

Abstract
BACKGROUND/AIMS
Studies in animal models and humans suggest a link between endotoxemia and non-alcoholic steatohepatitis. Since Kupffer cells are responsible for clearing endotoxin and are activated via endotoxin interaction with Toll-like receptor 4 (TLR-4), we examined the relationship between hepatic TLR-4 expression and Kupffer cell content during the genesis of steatohepatitis.
METHODS
Male C57BL/6, C3H/HouJ and TLR-4 mutant C3H/HeJ mice were fed control or methionine/choline-deficient diet (MCDD). In one group of C57BL/6 mice, Kupffer cells were depleted by weekly intravenous injections of clodronate liposomes. After 3 weeks, serum ALT activity and portal endotoxin levels were measured. Real-time PCR was used to examine mRNA expression of TLR-4, TLR-2, CD14, MD-2, TGFβ, TNFα CD36 PPAR-α, liver fatty acid binding protein (L-FABP) and collagen α1.
RESULTS
We observed histological evidence typical of steatohepatitis, portal endotoxemia and enhanced TLR-4 expression in wild type mice fed MCDD. In contrast, injury and lipid accumulation markers were significantly lower in TLR-4 mutant mice. Destruction of Kupffer cells with clodronate liposomes blunted histological evidence of steatohepatitis and prevented increases in TLR-4 expression.
CONCLUSIONS
These findings demonstrate the importance of TLR-4 signaling and underscore a direct link between TLR-4 and Kupffer cells in the pathogenesis of steatohepatitis.
Keywords: Toll-like receptors, non-alcoholic steatohepatitis, Clodronate liposomes, hepatic inflammation, fibrosis, endotoxin
1. Introduction
Non-alcoholic steatohepatitis (NASH) in the setting of obesity is believed to occur via a “two-hit” mechanism involving hepatic fat accumulation and oxidative stress. Studies in animal models suggest that gut-derived endotoxin mediates oxidative stress and the development of NASH. In support of the idea that gut-derived bacterial products play a causal role in the pathogenesis of NASH in genetically overweight mice, treatment with probiotic dietary supplements that sterilized the gut of endotoxin-bearing gram negative organisms prevented histological alterations and insulin resistance associated with steatohepatitis (1–3). In humans, severe steatohepatitis often occurs following jejuno-ileal bypass and in patients placed on total parenteral nutrition, both of which are situations believed to cause bacterial overgrowth and endotoxemia. In fact, Wigg et al. evaluated 22 patients with NASH for small bowel bacterial content and found that bacterial overgrowth was prevalent among NASH patients (4). In a study by Vanderhoof et al. liver injury and steatosis in a rodent model of jejuno-ileal bypass was prevented by therapies that minimized bacterial load (5). Thus, leakage of endotoxin from the gut is a likely stimulus of NASH.
Since blood leaving the gut empties directly into the portal vein, the liver is exposed to gut-derived endotoxin. As a result of endotoxemia, Kupffer cells, the resident hepatic macrophages, are activated via the Toll-like receptor 4 complex (TLR-4) on the cell surface. This receptor is a member of the Toll-like family of pattern recognition receptors that are of central importance during host defense against invading pathogens. TLR-4 interaction with endotoxin results in the release of a myriad of pro-inflammatory mediators that induce hepatic injury and fibrosis (6). In addition, cytokines have profound effects on lipid metabolism (7).
The present study investigated the potential importance of Kupffer cells and TLR-4 in the pathogenic mechanisms underlying NASH. Accordingly, mice were fed a methionine and choline deficient diet (MCDD), which results in hepatic microvascular dysfunction and pronounced pathological changes within 3–4 weeks (8–10). Administration of this diet in mice depleted of Kupffer cells via administration of liposome encapsulated clodronate as well as in mice deficient in TLR-4 signaling due to a spontaneous mutation (i.e. C3H/HeJ mice) provides evidence that TLR-4 expression on Kupffer cells is a critical component of diet-induced NASH.
2. Materials and methods
2.1. Animal treatment
Male C57BL/6, C3H/HouJ and C3H/HeJ mice were purchased from Jackson laboratories at 4–6 weeks of age. C3H/HouJ and C3H/HeJ mice are both substrains of the C3H parent strain, which was derived by breeding Bagg albino female with DBA male mice. They were given free access to water and food, and maintained on a 12 hour light/dark cycle. The control diet (CD) used in this study was a standard amino acid-defined formulation (Dyets, Inc. Bethlehem, PA). Steatohepatitis was induced by 3 weeks of feeding a variation of the base diet that was deficient in methionine and choline. All mice received humane care in accordance with the handling protocol approved by the Louisiana State University Health Sciences Center animal care and use committee.
2.2. Kupffer cell depletion
Two days prior to feeding, one experimental series of C57BL/6 mice (n= 6/dietary group) was treated with clodronate liposomes via tail vein injection to deplete Kupffer cells. The clodronate was a gift of Roche Diagnostics GmbH (Mannheim, Germany) and was encapsulated into liposomes as described previously (11). Each mouse received 200 µl of a 1 mg/ml suspension. This dose had been shown to selectively deplete Kupffer cells and some splenic macrophages within 24h (12, 13). Additional 5 mice (n= 5/diet) were administered liposome-encapsulated PBS as a control. Repopulation of Kupffer cells was prevented by weekly administration of clodronate liposomes.
