

SUMMARY
Activating mutations in the tyrosine kinase domain of receptor tyrosine kinases (RTKs) cause cancer and skeletal disorders. Comparison of the crystal structures of unphosphorylated and phosphorylated wild-type FGFR2 kinase domains with those of seven unphosphorylated pathogenic mutants reveals an autoinhibitory ‘molecular brake’, mediated by a triad of residues in the kinase hinge region of all FGFRs. Structural analysis shows that many other RTKs, including PDGFRs, VEGFRs, KIT, CSF1R, FLT3, TEK and TIE, are also subject to regulation by this brake. Pathogenic mutations activate FGFRs and other RTKs by disengaging the brake either directly or indirectly.
INTRODUCTION
Activating mutations in the tyrosine kinase domain of RTKs are responsible for many forms of human cancers including gastrointestinal stromal tumors (KIT and PDGFRα) (Corless et al., 2004; Corless et al., 2005; Medeiros et al., 2004), acute myeloid leukemia and acute promyelocytic leukemia (FLT3) (Gilliland, 2003; Liang et al., 2003; Schittenhelm et al., 2006; Yamamoto et al., 2001), juvenile hemangioma (VEGFR2/3) (Walter et al., 2002), glioblastoma (FGFR1) (Rand et al., 2005), endometrial cancer (FGFR2) (Pollock et al., 2007), multiple myeloma (FGFR3) (Chesi et al., 1997; Richelda et al., 1997), bladder and cervical cancers (FGFR3) (Cappellen et al., 1999; van Rhijn et al., 2002). Gain-of-function mutations in FGFR2 and FGFR3 kinase domains (FGFR2K and FGFR3K) also give rise to some of the most devastating forms of human craniosynostosis and dwarfism syndromes such as Crouzon syndrome (CS), Pfeiffer syndrome (PS), achondroplasia (ACH), hypochondroplasia (HCH), thanatophoric dysplasia type I (TDI), thanatophoric dysplasia type II (TDII) and severe achondroplasia with developmental delay and acanthosis nigricans (SADDAN) (Passos-Bueno et al., 1999; Webster and Donoghue, 1997; Wilkie, 2005; Wilkin et al., 2001). Other human diseases are also linked to activating mutations in kinase domain of RTKs such as inherited venous malformations (TEK) (Calvert et al., 1999; Vikkula et al., 1996), and lymphoedema (VEGFR3) (Karkkainen et al., 2000).
Screening of patients with craniosynostosis (premature fusion of skeletal sutures) has led to the identification of a total of nine distinct mutations in FGFR2K, accounting for ~10% of patients with craniosynostosis (de Ravel et al., 2005; Kan et al., 2002; McGillivray et al., 2005; Zankl et al., 2004). These mutations are K526E, N549H, N549T, E565A, E565G, K641R, K659N, G663E and R678G. The N549H, N549T, E565G, E565A and K641R mutations map onto the kinase hinge (the linker between β5 strand and αD helix) or its vicinity, whereas K659N and R678G map onto the activation loop (A-loop) or its vicinity (Fig. S1). The K526E mutation maps onto the αC helix in the kinase N-lobe. In addition, screening of patients with endometrial cancer also identified two activating mutations, N549K and K659E, in FGFR2K (Pollock et al., 2007). Mutations at asparagine residues, corresponding to N549 of FGFR2, in FGFR1, FGFR3 and PDGFRα are also implicated in human diseases. Specifically, mutations of N540 to T, S or K in FGFR3 lead to HCH, a mild form of dwarfism (Bellus et al., 1995; Deutz-Terlouw et al., 1998; Grigelioniene et al., 2000; Mortier et al., 2000), the N546K mutation in FGFR1 is found in glioblastomas (Rand et al., 2005), and the N659K mutation in PDGFRα results in gastrointestinal stromal tumors (Corless et al., 2005; Medeiros et al., 2004). Mutations of K650 in FGFR3, corresponding to K659 of FGFR2, give rise to different clinical entities of varying severity ranging from HCH (K650Q/N) to SADDAN (K650M) to neonatal lethal TDII (K650E) (Bellus et al., 2000). The TDII and SADDAN mutations in FGFR3 are also implicated in multiple myeloma (Chesi et al., 1997; Richelda et al., 1997), bladder and cervical cancers (Cappellen et al., 1999). Mutation of K650 to threonine has also been reported in some bladder cancers (van Rhijn et al., 2002)
In this report we compared the crystal structures of unphosphorylated and A-loop tyrosine phosphorylated wild-type FGFR2Ks with those of seven unphosphorylated mutant FGFR2Ks each harboring a distinct pathogenic mutation. We found that these mutations cause constitutive activation of FGFR2 kinase by disengaging a “molecular brake” that is conserved in a wide range of RTKs.
RESULTS AND DISCUSSION
The FGFR2K Pathogenic Mutations Activate the Kinase Domain Constitutively
We compared the intrinsic kinase activity of the unphosphorylated and A-loop tyrosine phosphorylated wild-type FGFR2Ks with that of the nine unphosphorylated mutant kinases by measuring the rates of tyrosine autophosphorylation (Fig. 1A) and peptide substrate phosphorylation (Fig. 1B). All nine FGFR2K mutants demonstrated elevated autophosphorylation activity relative to the unphosphorylated wild-type kinase, ranging from 7-fold to as high as 32-fold. Compared to unphosphorylated wild-type kinase, all mutant kinases also exhibited increased capacity to phosphorylate peptide substrate. We then selected E565A and K659N mutants as the representatives of kinase hinge and A-loop hotspot region mutants, respectively, to determine the steady-state kinetic parameters using two different peptide substrates. The data showed that these two mutations activate FGFR2 kinase primarily by increasing the Vmax (over 45-fold in the case of E565A) (Table S1). Our autophosphorylation and peptide substrate phosphorylation data clearly demonstrate that all nine pathogenic FGFR2K mutations activate the kinases constitutively. We also investigated the role of A-loop tyrosine phosphorylation in the mechanism of kinase activation by the pathogenic mutations. For this study we selected K526E, E565A and K659N mutants because each mutation maps to a distinct location in kinase domain i.e. the kinase hinge region, the αC helix and the A-loop respectively. To this end we purified A-loop phosphorylated version of K526E, E565A and K659N mutants, and compared the kinase activity of A-loop phosphorylated version of these mutants with that of these mutant kinases in unphosphorylated state. Unphosphorylated and phosphorylated wild-type kinase served as controls. As shown in Fig. 1C, A-loop phosphorylation further elevated the kinase activity of all three mutant kinases. However, the degree of kinase activation by phosphorylation for mutant kinases is lower (~2 to 7 fold) than that for wild-type kinase (~9 fold). Taken together, these data show that the pathogenic mutations promote more readily ligand-independent A-loop tyrosine phosphorylation.
Figure 1.
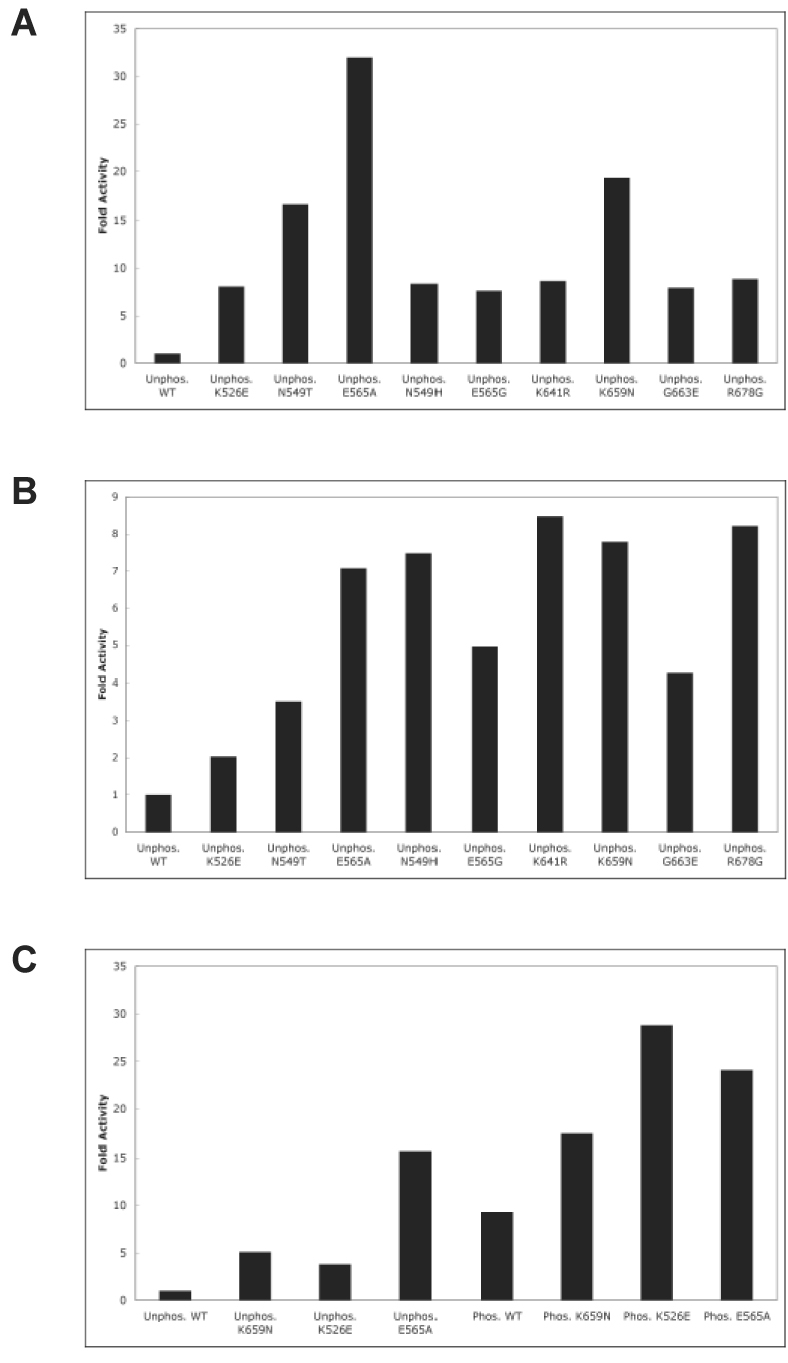
Pathogenic mutations activate FGFR2K constitutively in the absence of A-loop tyrosine phosphorylation. (A) Autophosphorylation assays: unphosphorylated wild-type or mutant FGFR2Ks were incubated with 1 mM ATP and autophosphorylation rates were determined using a continuous spectrophotometric assay. (B) Peptide phosphorylation assays: unphosphorylated wild-type or mutant FGFR2Ks were incubated with 100 µM ATP and 50 µM peptide substrate (KKEEEEYMMMMG). Rates of peptide phosphorylation were determined using the spectrophotometric assay. (C) The effect of A-loop phosphorylation on kinase activity of wild-type and mutant kinases. Unphosphorylated and A-loop tyrosine phosphorylated wild-type, K526E, K565 or K659N mutant FGFR2Ks were incubated with 500 µM ATP and 500 µM peptide substrate (KKEEEEYMMMMG). Rates of peptide phosphorylation were determined using the spectrophotometric assay.
