, and Satterthwaite's modification of the t-test (Sokal & Rohlf, 1995) was used when the F-ratio was significant. The chi-square statistic was used to test homogeneity of frequency distributions of dichotomous risk factor traits across genders.
A Cox proportional hazards model (Everitt, 1994) was used to compare the hazard of developing IHD in the sample of individuals who had a genotype identified in our previous study (Stengård et al. 2006) as a potential predictor of IHD, with the hazard of developing IHD in a reference group of individuals who did not have any of the three hypothesized high risk genotypes. The null hypothesis that the hazard of developing IHD in each of the high risk genotypes did not differ significantly from the hazard in the reference group (i.e., hazard ratio = 1) was tested by employing a Wald z-test statistic. Cox proportional hazards models, that considered the high risk genotypes, the three measures of dyslipidemia, other established risk factors and/or the structural isoform variations determined by the APOE gene, were used to address the question of whether an observed increase in the hazard of IHD persisted when the proposed covariates were considered in a risk assessment. We considered a nominal α = 0.05 level of probability to be a statistically significant test result. We did not employ any statistical procedure to correct biases attributable to multiple testing, because there is no such strategy that is appropriate for this situation.
Results
Description of the Sample
Gender-specific distributions of age, basic anthropometric characteristics, the three blood measures of lipid metabolism (HDL-C, TG, and T-C), and other established IHD risk factors, systolic and diastolic blood pressure, smoking status and diabetes are summarized in Table 1, separately for females and males. On average, females were significantly older (1.7 years) than males, while the variability in age was significantly greater in males. Females were significantly leaner than males. Females were less frequently diagnosed as having established risk factors (57.0%, 3.6%, and 62.4% for smoking, diabetes and hypertension, respectively) and were less likely to have dyslipidemic HDL-C or TG levels (5.9%, and 40.9% for low HDL-C and high TG, respectively) than males (70.2%, 7.6%, and 69.3% for smoking, diabetes and hypertension, and 20.3%, and 52.2% for low HDL-C and high TG, respectively). In contrast, the females had hypercholestrolinemia (89.4%) more often than the males (80.2%).
Table 1.
Description of the CCHS sample, for females and males separately
| Trait | Females (N = 3686) | Males (N = 2772) | (+) |
|---|---|---|---|
| Anthropometric character | |||
| Age (years) | |||
| Mean | 64.86 | 63.18 | *** |
| Variance | 95.8 | 102.57 | * |
| Weight (kg) | |||
| Mean | 67.11 | 80.73 | *** |
| Variance | 157.18 | 176.64 | *** |
| Height (cm) | |||
| Mean | 161.39 | 174.23 | *** |
| Variance | 42.36 | 48.64 | *** |
| BMI (kg/m2) | |||
| Mean | 25.77 | 26.57 | *** |
| Variance | 21.34 | 15.65 | *** |
| Blood Measure of Dyslipidemia | |||
| HDL-C (mg/dl) | |||
| Mean | 67.29 | 54.41 | *** |
| Variance | 397.46 | 297.36 | *** |
| HDL-C < 40 mg/dl (%) | 5.89 | 20.31 | *** |
| TG (mg/dl) | |||
| Mean | 158.66 | 191.24 | *** |
| Variance | 8436.36 | 26441.86 | *** |
| TG > 150 mg/dl (%) | 40.94 | 52.2 | *** |
| T-C (mg/dl) | |||
| Mean | 255.01 | 235.7 | *** |
| Variance | 2324.16 | 1915.26 | *** |
| T-C > 200mg/dl (%) | 89.39 | 80.16 | *** |
| Other Established Risk Factor Characters | |||
| Smokes (%) | 57.0 | 70.2 | *** |
| Diabetics (%) | 3.58 | 7.61 | *** |
| Systolic Blood Pressure (mmHg) | |||
| Mean | 142.56 | 144.48 | *** |
| Variance | 512.16 | 449.36 | *** |
| Diastolic Blood Pressure (mmHg) | |||
| Mean | 84.42 | 87.62 | *** |
| Variance | 139.12 | 143.75 | |
| Hypertensives (%) | 62.45 | 69.34 | *** |
+ Significant difference between males and females at the * α≤0.05, ** α≤0.01 and *** α≤0.001 level of probability.
The relative frequencies in the CCHS sample of the three hypothesized high risk 5′ APOE genotypes, identified in our earlier study of dyslipidemia using samples from multiple independent populations, are given in Table 2. The A560T832/A560G832 genotype was the most common (relative frequency = 0.35), and the A560T832/T560T832 genotype the least frequent (relative frequency = 0.06) in both genders.
Table 2.