2.3. Assessment of liver injury
At the time of sacrifice, a small section of each liver was preserved in zinc-buffered formalin. Routine H&E staining was performed to assess tissue architecture. Steatosis, inflammation and necrosis were scored by a one of the authors (P.A.) that was blinded to the study design. The absence of these histopathological features was scored as 0 and the most severe changes were given a score of 3. Blood samples were collected from the vena cava and serum stored at −80°C. Alanine aminotransferase (ALT) measured according to standard enzymatic assay (Thermo electron corporation, Waltham, MA).
2.4. Endotoxin determination
A heparinized blood sample was collected directly from the portal vein. The platelet-rich plasma fraction was prepared as described previously (14). Endotoxin was determined using a kinetic chromagenic assay (Cambrex, Walkersville, MD).
2.5. Immunohistochemistry
Prior to staining, epitope unmasking was performed by immersing sections in antigen retrieval solution A (BD Biosciences; San Jose, CA) and heating for 30 min. Antibodies directed against neutrophils (Gr-1) and macrophages (F4/80) obtained from AbD Serotec (Raleigh, NC) were applied at room temperature for 60 min followed by sequential application of a biotinylated chicken anti-rat secondary antibody streptavidin-conjugated horseradish peroxidase (30 min). Diaminobenzidine was applied for colorimetric detection of the target cells.
2.6. Hepatic triglyceride content
Approximately 200 mg of each liver was homogenized in PBS and then centrifuged at 12,000 × g for 15 min. After determination of the total protein content in each sample using the biuret method (Sigma Chemical Co.), the triglyceride content in the resulting supernatant was measured according to standard enzymatic assay (Thermo electron corporation; Medina, OH) normalized to the protein content.
2.7. Western blotting
Total protein (50µg) was separated on a 4–15% gel and transferred to nitrocellulose membranes. Membranes were blotted with anti-TLR4 (Cell Signaling Technology; Danvers, MA) or β-actin (AbD-Serotec) then incubated with an HRP-conjugated secondary antibody. Target proteins were visualized using ECL-Plus detection reagents (Amersham Biosciences; Piscataway, NJ) in a Chemidoc XRS documentation system (Bio-Rad Laboratories; Hercules, CA).
2.8. Reverse transcription and real-time PCR
Total RNA was extracted from frozen liver samples using the Qiagen RNeasy reagents. Each total RNA sample (250 ng) was reverse transcribed using TaqMan transcription buffer and multiscribe reverse transcriptase (Applied Biosystems; Foster City, CA). The relative mRNA expression of TLR-4, TLR-2, CD14, MD-2, TGFβ, TNFα CD36 PPAR-α, liver fatty acid binding protein (L-FABP) and collagen α1 was analyzed using pre-developed assays for real-time PCR (Applied Biosystems). In a separate tube, ribosomal 18s was amplified as a reference. Gene expression was quantified using a comparative critical threshold (CT) method as described previously (15).
2.9. Data Analysis
Statistical analysis was performed using student’s t-test or two-way ANOVA where appropriate with p < 0.05 as the level of significance. For each parameter tested, at least 4 observations per group were analyzed.
3. Results
3.1 Evidence of endotoxemia and toll-like receptor-4 signaling
The extent of endotoxemia in response to feeding MCDD was examined in the platelet-rich plasma fraction of portal blood samples. In mice fed control diet portal endotoxin levels were 33.9 ± 13.3 pg/ml. Feeding MCDD increased plasma endotoxin by approximately 3-fold (100.7 ± 26.7). To investigate activation of TLR-4 signaling, mRNA expression of components of the TLR-4 pathway were quantified via real-time PCR. TLR-4 expression was increased 5-fold by MCDD (Fig 1A). Western blot analysis confirmed the enhanced presence of TLR-4 protein (Fig. 1B). The expression of the TLR-4 accessory molecules MD-2 and CD14 were also increased significantly after feeding MCDD (Fig. 1A).
Figure 1. Effect of feeding MCDD on TLR-4 signaling molecules.

(A) For assessment of TLR-4, MD2 and CD14 expression, livers were collected from male C57BL/6 mice fed CD or MCDD for 3 weeks. Pre-developed assays for real-time PCR were used according to the manufacturer’s instructions (Applied Biosystems). Expression of each target mRNA was calculated relative to average values in the control group using a comparative CT method and presented as mean ± SEM of at least 4 observations/group. (B) Western blot analysis of hepatic TLR-4 expression relative to β-actin was used to confirm enhanced protein levels. Values are mean ± SEM of at least 4 observations per group. *p< 0.05, **p< 0.005 compared to control using Student’s t test.