Structural Basis by Which Tyrosine Phosphorylation on the A-loop Enhances FGFR2 Kinase Activity
We first solved the crystal structures of unphosphorylated wild-type FGFR2K as well as A-loop tyrosine phosphorylated wild-type FGFR2K in complex with a non-hydrolyzable ATP analogue and peptide substrate. The peptide substrate used for cocrystallization is derived from the C-terminal tail of FGFR2 (residues 764 to 778) and contains Y769, one of the major autophosphorylation sites of FGFR2. Both wild-type FGFR2Ks adopt the canonical bi-lobate structure of protein serine/tyrosine kinases (Hubbard and Till, 2000) (Fig. 2A and 2B). Comparison of the two structures reveals two critical changes that occur in the kinase domain upon A-loop tyrosine phosphorylation. At the global level, the N-lobe of the kinase undergoes an inward rotation (6.7 degrees) towards the C-lobe (Fig. S2A, Fig. S3A, Fig. S6 and Table S3), which coincides with a major rearrangement of the phosphorylated A-loop at the local level (Fig. 2C). These coupled structural changes align the catalytic residues from different regions including the A-loop, the catalytic loop and the αC helix, to promote peptide substrate and ATP binding and to increase catalytic efficiency. In the structure, peptide substrate binds into the active site in a similar fashion as in the two published structures of A-loop phsophorylated IRK (Hubbard, 1997) and IGF1R kinase (Favelyukis et al., 2001) in complex with substrate peptide. There is clear density for only a tetrapeptide region (767EEYL770) of the substrate and the remaining nine residues of the peptide have uninterpretable density. It should be noted that in both IRK and IGF1RK structures there are also only a few residues immediately adjacent to the substrate tyrosine are visible in electron density maps.
Figure 2.
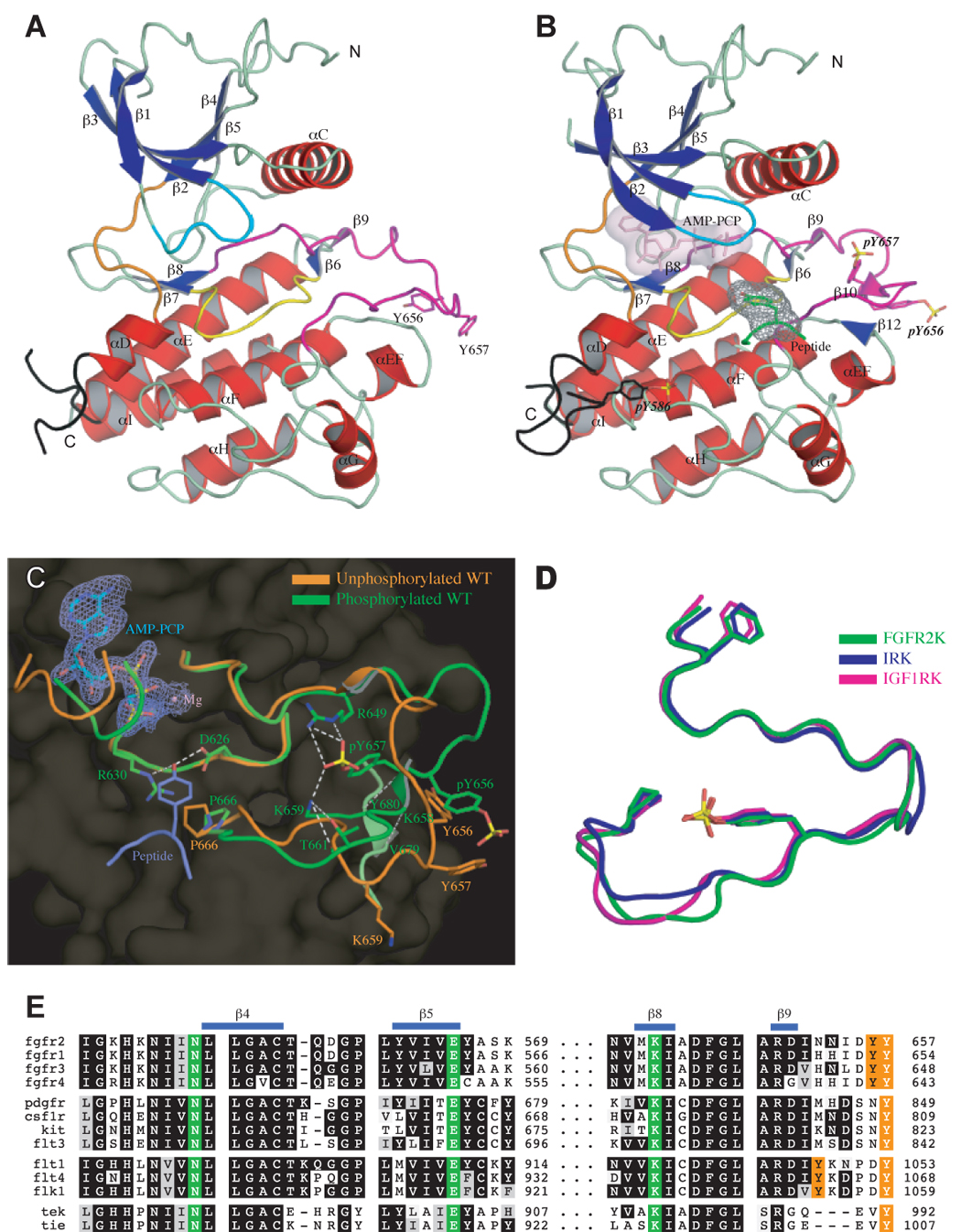
Structural basis for FGFR2K activation by phosphorylation of A-loop tyrosine. (A) and (B) The ribbon diagrams of unphosphorylated and A-loop tyrosine phosphorylated wild-type FGFR2K structures in the same orientation. Strands and helices are colored blue and red, respectively. The A-loop, catalytic loop, nucleotide-binding loop, kinase insert and kinase hinge are colored magenta, yellow, cyan, black, and orange, respectively. In the phosphorylated structure, the ATP analogue (in light pink) and the tyrosine side chain of the peptide substrate (in green) are rendered as sticks. The molecular surfaces of the ATP analogue and the substrate tyrosine are also shown as a solid semi-transparent surface and gray mesh, respectively. (C) Comparison of the A-loop conformations of unphosphorylated (in orange) and A-loop tyrosine phosphorylated wild-type (in green) FGFR2K structures. The ATP analogue (Cα in cyan) and substrate tyrosine (Cα in purple) are shown as sticks. The 2FO-FC electron density map contoured at 1σ for ATP analogue is shown as mesh in slate. Side chains of selected residues from the A-loop and the catalytic loop are shown as sticks. Atom colorings are as follows: oxygens in red, nitrogens in blue, phosphorus in yellow, and carbons are colored according to the kinase molecule to which they belong. Hydrogen bonds are shown as white dashed lines. (D) Comparison of the A-loop conformation of A-loop tyrosine phosphorylated FGFR2K (in green) with those of phosphorylated IRK (PDB ID: 1IR3) (in blue) and IGF1RK (PDB ID: 1K3A) (in magenta). The phosphotyrosine pY657 in FGFR2K fulfills the same function as pY1163 in IRK and pY1133 in IGF1RK. To assist the readers, the side chains of the phenylalanine from the conserved DFG motif at the N-terminus of the A-loop, and of the conserved proline at the C-terminus of the A-loop are also shown as sticks. Coloring scheme is the same as in (C). (E) The triad of residues (in green), which mediate the autoinhibitory molecular brake in FGFR, are also conserved in many other RTKs including PDGFs, CSF1R, KIT, FLT3, VEGFRs, Tie and Tek. Autophosphorylation sites in the A-loop are colored orange.
