

Abstract
The natural history of idiopathic pulmonary fibrosis (IPF) has been characterized as a steady, predictable decline in lung function over time. Recent evidence suggests that some patients may experience a more precipitous course, with periods of relative stability followed by acute deteriorations in respiratory status. Many of these acute deteriorations are of unknown etiology and have been termed acute exacerbations of IPF. This perspective is the result of an international effort to summarize the current state of knowledge regarding acute exacerbations of IPF. Acute exacerbations of IPF are defined as acute, clinically significant deteriorations of unidentifiable cause in patients with underlying IPF. Proposed diagnostic criteria include subjective worsening over 30 days or less, new bilateral radiographic opacities, and the absence of infection or another identifiable etiology. The potential pathobiological roles of infection, disordered cell biology, coagulation, and genetics are discussed, and future research directions are proposed.
Keywords: acute exacerbation, pulmonary fibrosis, diagnosis, definition
Idiopathic pulmonary fibrosis (IPF) is the most common form of idiopathic interstitial pneumonia (1, 2). No therapy has been proven effective, and median survival from the time of diagnosis is approximately 3 years (3–5). Historically, IPF has been described as a gradually progressive disease, characterized by steady worsening of symptoms, lung function, and gas exchange (6, 7). More recently, it has been recognized that some patients with IPF experience acute respiratory deterioration, suggesting that the clinical course may be less predictable (8). Many of these acute declines are idiopathic—that is, without clinically apparent infection, left heart failure, pulmonary embolism, or other identifiable cause. These episodes of idiopathic acute deterioration have been termed acute exacerbations of IPF (9, 10).
This perspective summarizes the current state of knowledge regarding acute exacerbations of IPF, proposes a definition and diagnostic criteria, and suggests areas for future investigation. It is an international perspective, combining the efforts of the Idiopathic Pulmonary Fibrosis Clinical Research Network (IPFnet), an NHLBI-sponsored collaborative formed to advance the clinical care of patients with IPF, with those of experienced individual investigators. It is the authors' strong belief that a consensus approach to defining acute exacerbations of IPF is essential to furthering our knowledge of this condition.
Significance of Acute Exacerbation
A number of studies have reported on the clinical significance of acute exacerbations of IPF (Table 1) (9–27). These studies are primarily retrospective case series and provide discordant estimates of incidence and mortality rates. This is likely due to differences in study design, particularly substantial variation in the case definition of acute exacerbation.
TABLE 1. PUBLISHED STUDIES OF ACUTE EXACERBATION.
| Publication | Study Design | No.* | Age (yr) | M/F | Incidence (%) | Mortality Rate (%) |
|---|---|---|---|---|---|---|
| Churg and colleagues, 2007 (27) | Retrospective review of lung biopsy cases | 9 | 62 | 3/6 | N/A | 22 |
| Tiitto and colleagues, 2006 (19) | Autopsy review | 9 | N/A | N/A | 21 | N/A |
| Okamoto and colleagues, 2006 (17) | Retrospective review of hospital admissions | 28 | 68 | 20/8 | 12.5 (2-yr incidence) | 86 |
| Kim and colleagues, 2006 (13) | Retrospective longitudinal cohort | 11 | 63 | 6/2 | 9.6 (2-yr incidence) | 78 |
| Kondoh and colleagues, 2006 (23) | Retrospective review of post-lung biopsy cases | 3 | 63 | 3/0 | N/A | 67 |
| Parambil and colleagues, 2005 (18) | Case series | 7 | 70 | 5/2 | N/A | 86 |
| Azuma and colleagues, 2005 (13) | Randomized controlled trial | 5 | N/A | N/A | 5 (9-mo incidence) | 20 |
| Kubo and colleagues, 2005 (16) | Randomized controlled trial | 32 | N/A | N/A | 57 (3-yr incidence) | 53 |
| Kondoh and colleagues, 2005 (15) | Prospective trial | 6 | 61 | 3/0 | 22 (5-yr incidence) | N/A |
| Homma and colleagues, 2005 (20) | Case series | 10 | 70 | 10/0 | N/A | 100 |
| Al-Hameed and Sharma, 2004 (21) | Retrospective review of ICU admissions | 25 | 69 | 23/2 | N/A | 96 |
| Rice and colleagues, 2003 (24) | Autopsy review | 12 | 66 | 10/2 | N/A | 100 |
| Ambrosini and colleagues, 2003 (12) | Case series | 5 | 70 | 4/1 | N/A | 80 |
| Saydain and colleagues, 2002 (25) | Retrospective review of ICU admissions | 15 | N/A | N/A | 47 | N/A |
| Stern and colleagues, 2001 (81) | Retrospective review of ICU admissions | 14 | N/A | N/A | 61 | >90 |
| Blivet and colleagues, 2001 (22) | Retrospective review of ICU admissions | 6 | N/A | N/A | 40 | 100 |
| Akira and colleagues, 1997 (11) | Case series | 17 | 63 | 14/3 | N/A | 53 |
| Kondoh and colleagues, 1993 (10) | Case series | 3 | 61 | 3/0 | N/A | 0 |
| Kondo and Saiki, 1989 (9) | Survey | 51 | N/A | N/A | 33 | 96 |
| Kondo and Saiki, 1989 (9) | Retrospective cohort | 4 | N/A | N/A | 18 | N/A |
Definition of abbreviations: ICU = intensive care unit; M/F = male/female; N/A = not available.