3.2 Liver histopathology in TLR-4 mutant mice
Additional experiments were performed in C3H/HeJ mice, which lack TLR-4 signaling due to a spontaneous point mutation. In wild type mice feeding MCDD resulted in extensive macrovesicular steatosis and necrosis typical of steatohepatitis (Fig. 2). These histopathological changes were largely prevented C3H/HeJ mice. To characterize the inflammatory infiltrate, sections of liver were subjected to immunohistochemical staining to identify the presence and distribution of neutrophils and macrophages. Feeding MCDD resulted in the clustering of neutrophils at sites of steatosis and injury in wild type mice (Fig 2E). Macrophage accumulation in close vicinity to injured hepatocytes was distinctly more pronounced (Fig. 2G). The infiltration of both neutrophils and macrophages was blunted in C3H/HeJ mice fed MCDD (Fig. 2F&H).
Figure 2. Hepatic Histopathology.
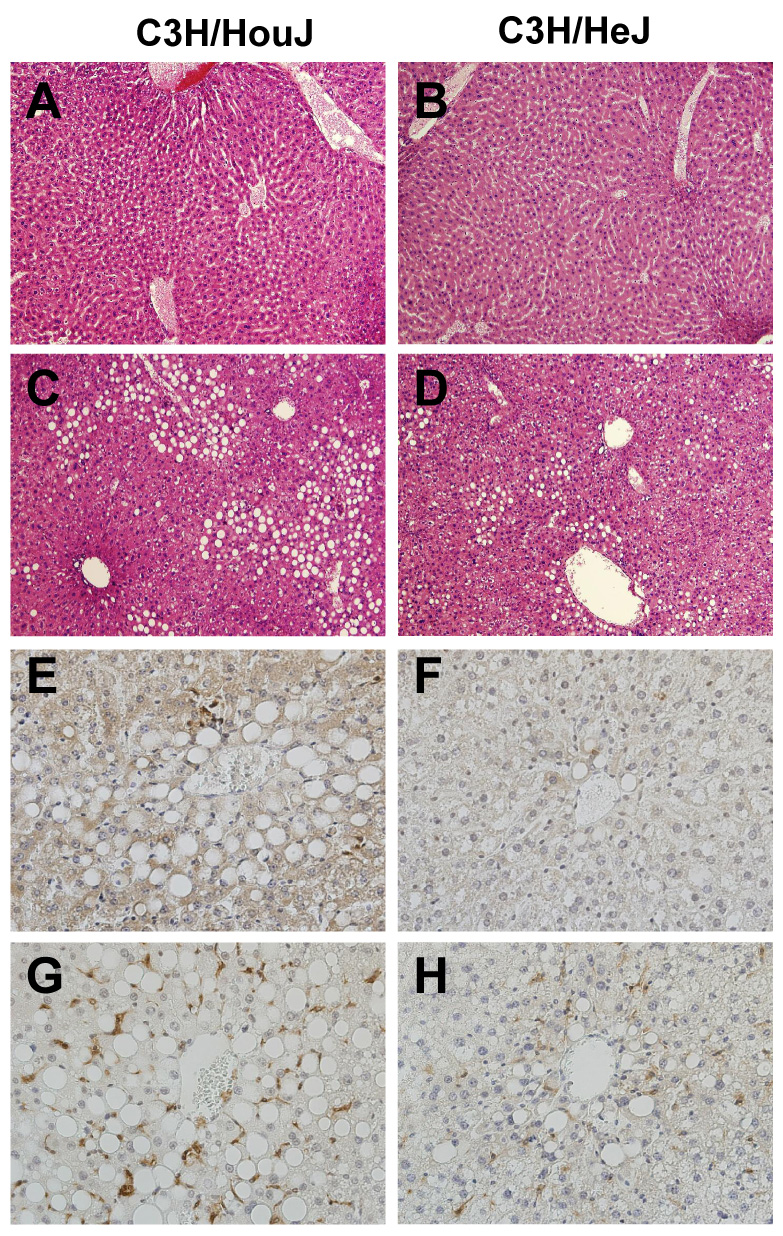
Representative H&E-stained photomicrographs of (A) C3H/HouJ + CD; (B)) C3H/HeJ + CD (C)) C3H/HouJ + MCDD (D) C3H/HeJ + MCDD; endotoxin sensitive (left panels) mice fed MCDD display obvious NASH. Injury was clearly diminished in endotoxin resistant mice (right panels). Additional sections of liver from mice fed MCDD were subjected to immunohistochemical staining for neutrophils (E&F) and Kupffer cells (F4/80; G&H). The appearance of brown staining indicates the presence of these cells in response to MCDD.
Serum ALT activity was measured to index injury and was increased significantly in wild type mice (Fig. 3). Consistent with histological findings, ALT activity in serum from C3H/HeJ mice fed MCDD was significantly lower than diet-matched wild type mice.
Figure 3. Hepatic triglyceride content and serum ALT activity.

Serum levels of the liver-specific injury marker ALT (A) and triglyceride levels (B) in 20% liver homogenates were measured using standard spectrophotometric assays. Statistical comparisons were made using two-way ANOVA. *p< 0.05 compared to PBS controls or #diet-matched C3H/HouJ mice.