The A-loop conformation in the A-loop tyrosine phosphorylated wild-type FGFR2K structure is stabilized primarily by hydrogen bonds between the phosphate group of pY657 and the side chains of R649 and K659, and between the side chain of K659 and the backbone and side chain atoms of T661 in the A-loop (Fig. 2C). Moreover, pY657 and K658 form a short β strand, which pairs with β12 strand formed by residues V679 and Y680 in the linker between αEF and αF helices (Fig. 2B and 2C). This extra β strand pairing provides additional stabilization of the active A-loop conformation. In contrast to pY657, phosphorylated Tyr-656 is solvent exposed and does not contribute to the activated A-loop conformation (Fig. 2B and 2C). The structural data are consistent with the mutagenesis data on the highly homologous FGFR1 kinase (Mohammadi et al., 1996). Mutation of Tyr-654 (equivalent to Tyr-657 of FGFR2) to Phe abrogates the kinase activity of FGFR1 kinase whereas mutation of Tyr-653 (equivalent to Tyr-656 of FGFR2) to Phe has no impact on kinase activity (Mohammadi et al., 1996).
The N549H, N549T, E565G, E565A and K641R Mutations Lead to Gain-of-Function by Directly Targeting a ‘Molecular Brake’ at the FGFR Kinase Hinge Region
In the unphosphorylated wild-type FGFR2K structure, the triad of residues E565 (in the kinase hinge), N549 (in the loop between the αC helix and the β4 strand) and K641 (in the β8 strand) interact with each other through a network of hydrogen bonds (Fig. 3A and Fig. S5). In the A-loop tyrosine phosphorylated wild-type FGFR2K structure, concomitant with the rearrangement of the A-loop and lobe closure, this network dissociates (Fig. 3B and Fig. S5). Therefore, we propose that this network of hydrogen bonds mediated by the triad of residues acts as a ‘molecular brake’ that inhibits movement of the N-lobe towards the C-lobe, thereby keeping the kinase in an autoinhibited state. Based on our model, mutation of any residue of the triad mediating the molecular brake, as found in human skeletal disorders and cancer, should disengage the ‘brake’, thereby relaxing the kinase towards its active state.
Figure 3.
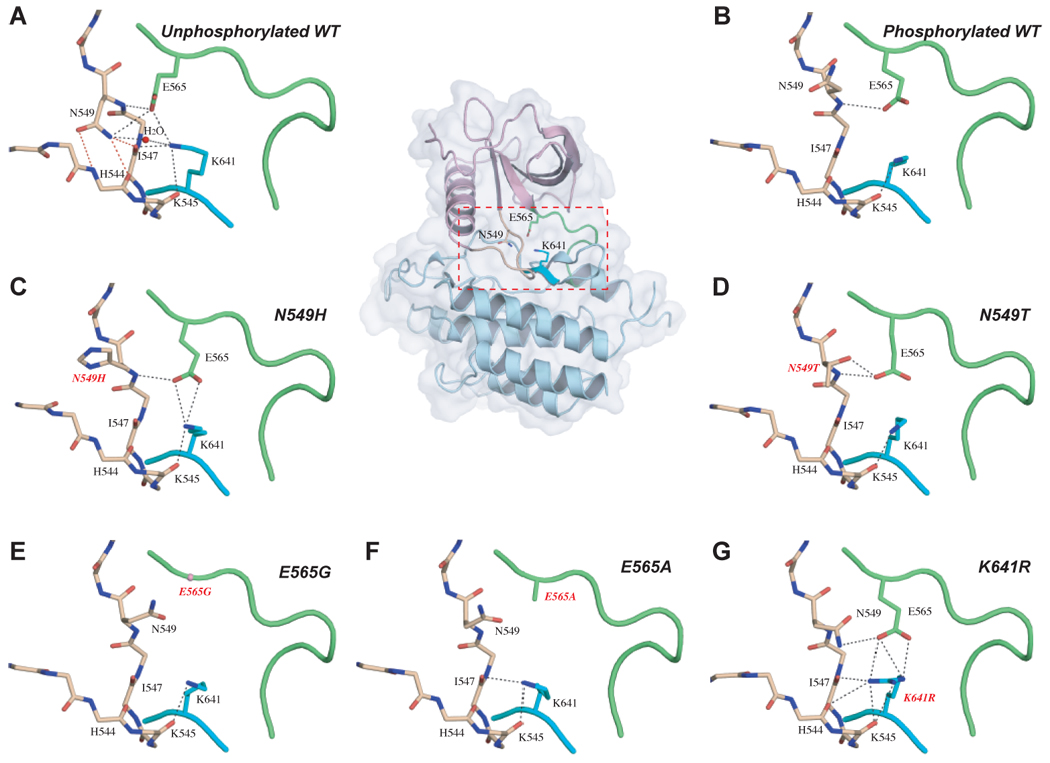
The molecular brake at the kinase hinge region of FGFR2K regulates the kinase activation and is disengaged either by A-loop tyrosine phosphorylation or directly by the pathogenic mutations. (A) In the unphosphorylated wild-type structure, residues N549, E565 and K641 form a network of hydrogen bonds in the kinase hinge region, which serves as a molecular brake to keep the enzyme in an inactive state. (B) The molecular brake is disengaged in the A-loop tyrosine phosphorylated wild-type FGFR2K structure. This molecular brake is also disengaged in the unphosphorylated mutant FGFR2K structures ((C) through (G)). To assist the readers, the whole unphosophorylated wild-type FGFR2K structure is also shown in cartoon and solid semi-transparent surface, and the kinase hinge region is boxed. Atom colorings are as follows: oxygens in red, nitrogens in blue, and carbons are colored according to the kinase region to which they belong. The kinase hinge, the αC-β4 loop (shown in sticks in (A) to (G)), and β8 strand are colored green, wheat and cyan, respectively. The rest of the N-lobe and C-lobe are colored light purple and light blue, respectively. The three critical hydrogen bonds between N549 and the backbone atoms of αC-β4 loop are highlighted by red dashed lines. The remaining hydrogen bonds are shown as black dashed lines.
To test our hypothesis, we solved the crystal structures of unphosphorylated N549H, N549T, E565G, E565A and K641R mutant FGFR2Ks. As anticipated, analysis of the hinge region of the N549H and N549T mutant kinases reveals the loss of the hydrogen bonds between mutated residue 549 and the backbone atoms of the loop between the αC helix and the β4 strand (Fig. 3C, Fig. 3D and Fig. S5). This relaxes the N-lobe to rotate towards the C-lobe at the same pivot point, as observed in the A-loop tyrosine phosphorylated wild-type FGFR2K (Fig. S3A, Fig. S6 and Table S3). In the E565G, E565A and K641R mutant structures, N549 also disengages from the backbone atoms of the αC-β4 loop, and instead turns towards E565 in the kinase hinge (Fig. 3E, Fig. 3F, Fig. 3G and Fig. S5), allowing a similar inward (towards the C-lobe) motion of the N-lobe in these three mutant structures (Fig. S3A, Fig. S6 and Table S3). This disengagement occurs because the mutated residues in these three structures are unable to assist N549 to hydrogen bond with the backbone atoms of the αC-β4 loop. Based on these data, the hydrogen bonds between N549 and the backbone atoms of the αC-β4 loop are the most critical constituents of the molecular brake. Taken together, our data show that these five pathogenic FGFR2K mutations lead to ligand-independent kinase activation by directly disengaging the molecular brake in the hinge region.
K526E Mutation Indirectly Disengages the Molecular Brake by Creating New Hydrogen Bonds Between the αC Helix and the A-loop
In the unphosphorylated wild-type FGFR2K structure, the side chain of K526 (located in the αC helix) engages in a hydrogen bond with D530 (data no shown). In the A-loop tyrosine phosphorylated structure, the side chain of K526 disengages from D530 and becomes completely solvent-exposed (data not shown). Thus, neither wild-type FGFR2K structure provides clues about the mode of action of this mutation. Analysis of our crystal structure of the unphosphorylated FGFR2 mutant kinase reveals that the αC helix mutation leads to ligand-independent kinase activation by indirectly disengaging the molecular brake at the kinase hinge region. The replacement of K526 by glutamate creates new hydrogen bonds between the mutant residue E526 and R664 in the A-loop (Fig. 4A). Superimposition of the C-lobe of the mutant kinase onto that of the unphosphorylated wild-type kinase shows that the N-lobe has rotated towards the C-lobe (Fig. S3A, Fig. S6 and Table S3). Analysis of the hinge region of the K526E mutant kinase shows that the side chain of N549 has disengaged from the backbone atoms of the αC-β4 loop, and instead has rotated towards E565 in the hinge indicating that the molecular brake has been loosened in this structure (Fig. 4B and Fig. S5). The disengagement of the molecular brake and the inward rotation of N-lobe, two hallmarks of the activated kinase, are induced indirectly by the hydrogen bonding between the mutant residue E526 and R664 in the A-loop.