Relative genotype frequencies in the CCHS sample, for females and males separately
| Relative Frequency (N) | ||
|---|---|---|
| Genotype Groups | Females | Males |
| 5′ Promoter Region | ||
| A560T832 / A560T832 | 0.16 (583) | 0.15 (416) |
| A560T832 / A560G832 | 0.35 (1290) | 0.36 (1010) |
| A560T832 / T560T832 | 0.06 (213) | 0.06 (174) |
| OTHERS | 0.43 (1600) | 0.43 (1172) |
Gender-Specific Utility of Different Measures of Dyslipidemia, Other Established Risk Factors, and the Exon Four Variation in the APOE Gene, in Predicting the Development of IHD
Estimates of hazard ratios (HRs) for IHD are summarized in Table 3, separately for each gender and each trait. The highest hazard was observed in diabetic females (HR 2.96, 95% CI = 1.96 to 4.48) and in females who had low HDL-C (HR 2.45, 95% CI = 1.66 to 3.60) when compared to the hazard in females in the respective reference groups. The hazard in females who smoked and in females who had high TG was moderately increased (HR 1.76, 95% CI = 1.36 to 2.29 and 1.54, 95% CI = 1.20 to 1.97, respectively). In males, smoking, diabetes, hypertension and high TG each predicted a moderate increase in the hazard of IHD (HR 1.59, 95% CI = 1.23 to 2.06; 1.59, 95% = CI 1.13 to 2.24; 1.72, 95% CI = 1.29 to 2.30 and 1.34, 95% CI = 1.07 to 1.63, respectively). Neither high T-C, nor the genotypes with the high risk E4 isoform, were predictors of an increased hazard of IHD in either gender.
Table 3.
Gender-specific hazard ratios of IHD, separately for each of the three blood measures of dyslipidemia, other established risk factors of IHD and the three isoforms of the APOE gene
| Trait | Females Hazard Ratio (95% C.I.) | Males Hazard Ratio (95% C.I.) |
|---|---|---|
| Blood Measure of Dyslipidemia | ||
| HDL-C | 2.45 *** | 1.28 |
| (1.66–3.60) | (0.99–1.66) | |
| TG | 1.54 *** | 1.34 ** |
| (1.20–1.97) | (1.07–1.68) | |
| T-C | 0.71 | 1.21 |
| (0.48–1.06) | (0.90–1.63) | |
| Other Established Risk Factors | ||
| Smoking | 1.76 *** | 1.59 *** |
| (1.36–2.29) | (1.23–2.06) | |
| Diabetes | 2.96 *** | 1.59 ** |
| (1.96–4.48) | (1.13–2.24) | |
| Hypertension | 1.29 | 1.72 *** |
| (0.96–1.74) | (1.29–2.30) | |
| ApoE Isoform Groups | ||
| E2/2, E3/2 | 0.91 | 0.81 |
| (0.62–1.34) | (0.57–1.15) | |
| E3/3 | 1 | 1 |
| (–) | (–) | |
| E4/2, E4/3, E4/4 | 1.13 | 0.9 |
| (0.86–1.49) | (0.70–1.16) | |
Significant at the * α ≤ 0.05, ** α ≤ 0.01, *** α ≤ 0.001 level of probability.
Gender-Specific Utility of Each of the Three High Risk 5′ Genotypes of APOE for Predicting the Development of IHD
The estimate of HRs for IHD is given in Table 4 for each of the three hypothesized high risk 5′ genotypes of APOE, separately for each gender. One of these genotypes, A560T832/T560T832, was a statistically significant predictor of an increase in the hazard of IHD (HR 1.69, 95% CI = 1.05 to 2.70) in females as compared with the hazard in females who did not have any of the proposed high risk genotypes. This increase was significant after fitting dyslipidemia, other established risk factors of IHD, or genotypes defined by variations in the two coding SNPs 3937 and 4075. The estimate of the HR for the A560T832/T560T832 genotype was 1.81 (95% CI = 1.12 to 2.95) when the contribution of all of these covariates was included in the Cox regression model.
Table 4.
Gender-specific hazard ratios of IHD, separately for the four 5′ genotype groups before and after the three blood measures of dyslipidemia, other established risk factors of IHD and the three isoforms of the APOE gene, are considered in the modeling separately for females (N = 3,686) and males (N = 2,772)
| Females | |||||
|---|---|---|---|---|---|
| Genotype Groups | N (# w/IHD) | Genotype Only | Genotype HDL-C, TG,T-C | Genotype, HDL-C,TG,T-C, Smoking, Diabetes, Hypertension | Genotype, HDL-C,TG,T-C, Smoking, Diabetes, Hypertension, APOE Isoforms |
| 5′ Promoter Region | |||||
| A560T832/A560T832 | 583 | 0.972 | 0.986 | 0.978 | 0.919 |
| (36) | 0.67–1.42 | 0.67–1.44 | 0.67–1.44 | 0.61–1.38 | |
| A560T832/A560G832 | 1290 | 1.164 | 1.199 | 1.253 | 1.200 |
| (95) | 0.88–1.54 | 0.91–1.58 | 0.95–1.66 | 0.89–1.61 | |
| A560T832/T560T832 | 213 | 1.686 * | 1.754 * | 1.910 * | 1.814 * |
| (21) | 1.05–2.70 | 1.10–2.81 | 1.19–3.07 | 1.12–2.95 | |
| OTHERS | 1600 | 1 | 1 | 1 | 1 |
| (105) | |||||
| Males | |||||
| Genotype Groups | N (# w/IHD) | Genotype Only | Genotype HDL-C,TG,T-C | Genotype, HDL-C,TG,T-C, Smoking, Diabetes, Hypertension | Genotype, HDL-C,TG,T-C, Smoking, Diabetes, Hypertension, APOE Isoforms |
| 5′ Promoter Region | |||||
| A560T832/A560T832 | 416 | 0.992 | 0.995 | 0.934 | 0.942 |
| (45) | 0.71–1.39 | 0.71–1.40 | 0.67–1.31 | 0.66–1.35 | |
| A560T832/A560G832 | 1010 | 1.098 | 1.111 | 1.089 | 1.083 |
| (120) | 0.86–1.40 | 0.87–1.42 | 0.85–1.40 | 0.84–1.40 | |
| A560T832/T560T832 | 174 | 0.991 | 1.021 | 0.979 | 0.954 |
| (18) | 0.61–1.62 | 0.62–1.67 | 0.60–1.61 | 0.58–1.58 | |
| OTHERS | 1172 | 1 | 1 | 1 | 1 |
| (136) |
Significant at the * α ≤ 0.05 level of probability.