3.3 Hepatic triglyceride content and evidence of improved PPAR-α signaling in TLR-4 mutant mice
Induction of steatohepatitis by feeding MCDD resulted in a 3-fold increase in hepatic triglyceride content; however, triglyceride accumulation was prevented in TLR-4 mutant C3H/HeJ mice. (Fig 3B). In an attempt to understand how TLR-4 might regulate lipid accumulation, we examined the expression of PPAR-α in wild type and TLR-4 mutant mice fed MCDD. Mutant mice had significantly greater PPAR-α expression after MCDD while levels in wild types did not change (Fig 4A). Moreover, expression of L-FABP, which is regulated by PPAR-a, was significantly higher in mutant mice fed MCDD (Fig. 4B). This data suggests that PPAR-α may be negatively regulated in response to TLR-4 signaling.
Figure 4. Hepatic PPAR-αand L-FABP expression.

Expression of PPAR-α and L-FABP mRNA was analyzed by real-time PCR and calculated relative to average values in the control group using a comparative CT method. Data are presented as mean ± SEM of 5 observations/group. Statistical comparisons were made using two-way ANOVA. *p< 0.05 compared to PBS controls or #diet-matched C3H/HouJ mice.
3.4 TNF-α and Collagen αI expression in TLR-4 mutant mice
In accordance with histological and biochemical findings, mRNA expression of injury markers were blunted in TLR-4 mutant mice. As shown in Fig 5, MCDD-induced increases in expression of the pro-inflammatory cytokine TNF-α as well as the matrix protein collagen αI was prevented in C3H/HeJ mice.
Figure 5. TNF-α and Collagen mRNA in C3H/HeJ mice.

Expression of TNF-α and Collagen α1 mRNA was analyzed by real-time PCR and calculated relative to average values in the control group using a comparative CT method. Data are presented as mean ± SEM of 5 observations/group. Statistical comparisons were made using two-way ANOVA. *p< 0.05 compared to PBS controls or #diet-matched C3H/HouJ mice.
3.5 Effect of Kupffer cell depletion on liver histopathology
We next examined the extent of injury in the absence of Kupffer cells. This was accomplished by weekly intravenous administration of liposome-encapsulated clodronate, which has been shown to induce apoptosis in liver and splenic macrophages (11, 13). Immunohistochemical staining of liver sections collected at the end of the study revealed the successful depletion of Kupffer cells, whereas injection of liposomes containing PBS had no effect on MCDD-induced macrophage accumulation (Fig. 6). In addition to markedly reducing inflammation, destruction of Kupffer cells attenuated the histological appearance of hepatic steatosis and necrosis as summarized in Table I.
Figure 6. Effect of clodronate liposomes on Kupffer cells.

To confirm destruction of Kupffer cells, livers of mice fed control diet or MCDD were preserved in zinc fixative and subjected to immunohistochemical staining with primary antibodies directed against macrophages (F4/80). Hematoxylin was used as a counter stain. Positive cells are stained brown. Photomicrographs are representative of 6 mice/dietary group.
Table I.
Effect of Clodronate liposomes on hepatic pathology
| Steatosis | Inflammation | Necrosis | |
|---|---|---|---|
| PBS | 0 ± 0.0 | 0 ± 0.0 | 0 ± 0.0 |
| Clodronate liposomes | 0.1 ± 0.1 | 0.2 ± 0.2 | 0 ± 0.0 |
| PBS + MCDD | 2.5 ± 0.0* | 3.0 ± 0.0* | 2.0 ± 0.0* |
| Clodronate liposomes + MCDD | 0.9 ± 0.2# | 0 ± 0.0# | 0 ± 0.0# |
Data are expressed as mean ± SEM; n= 4 observations per treatment group. Statistical comparisons were made using two-way ANOVA.
p< 0.05 compared to PBS controls.
p< 0.05 compared to MCDD + PBS.
3.6 Kupffer cell depletion blunts hepatic expression of TNF-α and markers of fibrosis
The influence of Kupffer cell depletion on the mRNA expression of various injury mediators was assessed using real-time PCR. Consistent with the observed inflammatory response to MCDD, expression of TNF-α was enhanced significantly in PBS-treated mice (Fig 7A). Although histological evidence of fibrosis was not detectable after just 3 weeks of MCDD feeding, there was significant induction of mRNA levels of the profibrogenic cytokine TGF-β as well as pro-collagen αI in PBS controls (Fig 7B&C). In mice treated with clodronate liposomes, feeding MCDD did not increase the expression of markers used to index fibrosis or inflammation.
Figure 7. Hepatic expression of (A) TNF-α, (B) TGF-β and (C) Collagen.

Livers were collected from male C57BL/6 mice fed control (CD) or methionine/choline deficient (MCDD) for 3 weeks. In addition, mice were administered clodronate liposomes (200 µg/mouse) to destroy Kupffer cells or PBS (phosphate-buffered saline) as a vehicle control as described in Methods. Pre-developed assays for real-time PCR were used to assess target mRNA levels. Statistical comparisons were made using two-way ANOVA (n= 5/group). *p< 0.05 compared to mice fed control diet; #diet-matched PBS (phosphate-buffered saline) controls.