Figure 4.
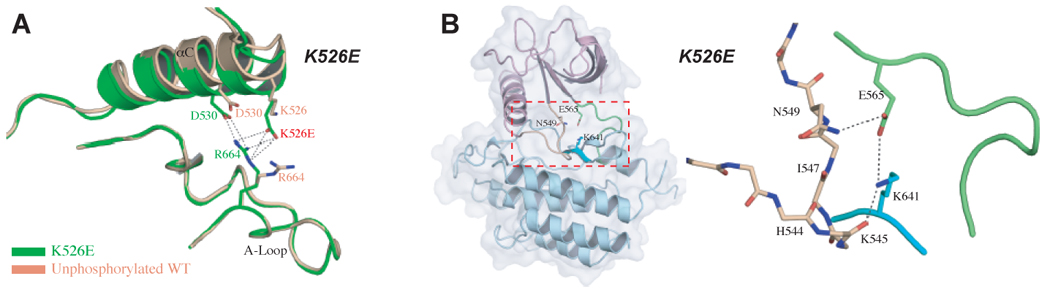
Molecular basis by which the K526E mutation activates FGFR2K in the absence of A-loop tyrosine phosphorylation. (A) The pathogenic K526E mutation (in green) creates new hydrogen bonds between the αC helix and the A-loop which contribute to the inward rotation of the N-lobe. Note that this hydrogen bonding does not occur in the unphosphorylated wild-type FGFR2K structure (in wheat) (B) The molecular brake in the hinge region of the unphosphorylated K526E mutant structure is disengaged indirectly due to the additional hydrogen bonds between the αC helix and the A-loop. To assist the readers, the whole unphosophorylated K526E mutant FGFR2K structure is also shown in cartoon and solid semi-transparent surface, and the kinase hinge region is boxed. Coloring scheme in (B) is as in Fig. 3.
The K659N Mutation Indirectly Disengages the Molecular Brake by Forcing the A-loop to Adopt the Active Conformation
In the unphosphorylated wild-type structure, the A-loop residue K659 is completely solvent-exposed and does not participate in any intramolecular interactions (Fig. 2C). In the A-loop tyrosine phosphorylated wild-type structure, however, this residue stabilizes the active conformation of the A-loop as discussed above (Fig. 2C). These observations suggest that a K659N mutation might negatively impact kinase activity of FGFR2 by hampering the A-loop’s ability to assume an active conformation. Analysis of our unphosphorylated K659N mutant kinase structure reveals that the A-loop of the mutant kinase adopts an active conformation in spite of the fact that the A-loop Y657 is not phosphorylated (Fig. 5A). Moreover, the superimposition of the C-lobe of the K569N mutant kinase onto those of the unphosphorylated wild-type kinase also reveals that the N-lobe of the mutant kinase has moved towards the C-lobe (Fig. S3A, Fig. S6 and Table S3). Analysis of the hinge region shows the disengagement of N549 from backbone atoms of the αC-β4 loop, indicating the loss of the molecular brake (Fig. 5B and Fig. S5). These local and global structural changes observed in the mutant structure are hallmarks of an activated kinase and are induced solely by the K659N mutation.
Figure 5.
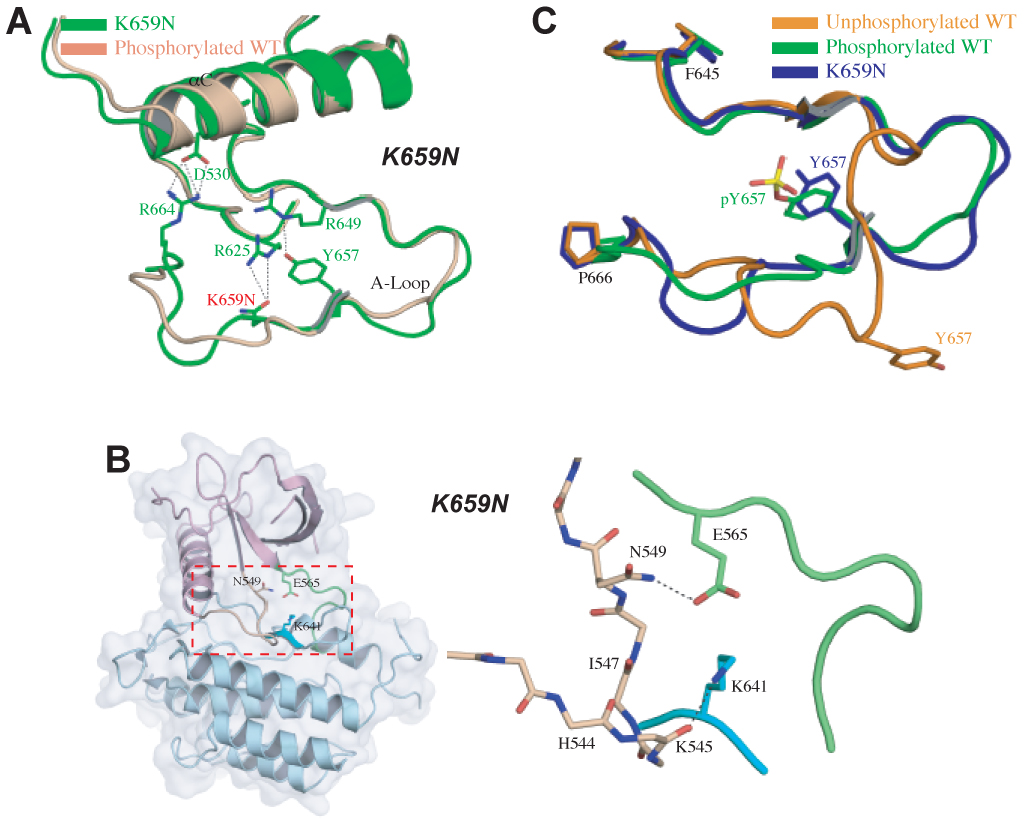
Molecular basis by which the K659N mutation activates FGFR2K in the absence of A-loop tyrosine phosphorylation. (A) Side chain of mutated N659 (in green) in the A-loop hydrogen-bonds with that of R625 in the catalytic loop forcing the A-loop to adopt its active conformation as seen in A-loop tyrosine phosphorylated wild-type FGFR2K structure (in wheat). (B) shows the disengagement of the molecular brake in the hinge region of K659N mutant structure. To assist the readers, the whole unphosophorylated K659E mutant FGFR2K structure is also shown in cartoon and solid semi-transparent surface, and the kinase hinge region is boxed. Coloring scheme in (B) is as in Fig. 3. (C) The K659N mutation drives the A-loop to adopt its active conformation in the absence of A-loop tyrosine phosphorylation. Unphosphorylated wild-type, A-loop phosphorylated wild-type and unphosphorylated K659N mutant FGFR2Ks are shown in orange, green and blue, respectively.
The active conformation of the A-loop in the K659N mutant kinase is induced by the formation of two hydrogen bonds between the side chain of the mutated residue N659 and the side chain of R625 in the catalytic loop (Fig. 5A). The side chain of A-loop Y657 occupies a very similar position as pY657 in the A-loop tyrosine phosphorylated wild-type FGFR2K (Fig. 5C). Unlike pY657 (Fig. 2C), however, the hydroxyl group of Y657 forms a direct hydrogen bond with the side chain of R649, further contributing to the active conformation of the A-loop (Fig. 5A).