In order to further characterize the observed gender specific utility of the three 5′ genotypes of APOE for predicting development of IHD, we next estimated probabilities of not having IHD at a particular age, by employing a stratified Cox regression model without covariates. The relevant survival curves for each of the three high risk 5′ genotypes of APOE and the reference group of individuals having all other 5′ genotypes are given in Fig. 2, separately for females and males. More IHD events were observed in males under 55 years of age than in females under that age. The decrease in the estimated probability of not having IHD with age was less steep in females than in males in each of the four genotype groups. In females, the decrease in the estimated probability of not having IHD by age was steepest in the high risk A560T832/T560T832 genotype, particularly in old age.
Figure 2.
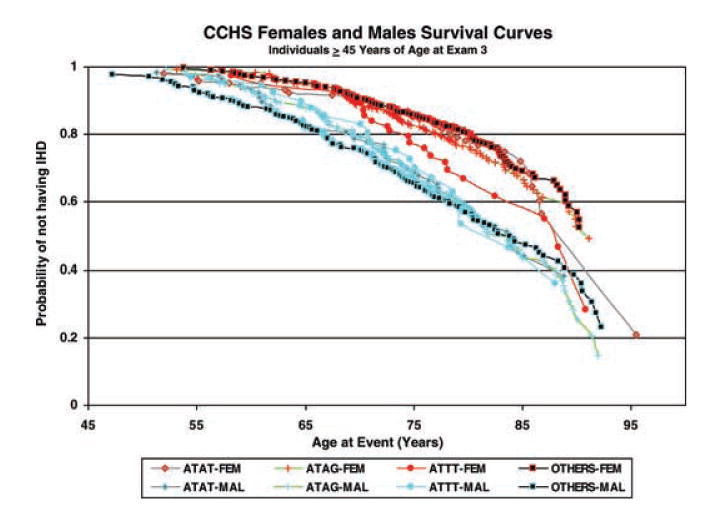
Gender and genotype-specific probabilities of not having IHD at a particular age by employing a stratified Cox regression model without covariates.
Discussion
Modeling Strategies
A promise of the postgenomic era is that fast and inexpensive gene-measurement technologies will provide DNA information that can be used in risk assessment, by medical and public health care delivery systems that are designed to treat and/or to prevent IHD and other common chronic diseases that have a complex multifactorial etiology (Sing et al. 2004; Guttmacher & Collins, 2005; Van Asselt et al. 2006). The application of such high throughput technologies is providing a plethora of variation in genes that influence biochemical and physiological traits that connect genome variation with variation in the risk for atherosclerosis (Crawford et al. 2004; Nickerson, 2005; Fullerton et al. 2000, 2004; Nickerson et al. 2000; Knoblauch et al. 2004; Nabel 2003). Statistical evaluations of the impact of these genomic variations on risk of disease have thus far been less successful. Many of the single-SNP genotype variations that have been identified as predictors of particular atherosclerotic risk factor traits and/or disease endpoints have not survived further testing in studies of independently ascertained samples (Ioannidis et al. 2001; Freimer & Sabatti, 2004). The hypothesized predictors may not survive further testing because there is heterogeneity of the phenotype-genotype relationship among populations, or because there is a lack of statistical power to detect the modest effects of single-SNP genotypes in the small samples that have been studied. It is also possible that a hypothesized genotype effect will not survive further testing in any population, because it was suggested from the study of only one population and the effect in that study was a false positive, i.e. a type I statistical error (Sing et al. 2004; Ioannidis et al. 2001; Freimer & Sabatti, 2004).
We have suggested that an ecological, multiple population, data-mining strategy be used to identify those hypothesized phenotype-genotype relationships that are less likely to be type I errors and more likely to be of utility for predicting risk of disease in an unstudied population (Stengård et al. 2006). This expectation has its roots in the composibility concept, proposed by the German mathematician and rational philosopher Leibniz (Russell, 1945). Previously we employed this strategy to select the two public non-coding SNPs of the APOE gene, located in the 5′ promoter region at positions 560 and 832, for consideration in the study of IHD reported here. These SNPs define a group of three genotypes, A560T832/A560T832, A560T832/A560G832 and A560T832/T560T832, that distinguish between high and low levels of HDL-C, TG and/or T-C in samples from four populations that represented gender, ethnic and geographic diversity, including the large population-based sample from the CCHS (Stengård et al. 2006). Here we tested the hypothesis that the three 5′ genotypes that predict dyslipidemia in the four populations are also predictors of IHD in the CCHS sample. We found that a significant increase (p<0.05) in the hazard of IHD was associated with one of the three hypothesized high risk 5′ genotypes of APOE, A560T832/T560T832, in Danish females when compared with the hazard in females who did not have any of the proposed high risk genotypes. This increase remained statistically significant after fitting the effects of dyslipidemia, other known risk factors of IHD, and genotypes defined by variations in the two coding SNPs at positions 3937 and 4075.