3.7 Effect of Kupffer cell depletion on Toll-like receptor expression
To further characterize the importance of the relationship between Kupffer cells and TLR-4 expression during MCDD-induced NASH, the effect of Kupffer cell depletion on TLR-4 expression was analyzed. In contrast to MCDD-fed mice administered liposome encapsulated PBS, TLR-4 expression in mice treated with clodronate liposomes was not increased significantly above CD-fed mice (Fig. 8A), suggesting that a significant fraction of the expression of this receptor is contributed by Kupffer cells. In addition to TLR-4, TLR-2 reportedly acts as a receptor for endotoxin as well as various other microbial products and lipoproteins (18–20). Transmission of intracellular signals following TLR-2 interaction with these products requires the scavenger receptor CD36 (21). Real-time PCR revealed that feeding MCDD caused a 5-fold and 7-fold increase in TLR-2 and CD36 expression, respectively, when compared to mice fed the control diet, an effect largely blocked by the destruction of Kupffer cells (Fig. 8B&C).
Figure 8. Effect of Kupffer cell destruction on toll-like receptor expression.

After 3 weeks of feeding MCDD the effect of Kupffer cell destruction on expression of TLR-4, CD36 and TLR-2 was examined using real-time PCR. Statistical comparisons were made using two-way ANOVA (n= 5/group). *p< 0.05 compared to mice fed control diet or #p< 0.05 compared to diet-matched PBS (phosphate-buffered saline) controls.
4. Discussion
Ligand interaction with the TLR-4 receptor complex results in the recruitment of multiple adaptor molecules to the cell membrane, eventually resulting in the degradation of IkB and the translocation of NFkB to the nucleus where it facilitates the transcription of inflammatory mediators. Previous studies have demonstrated the activation of NFkB and enhanced sensitivity to TLR-4 ligands in mice with steatohepatitis induced by feeding MCDD (22). In an attempt to clarify signals upstream of NFkB activation during NASH, we investigated the role of TLR-4 in the genesis of steatohepatitis. Our results indicated that MCDD-induced NASH was accompanied by induction of TLR-4 expression, as well as the TLR accessory molecules MD-2 and CD14. Injury and inflammation were markedly attenuated in TLR-4 mutant C3H/HeJ mice. Moreover, this strain was protected from matrix protein over-expression, demonstrating a role for TLR-4 signaling in MCDD-induced fibrogenesis. These findings provide strong evidence that TLR-4 signaling is, indeed, important for the pathogenesis of NASH. However, additional experiments are needed to clarify specifically how TLR-4 modulates lipid accumulation and fibrosis.
We observed prominent macrophage accumulation in close proximity to areas of injury in wild type mice fed MCDD. Therefore, one object of the current study was to examine the importance of Kupffer cells in NASH. Kupffer cells are a first line of defense against debris and microbial products expelled from the intestines. In fact, these cells clear approximately 90% of an intravenous dose of endotoxin. Several studies have demonstrated that Kupffer cells play a critical role in various forms of liver injury via paracrine interactions with hepatocytes. For example, depletion or inactivation of Kupffer cells diminished injury and fibrosis due to chronic carbon tetrachloride exposure early as well as early alcoholic steatohepatitis (ASH) (16, 17, 23), without affecting ethanol metabolism (24).
The involvement of Kupffer cells in alcoholic hepatitis is believed to be initiated by portal endotoxemia (23, 25). Based on the finding of portal endotoxemia in mice fed MCDD, it was postulated that activated Kupffer cells participate in the observed hepatic injury. Indeed, targeted deletion of Kupffer cells via liposome-encapsulated clodronate diminished MCDD-induced increases in the production of TNF-α, ICAM-1 and significantly impaired the development of hepatic injury. In support of the idea that the expression of TLR-4 on Kupffer cells is essential for injury, destruction of these cells also prevented increases in TLR-4 expression induced.
We have shown previously that Kupffer cells also play a causal role in fibrogenesis (17). Hepatic fibrosis may occur simultaneously with hepatitis and is characterized by the overproduction of extracellular matrix proteins such as Collagen type I (26). Products secreted from Kupffer cells isolated from fibrotic rats have been shown to stimulate the proliferation and phenotypic transformation of stellate cells into myofibroblast-like cell that produces collagen. TGF-β is one of the cytokines believed to be primarily responsible for these effects and also has been implicated in decreased degradation of matrix proteins (27). We found a significant induction of mRNA levels of collagen α1 and the profibrogenic cytokine TGF-β in wild type mice. Destruction of Kupffer cells completely prevented the induction of TGF-β mRNA expression, but only blunted collagen expression. Stellate cells express TLR-4 and can be activated by TLR-4 ligands. It is likely that the residual induction of collagen mRNA expression noted in clodronate-treated mice was due to direct stimulation of stellate cells via TLR-4.