In addition to a disengaged molecular brake at the hinge region, all unphosphorylated mutant kinases share yet another common feature, which is the formation of a salt bridge between D530 in the αC helix and R664 in the A-loop (Fig. 4A, 5A and S3B). These salt bridges further contribute to the inward motion of the N-lobe in all mutant kinases. Hence, our structural data reveals for the first time that the three key regulatory regions of the kinase domain (the kinase hinge, the αC helix and the A-loop) communicate with each other and act in concert to regulate tyrosine kinase activity of FGFR
Autoinhibition by the Molecular Brake is a Major Mechanism for RTK Regulation
All three residues of FGFR2 that mediate the inhibitory network of hydrogen bonds at the hinge region are fully conserved in all FGFRs (Fig. 2E), as well as in PDGFRs, VEGFRs, KIT, CSF1R, FLT3, TEK and TIE (Fig. 2E). This leads us to suggest that the autoinhibition by the molecular brake is a common regulatory mechanism in RTK regulation. Indeed, reanalysis of crystal structures of the kinase domains of FGFR1 (Mohammadi et al., 1996) (Fig. 6A), CSF1R (Schubert et al., 2006) (Fig. 6B), VEGFR2 (McTigue et al., 1999) (Fig. 6C), TEK (Shewchuk et al., 2000) (Fig. 6D) and c-KIT (Mol et al., 2004) (Fig. 6E) reveals the presence of the same inhibitory network of hydrogen bonds in the hinge region of these kinases in their inactivated state. For c-Kit, an “active” structure of kinase domain (Mol et al., 2003) has been reported. Comparison of hinge region of the “active” c-Kit (Fig. 6F) with that of inactive c-Kit (Fig. 6E) shows that in the “active” c-Kit strucuture the three key hydrogen bonds of the molecular brake between Asn-655 (residue equivalent to Asn-549 of the triad in FGFR2K) and backbone atoms of the αC-β4 loop are lost. This observation is consistent with the molecular brake hypothesis, and suggests that c-Kit kinase activity is also regulated by the molecular brake at the kinase hinge region. It is noteworthy that in the “active” c-Kit structure the A-loop tyrosine (Tyr-823) is not phosphorylated (Mol et al., 2003). Instead, the active A-loop conformation of c-Kit in the structure is stabilized by unique enzyme-substrate relationship between the two c-Kit enzymes in the asymmetric unit (Mol et al., 2003). Specifically, a phosphotyrosine residue from juxtamembrane region of the substrate-acting enzyme inserts into the active site of enzyme-acting c-Kit stabilizing the active A-loop conformation of the former enzyme. Interestingly, in the “active” c-Kit structure the unphosphoyrlated Tyr-823 directly hydrogen bonds with Arg-815 (Mol et al., 2003). This hydrogen bonding is equivalent to that between Tyr-657 and Arg-649 observed in our K659N mutant structure, and may also contribute to the active A-loop conformation in the “active” c-Kit structure.
Figure 6.
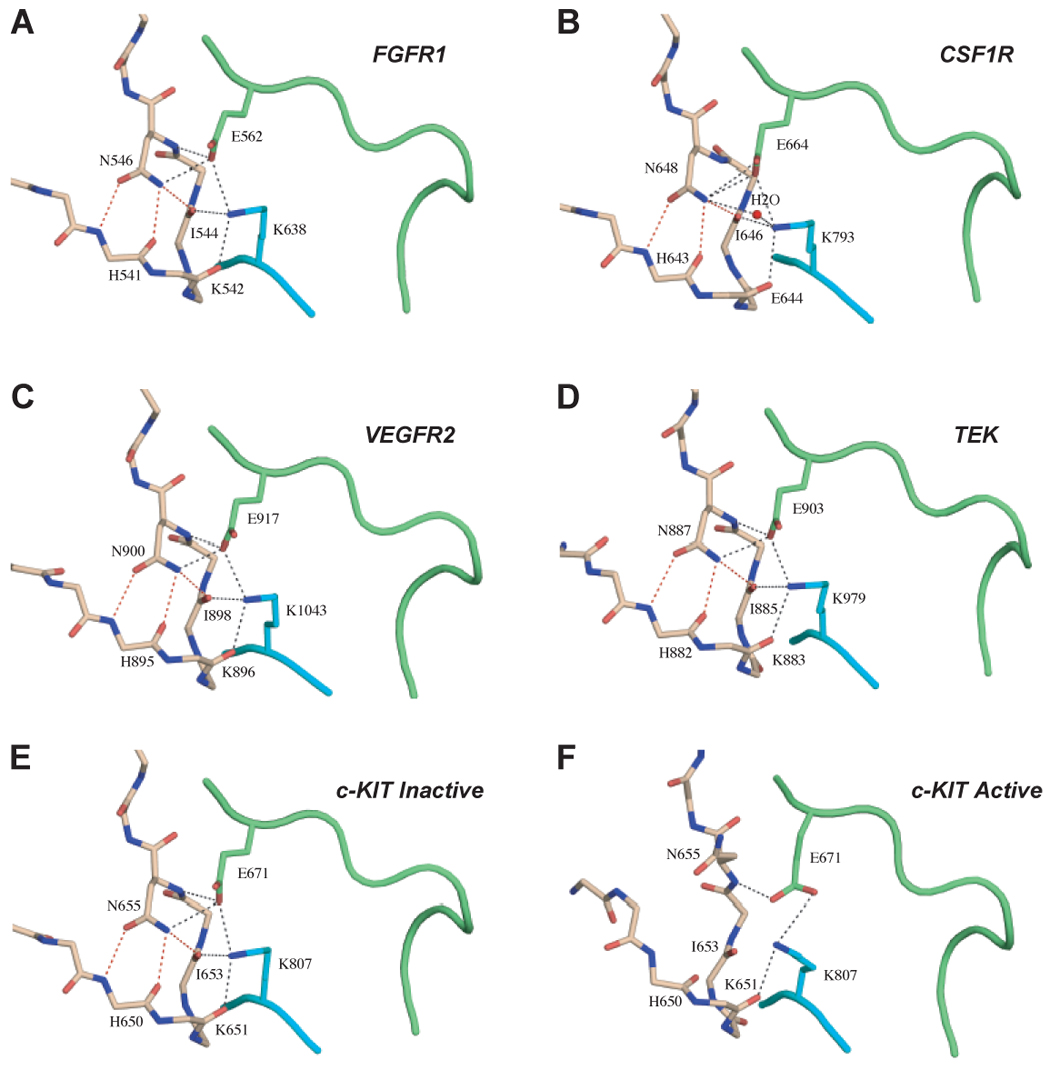
The autoinhibition by the molecular brake is a common regulatory mechanism for many RTKs. (A) to (E) show the presence of the engaged molecular brake at the kinase hinge region of unphosphorylated wild-type FGFR1 (PDB ID: 1FGK), CSF1R (PDB ID: 2I1M), VEGFR2 (PDB ID: 1VR2), TEK (PDB ID: 1FVR) and c-KIT (PDB ID: 1T45) kinases, respectively. (F) shows the disengagement of the molecular brake at the kinase hinge region of an “active” c-KIT kinase (PDB ID 1PKG). Coloring scheme is as in Fig. 3.
Our molecular brake hypothesis is further supported by the fact that pathogenic mutations at the N540 codon of FGFR3, N546 codon of FGFR1, and N659 codon of PDGFRα, which are homologous to the N549 codon of FGFR2, result in gain-of-function in human skeletal disorders (Webster and Donoghue, 1997) and cancer (Corless et al., 2005; Rand et al., 2005). Based on our structural data, the N549K mutations in FGFR2, the N540K/T/S mutations in FGFR3, N546K mutation in FGFR1 and N659K mutation in PDGFRα should disengage these residues from the backbone atoms of the αC-β4 loop, loosen the molecular brake, and cause gain-of-function in FGFR2, FGFR3, FGFR1 and PDGFRα in endometrial cancer, HCH, glioblastoma and gastrointestinal stromal tumors respectively. Moreover, the pathogenic mutation of K659E of FGFR2 and the pathogenic mutations of K650Q/N/M/E of FGFR3, corresponding to K659 of FGFR2, also should lead to receptor hyperactivation in human skeletal disorders (Bellus et al., 2000) and cancer (Cappellen et al., 1999; Chesi et al., 1997; Pollock et al., 2007; Richelda et al., 1997) by driving the A-loop to an active conformation and indirectly disengaging the brake at the kinase hinge region.
Conclusion
In this study we provided the structural basis for FGFR2 kinase activation by phosphorylation of the A-loop tyrosine, and the molecular basis by which seven different mutations in the kinase domain lead to pathogenic ligand-independent activation of FGFR2 in human skeletal disorders. The ensemble of 9 crystal structures disclosed an autoinhibitory mechanism in the regulation of FGFR kinases and many other RTKs. This autoinhibition is regulated by a molecular brake, mediated by a triad of residues in the kinase hinge region. Pathogenic mutations disengage this molecular brake either directly or indirectly to promote more readily ligand-independent autophosphorylation and activation.
The structure of A-loop tyrosine phosphorylated FGFR2K presented here is the first example of an A-loop phosphorylated activated RTK structure outside of the insulin receptor subfamily. Superimposition of the C-lobe of the A-loop tyrosine phosphorylated FGFR2K onto those of the A-loop tyrosine phosphorylated IRK (Hubbard, 1997) and IGF1RK (Favelyukis et al., 2001) reveals that the A-loops of all these activated kinases adopt a very similar conformation (Fig. 2D). Therefore, our data demonstrate that although the A-loops of different RTKs adopt different conformations in unphosphorylated state, they all assume a common conformation in the phosphorylated activated state. Superimposition of the β strands of N-lobe between phosphorylated and unphosphorylated FGFR2K shows that the αC helix in FGFR2 kinase does not shift independently of the other secondary structure elements of N-lobe (Fig. S2B). This is evidenced by that fact that the key salt bridge between K517 in the β3 strand and E534 in the αC helix is already present in the unphosphorylated wild-type FGFR2K structure (Fig. S2C). Hence, FGFR2K contrasts with the Cdks, Src family kinases and insulin/IGF receptor kinases whose αC helix can shift by as much as 10 Å relative to other N-lobe secondary structures upon activation.