Biological Plausibility of Selected Model
The role of the ApoE molecule in lipid metabolism serves as a rationale for evaluating the statistical associations between inter-individual differences in the risk of atherosclerotic disease endpoints and variation in the APOE gene. ApoE serves as a ligand for a low-density lipoprotein/ApoE receptor that modulates the clearance of circulating lipoprotein particles (Davignon et al. 1999). Variation in receptor affinity for the ApoE molecule depends on structural variation determined by coding SNPs located at positions 3937 and 4075 in exon four of the APOE gene. It has been suggested that this structural variation influences interindividual differences in both post-prandial cholesterol absorption (Davignon et al. 1999; Miettinen et al. 1992) and hepatic cholesterol uptake (Davignon et al. 1999) which, in turn, is expected to explain a fraction of the inter-individual differences in blood T-C concentration and variation in the risk of atherosclerotic disease endpoints (Davignon et al. 1999). The highest risk of atherosclerosis has been associated with the E4 isoform (Davignon et al. 1999; Stengård et al. 1995; Song et al. 2004). On average, the odds of cardiovascular disease are 1.43 times higher in individuals with this isoform than in individuals who do not have it (Song et al. 2004). In our study the estimate of the hazard of IHD was 1.13 times higher in females, and 0.90 lower in males, who had the E4 isoform. These estimates were not significantly different from one. The failure for the E4 isoform to predict significant variation in atherosclerotic endpoints in our study of the Copenhagen sample, and in some other studies (Song et al. 2004; Lehtinen et al. 1995), is consistent with the biological reality that the phenotypic effects of APOE genotype variation on risk of IHD depend on contexts indexed by factors such as smoking (Davignon et al. 1999; Talmud et al. 2005; Stengård et al. 1999), gender (Davignon et al. 1999; Frikke-Schmidt et al. 2000b) and population-specific genetic and environmental factors (Davignon et al. 1999; Stengård et al. 1995, 1999) that influence IHD risk.
Variations at positions 560 and 832 of the APOE promoter region influence transcriptional activity of the gene both in in vitro assays (Artiga et al. 1998) and in ex vivo measurements of the APOE mRNA expression level in post mortem brain tissue (Lambert et al. 2005). On average, the T560T832 haplotype has 60%, and the A560T832 haplotype 30%, lower promoter activity than the A560G832 haplotype (Artiga et al. 1998). These haplotype variations may confer in vivo differences in expression, inducibility and/or tissue specificity of the human APOE gene (Artiga et al. 1998). These adaptive responses to the internal metabolic environment serve as a biological explanation for the observed association of interindividual differences in blood lipid concentrations with variations in the 5′ regulatory region of the APOE gene across populations (Stengård et al. 2002, 2006). This inference is supported by studies that suggest that there are estrogen response elements in the vicinity of the 832 promoter SNP, and that variation in this site modulates transcriptional activity of the APOE gene in response to estrogen (Lambert et al. 2004). The observed gender-specific phenotype effects of the APOE gene found in our study are consistent with these observations. Our finding is also consistent with evidence that the natural history of cardiovascular diseases (Barrett-Connor & Khaw, 1984) and genetic effects, in general, are gender specific (Ober et al. 2003).
An increased risk of atherosclerotic disease endpoints in those individuals with the T832/T832 single-SNP genotype has been previously reported (Lambert et al. 2000; Viitanen et al. 2001; Ye et al. 2003). These studies reported that the odds of coronary artery disease or myocardial infarction in individuals with the T832 allele were approximately 1.25 to 1.60 times higher than odds of these atherosclerotic disease endpoints in individuals who did not have this allele. In our study the HR for IHD in individuals with the T832 allele was 1.14 for females and 1.08 for males. Neither one of these estimates was significantly different from one (p>0.05). There was, however, a subset of Danish females who had the T832 allele, those with the A560T832/T560T832 two-SNPs genotype, who had approximately 1.8 times higher hazard of IHD than females who did not have any of the three high risk genotypes, A560T832/A560T832, A560T832/A560G832 and A560T832/T560T832. The statistical significance of risk of atherosclerotic diseases in individuals with the T832 allele was not influenced by dyslipidemia in our study, or in other studies (Lambert et al. 2000; Viitanen et al. 2001; Ye et al. 2003). These findings are consistent with the 5′ genotypes having in vivo cellular effects which have phenotypic implications, such as reduced cholesterol efflux from macrophages and an abundant accumulation of lipid-laden foam cells, which can contribute to the development of atherosclerosis (Ye et al. 2003).