We used clodronate encapsulated in liposomes to target delivery specifically to macrophages. Cell culture experiments have demonstrated that clodronate induces cell death in macrophages via apoptosis (11). Free clodronate released from dying macrophages does not easily penetrate lipid bilayers and is removed quickly from circulation by the renal system. Therefore, it is unlikely that decrements in the expression of lipid trafficking and fibrogenic markers observed in mice treated with encapsulated clodronate were a result of a direct effect of this treatment on hepatic cell types other than Kupffer cells.
It is important to note that fat accumulation in the liver was nearly absent in mice depleted of Kupffer cells. Since the expression of enzymes responsible for lipid synthesis were reduced dramatically following 3 weeks of MCDD feeding (data not shown), we speculated that the observed steatosis was the result of enhanced lipid storage. The class B scavenger receptor CD36 has been shown to facilitate the uptake of long chain fatty acids by endothelial cells as well as macrophages, and recent data suggests that this receptor plays a role in hepatic steatosis induced by feeding a high fat diet (28, 29). Another member of the toll-like receptor family, TLR-2, has been shown to play a role in lipid trafficking via uptake of diacylated lipoproteins (30), a process that requires CD36 (21). Consistent with the idea that TLR-2 and CD36 participate in MCDD-induced lipid trafficking, mRNA levels of both receptors were enhanced in wild type mice. In addition, the absence of severe steatosis due to Kupffer cell depletion was associated with significantly lower expression of these receptors.
Several previous studies have investigated mechanisms underlying hepatic fat accumulation in mice fed MCDD. For example, Ip et al. demonstrated that mice deficient in the expression of PPAR-α nuclear hormone receptor developed more severe steatohepatitis when fed MCDD compared to wild type mice (32). Conversely, treatment with the PPAR-α agonist Wy-14643 prevented and reversed hepatic pathologies induced by MCDD (33). Resolution of steatohepatitis by agonist was attributed to the induction of genes that facilitate fatty acid breakdown (e.g. L-FABP). Herein, we found that TLR-4 mutant mice have significantly greater PPAR-α and L-FABP expression after MCDD, suggesting improved PPAR-α signaling. This data suggests that PPAR-α may be negatively regulated in response to TLR-4 signaling. Clearly, additional investigations are required to elucidate the roles of TLR-2 and TLR-4 in hepatic fat accumulation.
Taken together, the results of the present study provide insight into the importance of TLR-4 signaling and underscore the essential role of Kupffer cells in the development of NASH. In the setting of ASH the initial stimulus of TLR-4 signaling is believed to be gut-derived endotoxin. Herein we described a similar state of portal endotoxemia in response to feeding MCDD. A recent study by Brun et al. (34) demonstrated that feeding MCDD impaired the expression of junctional complex proteins between adjacent enterocytes, and significantly impaired gut barrier function. Thus, the presence of endotoxin in the portal blood supply was likely due to increased intestinal permeability. Although findings of portal endotoxemia support the idea that bacterial products could initiate TLR signaling, the involvement of gram positive microbial products and other endogenous TLR-4 ligands cannot be ruled out. Nevertheless, based on the present findings it appears likely that targeting the TLR-4 pathway specifically in macrophages could provide a potential therapeutic approach for the treatment of NASH.
4. Acknowledgments
This work was supported in part by federal funds from the National Heart Lung and Blood Institutes and the NHLBI (1K01HL084723-01) and the NIDDK (3P01DK43785-13S1).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Yang S, Lin H, Diehl AM. Fatty liver vulnerability to endotoxin-induced damage despite NF-kappaB induction and inhibited caspase 3 activation. Am J Physiol Gastrointest Liver Physiol. 2001 Aug;281(2):G382–G392. doi: 10.1152/ajpgi.2001.281.2.G382. [DOI] [PubMed] [Google Scholar]