The ensemble of our structural data provide much sought after snapshots of dynamics of RTK regulation by the three key regulatory regions of the kinase domain (the kinase hinge, the αC helix and the A-loop). The FGFR2K pathogenic mutations in the kinase hinge region disengage the molecular brake directly whereas mutations in the αC helix and the A-loop disengage it indirectly through long-range allosteric communication as depicted by the K659N mutant structure. The activating effect of the K659N mutation is relayed to the hinge region through a series of coupled intramolecular events consisting of reconfiguration of the A-loop to its active state, introduction of new set of interaction between the αC helix and the A-loop, and dissociation of the molecular brake. Taken together, these pathogenic mutations highlight the trajectory of a communication pathway between the A-loop, the αC helix and the kinase hinge and underscore that the three key regulatory regions of the kinase domain act in concert to regulate tyrosine kinase activity of RTKs (Fig. 7). We suggest that the unphosphorylated tyrosine kinase samples dynamically a spectrum of conformations. One end of the spectrum is populated by fully inhibited kinases with tightened molecular brake and the A-loop in the inactive conformation (represented by the unphsophorylated wild-type FGFR2K structure). The other end of the spectrum is populated by kinases with completely released molecular brake and the A-loop in the active conformation (depicted by the unphosphorylated K659N mutant structure). The latter state is ‘quasi stable’ in the absence of A-loop tyrosine phosphorylation, and the molecular brake at the kinase hinge region dominates, ‘swinging back’ the kinase to its inhibited state. This idea is supported by the fact that RTKs exhibit low level of basal tyrosine kinase activity in the absence of their ligands. Ligand binding and subsequent receptor dimerization increase the local concentration of the receptors at different autoinhibitory states, providing the few receptors in ‘quasi stable’ state with sufficient opportunity to transphosphorylate each other on the A-loop, holding them in the fully activated state (depicted by the A-loop tyrosine phosphorylated wild-type structure). The pathogenic mutations in the kinase hinge, the αC helix and the A-loop activate the kinases by loosening the molecular brake thereby shifting the equilibrium towards the activated state.
Figure 7.
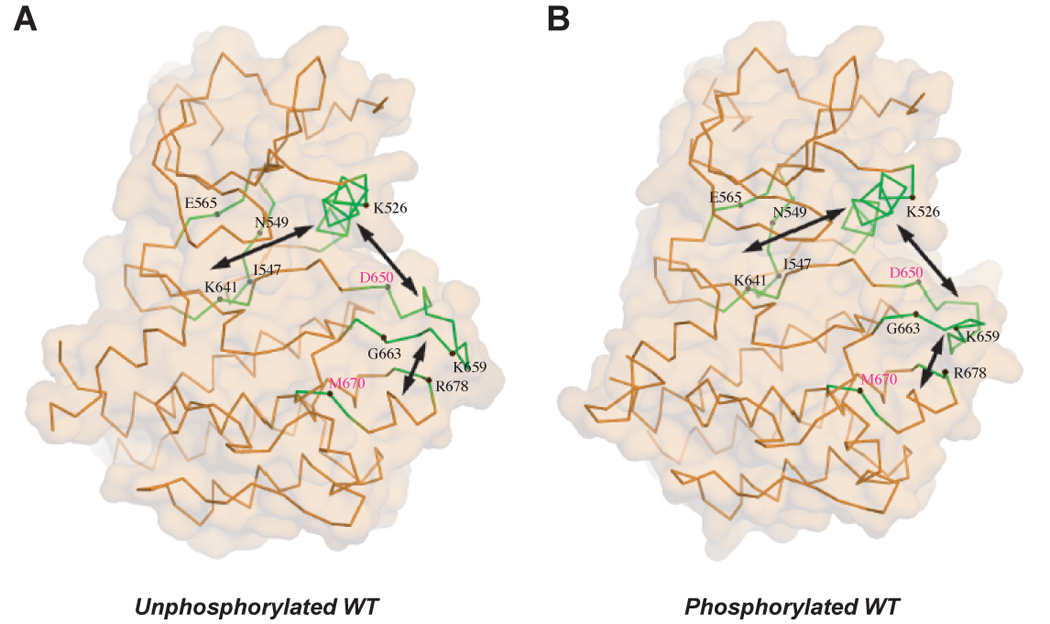
The pathogenic FGFR2K mutations highlight the existence of an allosteric communication between the three key regulatory regions of RTKs. The locations of mutated residues (labeled in black) are mapped onto the crystal structures of unphosphorylated (A) and phosphorylated (B) wild-type FGFR2 kinase. D650V and M670T mutations (residue location labeled in purple) also activate FGFR2K (unpublished results). D650V corresponds to the recurrent activating mutations D842V in PDGFRα, D816V in KIT and D835V in FLT3. M670T corresponds to the activating mutation of M918T in RET. The double headed arrows denote the reciprocal direction of communication between the A-loop, the αC helix and the kinase hinge.
EXPERIMENTAL PROCEDURES
Protein Expression, Purification and Crystallization
Wild-type and nine mutant FGFR2 kinase domains each harboring a distinct pathogenic mutation (K526E, N549H, N549T, E565G, E565A, K641R, K659N, G663E and R678G) were expressed with an N-terminal 6XHis-tag to aid in protein purification. The N- and C-terminal boundaries of the expressed FGFR2 kinase domain are Pro-458 and Glu-768, respectively. These boundaries were chosen based on the crystal structure of the unphosphorylated wild-type FGFR1 kinase domain (Mohammadi et al., 1996). The wild-type and mutant kinases were purified using sequential Ni2+-chelating, anion exchange and size exclusion chromatography. The purity of the proteins was estimated to be over 98% based on SDS-PAGE analysis. Traces of phosphorylation on wild-type and mutant kinases were removed by treating the proteins with alkaline phosphatase, and MALDI Q-TOF mass spectrometry was used to confirm that all purified kinases were not phosphorylated on the A-loop tyrosines. As examples, data for unphosphorylated wild-type and N549H and R678G mutant FGFR2Ks are shown in Fig. S4A to Fig. S4C. To generate A-loop tyrosine phosphorylated wild-type kinase, the purified wild-type FGFR2 kinase was mixed with 10 mM ATP and 5 mM MgCl2 and the completion of tyrosine autophosphorylation was monitored by native PAGE analysis. Phosphorylated wild-type kinase was then separated from excess of ATP on a size-exclusion column, followed by anion exchange chromatography to yield wild-type kinase that was homogenously phosphorylated on both Y656 and Y657 in the A-loop. The phosphorylation was confirmed by positive and negative mode MALDI Q-TOF mass spectrometry (Fig. S4D and S4E) after methyl esterification of tryptic peptides to improve detection of phosphopeptides (Xu et al., 2005). Wild-type (either unphosphorylated and A-loop tyrosine phosphorylated) and unphosphorylated mutant FGFR2K proteins were concentrated to about 10–100 mg/ml using Centricon-10. Prior to crystallization, the phosphorylated wild-type kinase was mixed with peptide substrate, ATP-analogue (AMP-PCP) and MgCl2 at a molar ratio of 1:1:3:15. The peptide substrate (15 amino acid long) is derived from the C-terminal tail of FGFR2 (residues 764 to 778) (Fig. S1) and contains Y769, one of the major autophosphorylation sites of FGFR2. Crystals of wild-type (either unphosphorylated and A-loop tyrosine phosphorylated) and unphosphorylated mutant kinases were grown by hanging drop vapor diffusion at 20 °C using crystallization buffer composed of 25 mM HEPES pH7.5, PEG4000 (15%–20%) and NH4SO3 (0.2–0.3 M). MALDI Q-TOF MS/MS analysis of the dissolved crystals showed stoichiometrical phosphorylation on Tyr-657 and heterogeneous phosphorylation on Tyr-656 (Fig. S4F and S4G). Mass spec analysis also showed heterogeneous phosphorylation on Tyr-586 in the kinase insert (Data not shown). Importantly, the mass spec data are consistent with the structure, which shows that phosphorylation on Tyr-657 is indispensable in stabilizing the active conformation of the A-loop. Phosphorylated Tyr-656 is solvent exposed and does not contribute to the activated A-loop conformation.
Data Collection and Structure Determination
Diffraction data were collected on single cryo-cooled crystals at beamlines X-4A and X-4C at the National Synchrotron Light Source, Brookhaven National Laboratory. Crystals were stabilized in mother liquor by stepwise increasing glycerol concentration to 20%, and then flash-frozen in dry nitrogen stream. All diffraction data were processed using HKL2000 Suite (Otwinowski and Minor, 1997). Molecular replacement solutions were found for unphosphorylated wild-type FGFR2K using unphosphorylated wild-type FGFR1K as the search model using the program AMoRe (Navaza, 1994). Subsequently, the unphosphorylated wild-type FGFR2K structure was used as search model for A-loop tyrosine phosphorylated wild-type and unphosphorylated mutant FGFR2 kinases. Rigid-body refinements of wild-type (either unphosphorylated and A-loop tyrosine phosphorylated) and unphosphorylated mutant kinases were performed using CNS (Brunger et al., 1998) by treating the N-lobe and C-lobe of the kinases as two separate entities. Model building was carried out using O (Jones et al., 1991) and iterative positional and B-factor refinements were done using CNS (Brunger et al., 1998). All refined structures display good geometry and Ramachandran statistics. Data collection and structure refinement statistic with PDB IDs of reported structures are listed in Table S2. Atomic superimpositions and calculations of lobe rotations were performed using program lsqkab (Kabsch, 1976) in CCP4 Suite (Collaborative, 1994) and structural representations were prepared using PyMol (DeLano, 2002). The Difference Distance Matrix Plot (DDMP) was used to compare conformational changes between wild-type and mutant kinases (Fleming, P.J., http://roselab.jhu.edu/ddmp/).