Utility of the 5′ Genotypes of APOE as Diagnostic and Screening Tests
The prevailing paradigm in translating medical research into the practice of medicine is to begin by reducing different human physiological and metabolic systems to their presumed essential component agents, modeling the relationships between these agents and the biological outcome of interest by physical, chemical and/or statistical methods, and testing the predictive utility of the hypothesized models in samples from different populations (Rea et al. 2006). Various expert panels are then convened to review the research findings and to formulate guidelines that are expected to help physicians to weight the benefits and risks of using a particular agent as a diagnostic or screening test in everyday clinical decision-making. European guidelines on cardiovascular disease prevention in clinical practice (De Backer et al. 2003) serve as an example of recommendations from such an expert panel. Usually such guidelines recommend those agents that can be used to predict an outcome of interest in a broad range of human populations. Our ecological, multi-population, data-mining strategy has the potential to identify such agents. It has, however, shortcomings that need to be considered and evaluated. For instance, it focuses only on the genetic and environmental agents that vary in all populations. Only ten out of the twenty APOE SNPs considered in building the 5′ genotype model segregate in all four of the populations studied (Fullerton et al. 2000; Nickerson et al. 2000; Stengård et al. 2002, 2006). And our strategy ignores inter-population differences in the relative frequencies of the SNPs that do segregate in all populations, and the heterogeneity of specific gene-gene and gene-environment interactions among populations. Guidelines that are based on diagnostic or screening tests that do not account for this heterogeneity of the genetic architecture of disease among human populations are not expected to be the best choices for predicting atherosclerotic disease endpoints in a particular individual in a particular population.
An evaluation of the expected benefits of a particular agent as a diagnostic or screening test can be made by estimating its positive and negative predictive values, +PV and −PV, respectively (Fletcher & Fletcher, 2005). The +PV reflects the probability of the onset of the disease of interest in individuals who have a particular value of the proposed agent, and the −PV reflects a probability that an individual does not have the disease if she/he does not have the value of that agent. The +PV is related to sensitivity and specificity of a particular diagnostic or screening test, and to the prevalence of the trait of interest, through a mathematical formula that is derived from the application of the Bayes' theorem of conditional probabilities (Fletcher & Fletcher, 2005). An estimate of +PV for the high risk A560T832/T560T832 genotype in middle-aged and elderly Danish females in this study is 0.099, which is slightly higher than the estimated cumulative incidence of IHD, 0.070, in the overall sample of Danish females in this age group. Such a modest difference between these two estimates is consistent with the small contribution of a single gene variation to predicting the onset of atherosclerosis in the population at large. The estimate of the +PV for an established risk factor such as smoking status, 0.079 in our study, argues that guidelines that use the A560T832/T560T832 genotype as their only risk indicator could be only slightly better than the one that use only smoking status in a risk assessment in middle aged and elderly Danish females. The estimate of the −PV for the reference 5′ genotypes in this study is 0.934, which does not differ from 0.930 for middle-aged and elderly Danish females. This comparison suggests that the three high risk 5′ genotypes of APOE will not be useful in ruling out an increased risk of IHD in this group of Danish females.
Mathematically +PV and −PV depend on the relative frequency of the disease endpoint of interest in the particular population of interest. One possibility for improving the yield of a diagnostic or screening test is to apply it in subpopulations where the risk of the related disease end-point is highest (Fletcher & Fletcher, 2005). We evaluated the latter possibility by estimating +PV for those diabetic females with the A560T832/T560T832 genotype. The estimate of the +PV for this subgroup is 0.375, which is 5 and 2 times higher than the estimates of 0.070 and 0.189 for the cumulative incidences of IHD in the overall sample of Danish females and in the subsample of diabetic Danish females, respectively.
There are many political, economic, social, and institutional realities that need to be considered for implementing a diagnostic or screening test in a particular population. Knowledge of the test's +PV (or −PV) is only one of them. A useful screening test is expected to be simple, cheap, safe and acceptable to both patients and clinicians (Fletcher & Fletcher, 2005). Efficacious and effective pharmacological and/or non-pharmacological interventions are also expected to be available if a screening test is implemented. Many of the established risk factors for atherosclerosis have been good candidates for screening tests. For example, information about an individual's smoking status is inexpensive, and a smokers' cardiovascular health can be improved by simply quitting smoking. Knowledge about one's A560T832/T560T832 genotype does not have such obvious therapeutic implications. Hence, smoking status must be considered to be a better indicator of IHD risk in the population at large than the A560T832/T560T832 genotype, despite the fact that it has a lower +PV than the proposed high risk genotype. There may, however, be subgroups of individuals, such as diabetic females, who will benefit from the A560T832/T560T832 genotype testing. Our observation in the CCHS, that approximately 40% of diabetic women who have this high risk genotype are expected to experience IHD within the next eight years, suggests that this subset of individuals should receive more focused management of the established risk factors for atherosclerosis than information about diabetes alone would indicate.