- 2.Yang SQ, Lin HZ, Lane MD, Clemens M, Diehl AM. Obesity increases sensitivity to endotoxin liver injury: implications for the pathogenesis of steatohepatitis. Proc Natl Acad Sci U S A. 1997 Mar 18;94(6):2557–2562. doi: 10.1073/pnas.94.6.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li Z, Yang S, Lin H, Huang J, Watkins PA, Moser AB, et al. Probiotics and antibodies to TNF inhibit inflammatory activity and improve nonalcoholic fatty liver disease. Hepatol. 2003 Feb;37(2):343–350. doi: 10.1053/jhep.2003.50048. [DOI] [PubMed] [Google Scholar]
- 4.Wigg AJ, Roberts-Thomson IC, Dymock RB, McCarthy PJ, Grose RH, Cummins AG. The role of small intestinal bacterial overgrowth, intestinal permeability, endotoxaemia, and tumour necrosis factor alpha in the pathogenesis of nonalcoholic steatohepatitis. Gut. 2001 Feb;48(2):206–211. doi: 10.1136/gut.48.2.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vanderhoof JA, Metz MJ, Tuma DJ, Antonson DL, Sorrell MF. Effect of improved absorption on development of jejunoileal bypass- induced liver dysfunction in rats. Dig Dis Sci. 1980 Aug;25(8):581–586. doi: 10.1007/BF01318870. [DOI] [PubMed] [Google Scholar]
- 6.Decker K. Biologically active products of stimulated liver macrophages (Kupffer cells) Eur J Biochem. 1990;192:245–261. doi: 10.1111/j.1432-1033.1990.tb19222.x. [DOI] [PubMed] [Google Scholar]
- 7.Hardardottir I, Grunfeld C, Feingold KR. Effects of endotoxin and cytokines on lipid metabolism. Curr Opin Lipidol. 1994 Jun;5(3):207–215. doi: 10.1097/00041433-199405030-00008. [DOI] [PubMed] [Google Scholar]
- 8.McCuskey RS, Ito Y, Robertson GR, McCuskey MK, Perry M, Farrell GC. Hepatic microvascular dysfunction during evolution of dietary steatohepatitis in mice. Hepatol. 2004 Aug;40(2):386–393. doi: 10.1002/hep.20302. [DOI] [PubMed] [Google Scholar]
- 9.Kirsch R, Clarkson V, Shephard EG, Marais DA, Jaffer MA, Woodburne VE, et al. Rodent nutritional model of non-alcoholic steatohepatitis: species, strain and sex difference studies. J Gastroenterol Hepatol. 2003 Nov;18(11):1272–1282. doi: 10.1046/j.1440-1746.2003.03198.x. [DOI] [PubMed] [Google Scholar]
- 10.Sahai A, Malladi P, Pan X, Paul R, Melin-Aldana H, Green RM, et al. Obese and diabetic db/db mice develop marked liver fibrosis in a model of nonalcoholic steatohepatitis: role of short-form leptin receptors and osteopontin. Am J Physiol Gastrointest Liver Physiol. 2004 Nov;287(5):G1035–G1043. doi: 10.1152/ajpgi.00199.2004. [DOI] [PubMed] [Google Scholar]
- 11.van RN, Sanders A. Liposome mediated depletion of macrophages: mechanism of action, preparation of liposomes and applications. J Immunol Methods. 1994 Sep 14;174(1–2):83–93. doi: 10.1016/0022-1759(94)90012-4. [DOI] [PubMed] [Google Scholar]
- 12.van RN, Sanders A. Kupffer cell depletion by liposome-delivered drugs: comparative activity of intracellular clodronate, propamidine, and ethylenediaminetetraacetic acid. Hepatol. 1996 May;23(5):1239–1243. doi: 10.1053/jhep.1996.v23.pm0008621159. [DOI] [PubMed] [Google Scholar]
- 13.van RN, van NR. Elimination of phagocytic cells in the spleen after intravenous injection of liposome-encapsulated dichloromethylene diphosphonate. An enzyme-histochemical study. Cell Tissue Res. 1984;238(2):355–358. doi: 10.1007/BF00217308. [DOI] [PubMed] [Google Scholar]
- 14.Rivera CA, Thurman RG. Tips for measuring endotoxin in plasma. Alcohol Clin Exp Res. 1998 Dec;22(9):2192–2194. doi: 10.1111/j.1530-0277.1998.tb05933.x. [DOI] [PubMed] [Google Scholar]
- 15.Rivera CA, Abrams SH, Tcharmtchi HM, Allman M, Finegold MJ, Smith WC. Feeding A Corn Oil/Sucrose-Enriched Diet Enhances Steatohepatitis in Sedentary Rats. Am J Physiol Gastrointest Liver Physiol. 2005 Oct 13; doi: 10.1152/ajpgi.00229.2005. [DOI] [PubMed] [Google Scholar]
- 16.Adachi Y, Bradford BU, Gao W, Bojes HK, Thurman RG. Inactivation of Kupffer cells prevents early alcohol-induced liver injury. Hepatol. 1994 Aug;20(2):453–460. [PubMed] [Google Scholar]
- 17.Rivera CA, Bradford BU, Hunt KJ, Adachi Y, Schrum LW, Koor DR, et al. Attenuation of CCl(4)-induced hepatic fibrosis by GdCl(3) treatment or dietary glycine. Am J Physiol Gastrointest Liver Physiol. 2001 Jul;281(1):G200–G207. doi: 10.1152/ajpgi.2001.281.1.G200. [DOI] [PubMed] [Google Scholar]
- 18.Schwandner R, Dziarski R, Wesche H, Rothe M, Kirschning CJ. Peptidoglycan-and lipoteichoic acid-induced cell activation is mediated by toll-like receptor 2. J. Biol Chem. 1999 Jun 18;274(25):17406–17409. doi: 10.1074/jbc.274.25.17406. [DOI] [PubMed] [Google Scholar]