Kinase Assays
A continuous spectrophotometric assay (Barker et al., 1995) was used to measure the kinase activity of FGFR2K. The assays were carried out at 30°C in 100 mM Tris-HCl (pH7.5), 10 mM MgCl2, 1 mM phosphoenolpyruvate, 0.28 mM NADH, 89 units/ml pyruvate kinase, 124 units/ml lactate dehydrogenase, in a total volume of 50µl. Assays of FGFR2K autophosphorylation were carried out with 1 µM enzyme and 1 mM ATP. For FGFR2 peptide substrate phosphorylation assays, 1 µM enzyme, 100 µM ATP, and 50 µM peptide substrate (KKEEEEYMMMM) were used. To test the effect of A-loop phosphorylation on kinase activity of wild-type and mutant kinases, assays of FGFR2K peptide substrate phosphorylation were carried out with 1 µM enzyme, 500 µM ATP, and 500 µM peptide substrate (KKEEEEYMMMM). For determinations of Km, ATP, reactions contained a peptide concentration of 2 mM and ATP at varying concentrations of 10–4000 µM. For determinations of Km, peptide, reactions contained an ATP concentration of 1 mM and peptide substrate at varying concentrations of 10–2500 µM. Data were recorded every 6 seconds. The kinetic parameters were determined by fitting initial rate data to the Michaelis-Menten equation. Phosphorylation of the STAT1 peptide was measured by the phosphocellulose binding assay (Casnellie, 1991) under similar conditions.
Supplemental Data
Supplemental Data include six figures and three tables.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to Drs. R. Abramowitz and J. Schwanof for synchrotron beamline assistance, and Drs. J. Kuriyan, X.-P. Kong, S.R. Hubbard, and R. Goetz and Mr. Andrew Beenken for comments on the manuscript. Beamlines X-4A and X-4C at the National Synchrotron Light Source, Brookhaven National Laboratory, a DOE facility, are supported by the New York Structural Biology Consortium. This work was supported by National Institutes of Health grants DE13686 (to M.M.), and CA58530 (to W.T.M.). The NYU Protein Analysis Facility is supported by NIH Shared Instrumentation Grant S10 RR017990, NINDS grant P30 NS050276, and NCI core grant P30 CA016087 (to T.A.N.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Barker SC, Kassel DB, Weigl D, Huang X, Luther MA, Knight WB. Characterization of pp60c-src tyrosine kinase activities using a continuous assay: autoactivation of the enzyme is an intermolecular autophosphorylation process. Biochemistry. 1995;34:14843–14851. doi: 10.1021/bi00045a027. [DOI] [PubMed] [Google Scholar]
- Bellus GA, McIntosh I, Smith EA, Aylsworth AS, Kaitila I, Horton WA, Greenhaw GA, Hecht JT, Francomano CA. A recurrent mutation in the tyrosine kinase domain of fibroblast growth factor receptor 3 causes hypochondroplasia. Nat Genet. 1995;10:357–359. doi: 10.1038/ng0795-357. [DOI] [PubMed] [Google Scholar]
- Bellus GA, Spector EB, Speiser PW, Weaver CA, Garber AT, Bryke CR, Israel J, Rosengren SS, Webster MK, Donoghue DJ, Francomano CA. Distinct missense mutations of the FGFR3 lys650 codon modulate receptor kinase activation and the severity of the skeletal dysplasia phenotype. Am J Hum Genet. 2000;67:1411–1421. doi: 10.1086/316892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- Calvert JT, Riney TJ, Kontos CD, Cha EH, Prieto VG, Shea CR, Berg JN, Nevin NC, Simpson SA, Pasyk KA, et al. Allelic and locus heterogeneity in inherited venous malformations. Hum Mol Genet. 1999;8:1279–1289. doi: 10.1093/hmg/8.7.1279. [DOI] [PubMed] [Google Scholar]
- Cappellen D, De Oliveira C, Ricol D, de Medina S, Bourdin J, Sastre-Garau X, Chopin D, Thiery JP, Radvanyi F. Frequent activating mutations of FGFR3 in human bladder and cervix carcinomas. Nat Genet. 1999;23:18–20. doi: 10.1038/12615. [DOI] [PubMed] [Google Scholar]
- Casnellie JE. Assay of protein kinases using peptides with basic residues for phosphocellulose binding. Methods Enzymol. 1991;200:115–120. doi: 10.1016/0076-6879(91)00133-h. [DOI] [PubMed] [Google Scholar]
- Chesi M, Nardini E, Brents LA, Schrock E, Ried T, Kuehl WM, Bergsagel PL. Frequent translocation t(4;14)(p16.3;q32.3) in multiple myeloma is associated with increased expression and activating mutations of fibroblast growth factor receptor 3. Nat Genet. 1997;16:260–264. doi: 10.1038/ng0797-260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collaborative. The CCP4 suite: programs for protein crystallography. Acta Crystallographica Section D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Corless CL, Fletcher JA, Heinrich MC. Biology of gastrointestinal stromal tumors. J Clin Oncol. 2004;22:3813–3825. doi: 10.1200/JCO.2004.05.140. [DOI] [PubMed] [Google Scholar]
- Corless CL, Schroeder A, Griffith D, Town A, McGreevey L, Harrell P, Shiraga S, Bainbridge T, Morich J, Heinrich MC. PDGFRA mutations in gastrointestinal stromal tumors: frequency, spectrum and in vitro sensitivity to imatinib. J Clin Oncol. 2005;23:5357–5364. doi: 10.1200/JCO.2005.14.068. [DOI] [PubMed] [Google Scholar]
- de Ravel TJ, Taylor IB, Van Oostveldt AJ, Fryns JP, Wilkie AO. A further mutation of the FGFR2 tyrosine kinase domain in mild Crouzon syndrome. Eur J Hum Genet. 2005;13:503–505. doi: 10.1038/sj.ejhg.5201325. [DOI] [PubMed] [Google Scholar]
- DeLano WL. The PyMOL User's Manual. San Carlos: DeLano Scientific; 2002. [Google Scholar]
- Deutz-Terlouw PP, Losekoot M, Aalfs CM, Hennekam RC, Bakker E. Asn540Thr substitution in the fibroblast growth factor receptor 3 tyrosine kinase domain causing hypochondroplasia. Hum Mutat. 1998 Suppl 1:S62–S65. doi: 10.1002/humu.1380110122. [DOI] [PubMed] [Google Scholar]
- Favelyukis S, Till JH, Hubbard SR, Miller WT. Structure and autoregulation of the insulin-like growth factor 1 receptor kinase. Nat Struct Biol. 2001;8:1058–1063. doi: 10.1038/nsb721. [DOI] [PubMed] [Google Scholar]
- Gilliland DG. FLT3-activating mutations in acute promyelocytic leukaemia: a rationale for risk-adapted therapy with FLT3 inhibitors. Best Pract Res Clin Haematol. 2003;16:409–417. doi: 10.1016/s1521-6926(03)00063-x. [DOI] [PubMed] [Google Scholar]
- Grigelioniene G, Eklof O, Laurencikas E, Ollars B, Hertel NT, Dumanski JP, Hagenas L. Asn540Lys mutation in fibroblast growth factor receptor 3 and phenotype in hypochondroplasia. Acta Paediatr. 2000;89:1072–1076. doi: 10.1080/713794579. [DOI] [PubMed] [Google Scholar]
- Hubbard SR. Crystal structure of the activated insulin receptor tyrosine kinase in complex with peptide substrate and ATP analog. Embo J. 1997;16:5572–5581. doi: 10.1093/emboj/16.18.5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard SR, Till JH. Protein tyrosine kinase structure and function. Annu Rev Biochem. 2000;69:373–398. doi: 10.1146/annurev.biochem.69.1.373. [DOI] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A. 1991;47(Pt 2):110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- Kabsch W. A solution for the best rotation to relate two sets of vectors. Acta Crystallographica Section A. 1976;32:922–923. [Google Scholar]
- Kan SH, Elanko N, Johnson D, Cornejo-Roldan L, Cook J, Reich EW, Tomkins S, Verloes A, Twigg SR, Rannan-Eliya S, et al. Genomic screening of fibroblast growth-factor receptor 2 reveals a wide spectrum of mutations in patients with syndromic craniosynostosis. Am J Hum Genet. 2002;70:472–486. doi: 10.1086/338758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karkkainen MJ, Ferrell RE, Lawrence EC, Kimak MA, Levinson KL, McTigue MA, Alitalo K, Finegold DN. Missense mutations interfere with VEGFR-3 signalling in primary lymphoedema. Nat Genet. 2000;25:153–159. doi: 10.1038/75997. [DOI] [PubMed] [Google Scholar]