Conclusions
Our study suggests that both regulatory and structural variations should be considered when evaluating the utility of variation in the APOE gene for predicting atherosclerotic disease endpoints. We found that Danish females with the A560T832/T560T832 genotype are at increased risk for developing IHD. This increase remained after fitting the effects of dyslipidemia, other established risk factors of IHD, or isoforms defined by variations in the two non-synonymous coding SNPs in the APOE gene at positions 3937 and 4075. However, the low positive predictive value of this 5′ genotype suggests that it would be a poor choice as a screening test in the population at large. By contrast, there may be clinically defined subpopulations, such as diabetic Danish females, who may benefit from gene testing. Extrapolation of our findings to other unstudied populations needs to be done with caution, because there may be context(s) in other populations, such as male gender in Denmark, where variation in the 5′ promoter region of the APOE gene does not contribute significantly to prediction of IHD. Our findings do justify, however, consideration of the 5′ genotypes of the APOE gene in studies designed to identify predictors of IHD and/or sets of agents that have the greatest potential as diagnostic and/or screening tests in a particular population of interest. The characterization of which individuals in which population can benefit from a particular gene test is a challenge we face in the post-genome era.
Acknowledgments
We wish to thank Ken Weiss for his devotion to carrying out the statistical analyses and Tom Rea for his expert insights into the role of lipid metabolism in atherosclerosis. We are especially grateful for Lynn Illeck's efforts in assembling this manuscript for publication. This work was supported by National Institutes of Health Grants HL-072905, GM-066509, HL-051021, HL-039107, HL-058238, HL-058239 and HL-058240.
References
- Artiga MJ, Bullido MJ, Sastre I, et al. Allelic polymorphisms in the transcriptional regulatory region of apolipoprotein E gene. FEBS Lett. 1998;421:105–108. doi: 10.1016/s0014-5793(97)01543-3. [DOI] [PubMed] [Google Scholar]
- Barrett-Connor E, Khaw KT. Family history of heart attack as an independent predictor of death due to cardiovascular disease. Circulation. 1984;69:1065–1069. doi: 10.1161/01.cir.69.6.1065. [DOI] [PubMed] [Google Scholar]
- Crawford DC, Carlson CS, Rieder MJ, et al. Haplotype diversity across 100 candidate genes for inflammation, lipid metabolism, and blood pressure regulation in two populations. Am J Hum Genet. 2004;74:610–622. doi: 10.1086/382227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davignon J, Cohn JS, Mabile L, et al. Apolipoprotein E and atherosclerosis: insight from animal and human studies. Clin Chim Acta. 1999;286:115–143. doi: 10.1016/s0009-8981(99)00097-2. [DOI] [PubMed] [Google Scholar]
- Davignon J, Gregg RE, Sing CF. Apolipoprotein E polymorphism and atherosclerosis. Arteriosclerosis. 1988;8:1–21. doi: 10.1161/01.atv.8.1.1. [DOI] [PubMed] [Google Scholar]
- De Backer G, Ambrosioni E, Borch-Johnsen K, et al. European guidelines on cardiovascular disease prevention in clinical practice: Third joint task force of European and other societies on cardiovascular disease prevention in clinical practice. Eur Heart J. 2003;24:1601–1610. doi: 10.1016/s0195-668x(03)00347-6. [DOI] [PubMed] [Google Scholar]
- Everitt BS. Statistical methods for medical investigations. New York, NY: Halsted Press; 1994. [Google Scholar]
- Fletcher RW, Fletcher SW. Clinical epidemiology; the essentials. Philadelphia, PA: Lippincott Williams & Wilkins; 2005. [Google Scholar]
- Freimer N, Sabatti C. The use of pedigree, sib-pair and association studies of common diseases for genetic mapping and epidemiology. Nat Genet. 2004;36:1045–1051. doi: 10.1038/ng1433. [DOI] [PubMed] [Google Scholar]
- Frikke-Schmidt R, Nordestgaard BG, Agerholm-Larsen B, et al. Context-dependent and invariant associations between lipids, lipoproteins, and apolipoproteins and apolipoprotein E genotype. J Lipid Res. 2000a;41:1812–1822. [PubMed] [Google Scholar]
- Frikke-Schmidt R, Sing CF, Nordestgaard BG, et al. Subsets of SNPs define rare genotype classes that predict ischemic heart disease. Hum Genet. 2007;120:865–877. doi: 10.1007/s00439-006-0233-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frikke-Schmidt R, Sing CF, Nordestgaard BG, et al. Gender- and age-specific contributions of additional DNA sequence variation in the 5′ regulatory region of the APOE gene to prediction of measures of lipid metabolism. Hum Genet. 2004;115:331–345. doi: 10.1007/s00439-004-1165-z. [DOI] [PubMed] [Google Scholar]