- 19.Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, et al. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity. 1999 Oct;11(4):443–451. doi: 10.1016/s1074-7613(00)80119-3. [DOI] [PubMed] [Google Scholar]
- 20.Werts C, Tapping RI, Mathison JC, Chuang TH, Kravchenko V, Saint G, I, et al. Leptospiral lipopolysaccharide activates cells through a TLR2-dependent mechanism. Nat Immunol. 2001 Apr;2(4):346–352. doi: 10.1038/86354. [DOI] [PubMed] [Google Scholar]
- 21.Hoebe K, Georgel P, Rutschmann S, Du X, Mudd S, Crozat K, et al. CD36 is a sensor of diacylglycerides. Nature. 2005 Feb 3;433(7025):523–527. doi: 10.1038/nature03253. [DOI] [PubMed] [Google Scholar]
- 22.Dela PA, Leclercq I, Field J, George J, Jones B, Farrell G. NF-kappaB activation, rather than TNF, mediates hepatic inflammation in a murine dietary model of steatohepatitis. Gastroenterology. 2005 Nov;129(5):1663–1674. doi: 10.1053/j.gastro.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 23.Iimuro Y, Gallucci RM, Luster MI, Kono H, Thurman RG. Antibodies to tumor necrosis factor alfa attenuate hepatic necrosis and inflammation caused by chronic exposure to ethanol in the rat. Hepatol. 1997 Dec;26(6):1530–1537. doi: 10.1002/hep.510260621. [DOI] [PubMed] [Google Scholar]
- 24.Koop DR, Klopfenstein B, Iimuro Y, Thurman RG. Gadolinium chloride blocks alcohol-dependent liver toxicity in rats treated chronically with intragastric alcohol despite the induction of CYP2E1. Mol Pharmacol. 1997 Jun;51(6):944–950. doi: 10.1124/mol.51.6.944. [DOI] [PubMed] [Google Scholar]
- 25.Nanji AA, Khettry U, Sadrzadeh SM, Yamanaka T. Severity of liver injury in experimental alcoholic liver disease. Correlation with plasma endotoxin, prostaglandin E2, leukotriene B4, and thromboxane B2. Am J Pathol. 1993 Feb;142(2):367–373. [PMC free article] [PubMed] [Google Scholar]
- 26.Rojkind M, Giambrone MA, Biempica L. Collagen types in normal and cirrhotic liver. Gastroenterology. 1979 Apr;76(4):710–719. [PubMed] [Google Scholar]
- 27.Ignotz RA, Massague J. Transforming growth factor-beta stimulates the expression of fibronectin and collagen and their incorporation into the extracellular matrix. J Biol Chem. 1986 Mar 25;261(9):4337–4345. [PubMed] [Google Scholar]
- 28.Inoue M, Ohtake T, Motomura W, Takahashi N, Hosoki Y, Miyoshi S, et al. Increased expression of PPARgamma in high fat diet-induced liver steatosis in mice. Biochem Biophys Res Commun. 2005 Oct 14;336(1):215–222. doi: 10.1016/j.bbrc.2005.08.070. [DOI] [PubMed] [Google Scholar]
- 29.Zhang YL, Hernandez-Ono A, Siri P, Weisberg S, Conlon D, Graham MJ, et al. Aberrant hepatic expression of PPARgamma2 stimulates hepatic lipogenesis in a mouse model of obesity, insulin resistance, dyslipidemia, and hepatic steatosis. J Biol Chem. 2006 Dec 8;281(49):37603–37615. doi: 10.1074/jbc.M604709200. [DOI] [PubMed] [Google Scholar]
- 30.Takeuchi O, Sato S, horiuchi T, Hoshino K, Takeda K, Dong Z, et al. Cutting edge: role of Toll-like receptor 1 in mediating immune response to microbial lipoproteins. J Immunol. 2002 Jul 1;169(1):10–14. doi: 10.4049/jimmunol.169.1.10. [DOI] [PubMed] [Google Scholar]
- 31.Szabo G, Velayudham A, Romics L, Jr, Mandrekar P. Modulation of nonalcoholic steatohepatitis by pattern recognition receptors in mice: the role of toll-like receptors 2 and 4. Alcohol Clin Exp Res. 2005 Nov;29(11 Suppl):140S–145S. doi: 10.1097/01.alc.0000189287.83544.33. [DOI] [PubMed] [Google Scholar]
- 32.Ip E, Farrell GC, Robertson G, Hall P, Kirsch R, Leclercq I. Central role of PPARalpha-dependent hepatic lipid turnover in dietary steatohepatitis in mice. Hepatol. 2003 Jul;38(1):123–132. doi: 10.1053/jhep.2003.50307. [DOI] [PubMed] [Google Scholar]
- 33.Ip E, Farrell G, Hall P, Robertson G, Leclercq I. Administration of the potent PPARalpha agonist, Wy-14,643, reverses nutritional fibrosis and steatohepatitis in mice. Hepatol. 2004 May;39(5):1286–1296. doi: 10.1002/hep.20170. [DOI] [PubMed] [Google Scholar]
- 34.Brun P, Castagliuolo I, Di L, V, Buda A, Pinzani M, Palu' G, et al. Increased intestinal permeability in obese mice: new evidences in the pathogenesis of nonalcoholic steatohepatitis. Am J Physiol Gastrointest Liver Physiol. 2006 Oct 5; doi: 10.1152/ajpgi.00024.2006. [DOI] [PubMed] [Google Scholar]