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. Journal of Applied Crystallography. 1993;26:283–291. [Google Scholar]
- Liang DC, Shih LY, Hung IJ, Yang CP, Chen SH, Jaing TH, Liu HC, Wang LY, Chang WH. FLT3-TKD mutation in childhood acute myeloid leukemia. Leukemia. 2003;17:883–886. doi: 10.1038/sj.leu.2402928. [DOI] [PubMed] [Google Scholar]
- McGillivray G, Savarirayan R, Cox TC, Stojkoski C, McNeil R, Bankier A, Bateman JF, Roscioli T, Gardner RJ, Lamande SR. Familial scaphocephaly syndrome caused by a novel mutation in the FGFR2 tyrosine kinase domain. J Med Genet. 2005;42:656–662. doi: 10.1136/jmg.2004.027888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McTigue MA, Wickersham JA, Pinko C, Showalter RE, Parast CV, Tempczyk-Russell A, Gehring MR, Mroczkowski B, Kan CC, Villafranca JE, Appelt K. Crystal structure of the kinase domain of human vascular endothelial growth factor receptor 2: a key enzyme in angiogenesis. Structure. 1999;7:319–330. doi: 10.1016/s0969-2126(99)80042-2. [DOI] [PubMed] [Google Scholar]
- Medeiros F, Corless CL, Duensing A, Hornick JL, Oliveira AM, Heinrich MC, Fletcher JA, Fletcher CD. KIT-negative gastrointestinal stromal tumors: proof of concept and therapeutic implications. Am J Surg Pathol. 2004;28:889–894. doi: 10.1097/00000478-200407000-00007. [DOI] [PubMed] [Google Scholar]
- Mohammadi M, Schlessinger J, Hubbard SR. Structure of the FGF receptor tyrosine kinase domain reveals a novel autoinhibitory mechanism. Cell. 1996;86:577–587. doi: 10.1016/s0092-8674(00)80131-2. [DOI] [PubMed] [Google Scholar]
- Mol CD, Dougan DR, Schneider TR, Skene RJ, Kraus ML, Scheibe DN, Snell GP, Zou H, Sang BC, Wilson KP. Structural basis for the autoinhibition and STI-571 inhibition of c-Kit tyrosine kinase. J Biol Chem. 2004;279:31655–31663. doi: 10.1074/jbc.M403319200. [DOI] [PubMed] [Google Scholar]
- Mol CD, Lim KB, Sridhar V, Zou H, Chien EY, Sang BC, Nowakowski J, Kassel DB, Cronin CN, McRee DE. Structure of a c-kit product complex reveals the basis for kinase transactivation. J Biol Chem. 2003;278:31461–31464. doi: 10.1074/jbc.C300186200. [DOI] [PubMed] [Google Scholar]
- Mortier G, Nuytinck L, Craen M, Renard JP, Leroy JG, de Paepe A. Clinical and radiographic features of a family with hypochondroplasia owing to a novel Asn540Ser mutation in the fibroblast growth factor receptor 3 gene. J Med Genet. 2000;37:220–224. doi: 10.1136/jmg.37.3.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navaza J. AMoRe: an automated package for molecular replacement. Acta Crystallographica Section A. 1994;50:157–163. [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. Vol 276. New York: Academic Press; 1997. [DOI] [PubMed] [Google Scholar]
- Passos-Bueno MR, Wilcox WR, Jabs EW, Sertie AL, Alonso LG, Kitoh H. Clinical spectrum of fibroblast growth factor receptor mutations. Hum Mutat. 1999;14:115–125. doi: 10.1002/(SICI)1098-1004(1999)14:2<115::AID-HUMU3>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Pollock PM, Gartside MG, Dejeza LC, Powell MA, Mallon MA, Davies H, Mohammadi M, Futreal PA, Stratton MR, Trent JM, Goodfellow PJ. Frequent activating FGFR2 mutations in endometrial carcinomas parallel germline mutations associated with craniosynostosis and skeletal dysplasia syndromes. Oncogene advance online publication. 2007 doi: 10.1038/sj.onc.1210529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand V, Huang J, Stockwell T, Ferriera S, Buzko O, Levy S, Busam D, Li K, Edwards JB, Eberhart C, et al. Sequence survey of receptor tyrosine kinases reveals mutations in glioblastomas. Proc Natl Acad Sci U S A. 2005;102:14344–14349. doi: 10.1073/pnas.0507200102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richelda R, Ronchetti D, Baldini L, Cro L, Viggiano L, Marzella R, Rocchi M, Otsuki T, Lombardi L, Maiolo AT, Neri A. A novel chromosomal translocation t(4; 14)(p16.3; q32) in multiple myeloma involves the fibroblast growth-factor receptor 3 gene. Blood. 1997;90:4062–4070. [PubMed] [Google Scholar]
- Schittenhelm MM, Yee KW, Tyner JW, McGreevey L, Haley AD, Town A, Griffith DJ, Bainbridge T, Braziel RM, O'Farrell AM, et al. FLT3 K663Q is a novel AML-associated oncogenic kinase: Determination of biochemical properties and sensitivity to Sunitinib (SU11248) Leukemia. 2006;20:2008–2014. doi: 10.1038/sj.leu.2404374. [DOI] [PubMed] [Google Scholar]
- Schubert C, Schalk-Hihi C, Struble GT, Ma HC, Petrounia IP, Brandt B, Deckman IC, Patch RJ, Player MR, Spurlino JC, Springer BA. Crystal structure of the tyrosine kinase domain of colony-stimulating factor-1 receptor (cFMS) in complex with two inhibitors. J Biol Chem. 2006;28:28. doi: 10.1074/jbc.M608183200. [DOI] [PubMed] [Google Scholar]
- Shewchuk LM, Hassell AM, Ellis B, Holmes WD, Davis R, Horne EL, Kadwell SH, McKee DD, Moore JT. Structure of the Tie2 RTK domain: self-inhibition by the nucleotide binding loop, activation loop, and C-terminal tail. Structure. 2000;8:1105–1113. doi: 10.1016/s0969-2126(00)00516-5. [DOI] [PubMed] [Google Scholar]
- van Rhijn BW, van Tilborg AA, Lurkin I, Bonaventure J, de Vries A, Thiery JP, van der Kwast TH, Zwarthoff EC, Radvanyi F. Novel fibroblast growth factor receptor 3 (FGFR3) mutations in bladder cancer previously identified in non-lethal skeletal disorders. Eur J Hum Genet. 2002;10:819–824. doi: 10.1038/sj.ejhg.5200883. [DOI] [PubMed] [Google Scholar]
- Vikkula M, Boon LM, Carraway KL, 3rd, Calvert JT, Diamonti AJ, Goumnerov B, Pasyk KA, Marchuk DA, Warman ML, Cantley LC, et al. Vascular dysmorphogenesis caused by an activating mutation in the receptor tyrosine kinase TIE2. Cell. 1996;87:1181–1190. doi: 10.1016/s0092-8674(00)81814-0. [DOI] [PubMed] [Google Scholar]
- Walter JW, North PE, Waner M, Mizeracki A, Blei F, Walker JW, Reinisch JF, Marchuk DA. Somatic mutation of vascular endothelial growth factor receptors in juvenile hemangioma. Genes Chromosomes Cancer. 2002;33:295–303. doi: 10.1002/gcc.10028. [DOI] [PubMed] [Google Scholar]
- Webster MK, Donoghue DJ. FGFR activation in skeletal disorders: too much of a good thing. Trends Genet. 1997;13:178–182. doi: 10.1016/s0168-9525(97)01131-1. [DOI] [PubMed] [Google Scholar]
- Wilkie AO. Bad bones, absent smell, selfish testes: the pleiotropic consequences of human FGF receptor mutations. Cytokine Growth Factor Rev. 2005;16:187–203. doi: 10.1016/j.cytogfr.2005.03.001. [DOI] [PubMed] [Google Scholar]
- Wilkin DJ, Hecht JT, Francomano CA. Achondroplasia and Pseudoachondroplasia. Vol 4. New York: McGraw-Hill; 2001. [Google Scholar]
- Xu CF, Lu Y, Ma J, Mohammadi M, Neubert TA. Identification of phosphopeptides by MALDI Q-TOF MS in positive and negative ion modes after methyl esterification. Mol Cell Proteomics. 2005;4:809–818. doi: 10.1074/mcp.T400019-MCP200. [DOI] [PubMed] [Google Scholar]
- Yamamoto Y, Kiyoi H, Nakano Y, Suzuki R, Kodera Y, Miyawaki S, Asou N, Kuriyama K, Yagasaki F, Shimazaki C, et al. Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood. 2001;97:2434–2439. doi: 10.1182/blood.v97.8.2434. [DOI] [PubMed] [Google Scholar]
- Zankl A, Jaeger G, Bonafe L, Boltshauser E, Superti-Furga A. Novel mutation in the tyrosine kinase domain of FGFR2 in a patient with Pfeiffer syndrome. Am J Med Genet A. 2004;131:299–300. doi: 10.1002/ajmg.a.30366. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.