- Frikke-Schmidt R, Tybjærg-Hansen A, Steffensen R, et al. Apolipoprotein E genotype: epsilon32 women are protected while epsilon43 and epsilon44 men are susceptible to ischemic heart disease: the Copenhagen City Heart Study. J Am Coll Cardiol. 2000b;35:1192–1199. doi: 10.1016/s0735-1097(00)00520-9. [DOI] [PubMed] [Google Scholar]
- Fullerton SM, Buchanan AV, Sonpar V, et al. The effects of scale: variation in the APOA1/C3/A4/A5 gene cluster. Hum Genet. 2004;115:36–56. doi: 10.1007/s00439-004-1106-x. [DOI] [PubMed] [Google Scholar]
- Fullerton SM, Clark AG, Weiss KM, et al. Apolipoprotein E variation at the sequence haplotype level: implications for the origin and maintenance of a major human polymorphism. Am J Hum Genet. 2000;67:881–900. doi: 10.1086/303070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttmacher AE, Collins FS. Realizing the promise of genomics in biomedical research. JAMA. 2005;294:1399–1402. doi: 10.1001/jama.294.11.1399. [DOI] [PubMed] [Google Scholar]
- Hixson JE, Vernier DT. Restriction isotyping of human apolipoprotein E by gene amplification and cleavage with Hha I. J Lipid Res. 1990;31:545–548. [PubMed] [Google Scholar]
- Ioannidis JP, Ntzani EE, Trikalinos TA, et al. Replication validity of genetic association studies. Nat Genet. 2001;29:306–309. doi: 10.1038/ng749. [DOI] [PubMed] [Google Scholar]
- Knoblauch H, Bauerfeind A, Toliat MR, et al. Haplotypes and SNPs in 13 lipid-relevant genes explain most of the genetic variance in high-density lipoprotein and low-density lipoprotein cholesterol. Mol Genet. 2004;13:993–1004. doi: 10.1093/hmg/ddh119. [DOI] [PubMed] [Google Scholar]
- Lambert JC, Brousseau T, Defosse V, et al. Independent association of an APOE gene promoter polymorphism with increased risk of myocardial infarction and decreased APOE plasma concentrations-the ECTIM study. Hum Mol Genet. 2000;9:57–61. doi: 10.1093/hmg/9.1.57. [DOI] [PubMed] [Google Scholar]
- Lambert JC, Coyle N, Lendon C. The allelic modulation of apolipoprotein E expression by oestrogen: potential relevance for Alzheimer's disease. J Med Genet. 2004;41:104–112. doi: 10.1136/jmg.2003.005033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert JC, Mann D, Richard F, et al. Is there a relation between APOE expression and brain amyloid load in Alzheimer's disease? J Neurol Neurosurg Psychiatry. 2005;76:928–933. doi: 10.1136/jnnp.2004.048983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehtinen S, Lehtimäki T, Sisto T, et al. Apolipoprotein E polymorphism, serum lipids, myocardial infarction and severity of angiographically verified coronary artery disease in men and women. Atherosclerosis. 1995;114:83–91. doi: 10.1016/0021-9150(94)05469-y. [DOI] [PubMed] [Google Scholar]
- Lussier-Cacan S, Bolduc A, Xhignesse M, et al. Impact of alcohol intake on measures of lipid metabolism depends on context defined by gender, body mass index, cigarette smoking, and Apolipoprotein E genotype. Arterioscler Thromb Vasc Biol. 2002;22:824–831. doi: 10.1161/01.atv.0000014589.22121.6c. [DOI] [PubMed] [Google Scholar]
- Mahley RW. Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science. 1988;240:622–630. doi: 10.1126/science.3283935. [DOI] [PubMed] [Google Scholar]
- Mahley RW, Rall SC., Jr . Apolipoprotein E: far more than a lipid transport protein. In: Lander E, Page D, Lifton E, editors. Annual review of genomics and human genetics. Palo Alto, CA: Annual Reviews Inc; 2000. pp. 507–538. [DOI] [PubMed] [Google Scholar]
- Miettinen TA, Gylling H, Vanhanen H, et al. Cholesterol absorption, elimination, and synthesis related to LDL kinetics during varying fat intake in men with different apoprotein E phenotypes. Arterioscler Thromb. 1992;12:1044–1052. doi: 10.1161/01.atv.12.9.1044. [DOI] [PubMed] [Google Scholar]
- Nabel EG. Cardiovascular disease. N Engl J Med. 2003;349:60–72. doi: 10.1056/NEJMra035098. [DOI] [PubMed] [Google Scholar]
- National Cholesterol Education Program National Heart, Lung, and Blood Institute. Third Report of the National Cholesterol Education Program (NCEP) Expert panel on detection, evaluation, and treatment of high blood cholesterol in adults (Adult Treatment Panel III) final report. Washington, DC: 2002. Sep, NIH Publication No 02-5215. [PubMed] [Google Scholar]
- Nelson MR, Kardia SL, Ferrell R, et al. A CPM to identify multilocus genotypic partitions that predict quantitative trait variation. Genome Res. 2001;11:458–470. doi: 10.1101/gr.172901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickerson DA. Department of Genome Sciences University of Washington. 2005. Nickerson Group; MDECODE; SNP data: //droog.gs.washington.edu/mdecode. [Google Scholar]
- Nickerson DA, Taylor SL, Fullerton SM, et al. Sequence diversity and large scale typing of SNPs in the human apolipoprotein E gene. Genome Res. 2000;10:1532–1545. doi: 10.1101/gr.146900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ober C, Aldrich CL, Chervoneva I, et al. Variation in the HLA-G promoter region influences miscarriage rates. Am J Hum Genet. 2003;72:1425–1435. doi: 10.1086/375501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rea TJ, Brown CM, Sing CF. Complex adaptive system models and the gene analysis of plasma HDL-Cholesterol concentration. Perspect Biol Med. 2006;49:490–503. doi: 10.1353/pbm.2006.0063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell B. A history of western philosophy. New York, NY: Simon and Schuster; 1945. [Google Scholar]
- Schnohr P, Jensen G, Lange P, et al. The Copenhagen City Heart Study. Tables with data from the third examination 1991-1994. Eur Heart J. 2001;3 H:1–83. [Google Scholar]
- Sing CF, Stengård JH, Kardia SLR. Dynamic relationships between the genome and exposures to environments as causes of common human diseases. In: Simopoulos AP, Ordovas JM, editors. Nutrigenetics and Nutrigenomics (World Review of Nutrition and Dietetics) Vol. 93. Switzerland: Karger; 2004. pp. 77–91. [DOI] [PubMed] [Google Scholar]
- Sokal RR, Rohlf FJ. Biometry: the principles and practice of statistics in biological research. 3rd. New York, NY: WH Freeman and Company; 1995. [Google Scholar]
- Song Y, Stampfer MJ, Liu S. Meta-analysis: apolipoprotein e genotypes and risk for coronary heart disease. Ann Intern Med. 2004;141:137–147. doi: 10.7326/0003-4819-141-2-200407200-00013. [DOI] [PubMed] [Google Scholar]
- Sposito AC, Gonbert S, Turpin G, et al. Common promoter C516T polymorphism in the ApoB gene is an independent predictor of carotid atherosclerotic disease in subjects presenting a broad range of plasma cholesterol levels. Arterioscler Thromb Vasc Biol. 2004;24:2192–2195. doi: 10.1161/01.ATV.0000144810.10164.50. [DOI] [PubMed] [Google Scholar]
- Steinberg D. An interpretive history of the cholesterol controversy: part 1. J Lipid Res. 2004;45:1583–1593. doi: 10.1194/jlr.R400003-JLR200. [DOI] [PubMed] [Google Scholar]
- Stengård JH, Clark AG, Weiss KM, et al. Contributions of 18 additional DNA sequence variations in the gene encoding apolipoprotein E to explaining variation in quantitative measures of lipid metabolism. Am J Hum Genet. 2002;S71:501–517. doi: 10.1086/342217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stengård JH, Kardia SLR, Hamon SC, et al. Contribution of regulatory and structural variations in APOE to predicting dyslipidemia. J Lipid Res. 2006;47:318–328. doi: 10.1194/jlr.M500491-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stengård JH, Kardia SLR, Tervahauta M, et al. Utility of the predictors of coronary heart disease mortality in a longitudinal study of elderly Finnish men aged 65 to 84 years is dependent on context defined by Apo E genotype and area of residence. Clin Genet. 1999;56:367–377. doi: 10.1034/j.1399-0004.1999.560505.x. [DOI] [PubMed] [Google Scholar]
- Stengård JH, Zebra KE, Pekkanen J, et al. Apolipoprotein E polymorphism predicts death from coronary heart disease in a longitudinal study of elderly Finnish men. Circulation. 1995;91:265–269. doi: 10.1161/01.cir.91.2.265. [DOI] [PubMed] [Google Scholar]
- Talmud PJ, Stephens JW, Hawe E, et al. The significant increase in cardiovascular disease risk in APOE ∈ 4 carriers is evident only in men who smoke: Potential relationship between reduced antioxidant status and ApoE4. Ann Hum Genet. 2005;69:1–10. doi: 10.1111/j.1529-8817.2005.00205.x. [DOI] [PubMed] [Google Scholar]
- Utermann G, Hees M, Steinmetz A. Polymorphism of apolipoprotein E and occurrence of dysbetalipoproteinemia in man. Nature. 1977;269:604–607. doi: 10.1038/269604a0. [DOI] [PubMed] [Google Scholar]
- van Asselt KM, Kok HS, van der Schouw YT, et al. Role of genetic analyses in cardiology: Part II: Heritability estimation for gene searching in multifactorial diseases. Circulation. 2006;113:1136–1139. doi: 10.1161/CIRCULATIONAHA.105.563197. [DOI] [PubMed] [Google Scholar]
- Viiri LE, Loimaala A, Nenonen A, et al. The association of the apolipoprotein E gene promoter polymorphisms and haplotypes with serum lipid and lipoprotein concentrations. Atherosclerosis. 2005;179:161–167. doi: 10.1016/j.atherosclerosis.2004.10.004. [DOI] [PubMed] [Google Scholar]
- Viitanen L, Pihlajamäki J, Miettinen R, et al. Apolipoprotein E gene promoter (-219G/T) polymorphism is associated with premature coronary heart disease. J Mol Med. 2001;79:732–737. doi: 10.1007/s001090100265. [DOI] [PubMed] [Google Scholar]
- Ye S, Dunleavey L, Bannister W, et al. Independent effects of the -219G > T and ∈2/∈3/∈4 polymorphisms in the apolipoprotein E gene on coronary artery disease: the Southampton Atherosclerosis Study. Eur J Hum Genet. 2003;11:437–443. doi: 10.1038/sj.ejhg.5200983. [DOI] [PubMed] [Google Scholar]