Number of acute exacerbations reported.
Three recent studies provide information regarding the incidence and mortality of acute exacerbations of IPF. A randomized, controlled trial of anticoagulant therapy in IPF included rehospitalization (which included acute exacerbation) as a secondary endpoint (16). Of 56 patients followed for approximately 3 years, 32 (57%) were rehospitalized for acute exacerbation and 53% of those patients died. Importantly, subjects in this study were initially enrolled during an admission to the hospital, suggesting the population recruited was enriched for previous acute exacerbation or progressive disease. A retrospective review of an observational cohort of 147 patients with IPF identified 11 who met criteria for acute exacerbation (14). The 2-year incidence of acute exacerbation was reported at 9.6%, and mortality at 78%. The time to development of acute exacerbation from the subjects' initial visits was between 3 and 60 months. Identifying acute exacerbations in this study was hindered by the retrospective analysis, as illustrated by 14 additional cases of acute worsening that could not be included due to lack of data. Finally, acute exacerbation was included as a secondary endpoint in a randomized, double-blinded, placebo-controlled trial of pirfenidone for the treatment of IPF (13). Of 107 patients followed for 6 months, 5 developed acute exacerbation, 1 of whom died. All cases were in the placebo group, suggesting that the incidence in untreated IPF might be higher (14%).
Current Diagnosis and Management of Acute Exacerbation
There is no established consensus approach to the diagnosis of acute exacerbation of IPF. Although specific criteria vary considerably among the published case series, most studies require a combination of the following: worsening of dyspnea within days to weeks (generally < 30 d); evidence of abnormal gas exchange as defined by a low partial pressure of arterial oxygen (PaO2)/percentage of inspired oxygen (FiO2) ratio or a decrease in PaO2; new radiographic opacities; and an absence of an alternative explanation, such as infection, left heart failure, or pulmonary embolism (9, 10–20). There is variation in the rigor with which this last criterion, particularly with regard to the evaluation for possible infection, has been approached.
Treatment of acute exacerbation of IPF has generally consisted of high-dose corticosteroids, but there are no data from controlled trials to prove their efficacy. Cyclosporin A has been studied, but no convincing evidence of benefit has been demonstrated (17, 20). Data from randomized clinical trials involving patients with IPF suggest possible roles for anticoagulant and antifibrotic therapies. Fifty-six subjects randomized to anticoagulant therapy over 3 years had a lower mortality from acute exacerbation than subjects who were untreated (18 vs. 71%) (16). This study was limited by several factors that prevent definitive conclusions (28). Most important, the study was not blinded and had a large and differential dropout of 25% in the treatment group that eliminated the benefits of randomization. A recent placebo-controlled trial of pirfenidone showed no difference in the primary endpoint of change in lowest oxygen saturation with exercise, but it was stopped early because of a statistically significant decrease in the risk of acute exacerbation in the pirfenidone-treated arm, suggesting a potential preventive role for this agent (13). Although this difference was statistically significant, there were only five acute exacerbation events on which this analysis was based, and confirmation of this potential preventive effect in a larger, longer term study is necessary.
Recognized Components of Acute Exacerbation
Risk Factors
Acute exacerbations of IPF appear to occur at any time during the course of disease, and for some patients, may be the presenting manifestation of their disease. Importantly, the risk of an exacerbation does not appear to be linked to the level of pulmonary function derangement (14), although in one prospective series, patients with lower forced vital capacity had more total and respiratory hospitalizations during subsequent follow-up (29). There is no clear association with age or smoking history, but acute exacerbations seem to be more common in men. Several reports suggest that surgical lung biopsy may be a risk factor, although distinguishing these cases from post-surgical acute respiratory distress syndrome is difficult (30–34).
Clinical Features
Acute to subacute worsening of dyspnea is invariable in patients with acute exacerbation of IPF. Development of new or worsening dyspnea generally occurs within 30 days, although certain series have reported longer time courses (11, 24). Cough, fever, and flulike symptoms are additional findings (12, 14, 18). Many patients present with severe hypoxemia, and respiratory failure requiring mechanical ventilation is common.
Laboratory Findings
Significant gas exchange abnormalities have been almost universally described (10, 12–20). The most common criteria for abnormal gas exchange have been a PaO2/FiO2 ratio of less than 225 and a decrease over time in PaO2 of 10 mm Hg or greater. Systematic studies of serum and bronchoalveolar lavage fluid (BALF) from patients with acute exacerbations of IPF are not available. Exaggerated ST2 protein, IL-8, and α-defensin levels have been reported in some patients with acute exacerbations, suggesting the importance of activated T cells and neutrophils (35–38). These levels correlate negatively with lung function parameters. An increase in BALF neutrophils has also been reported (12, 14, 18). Serum KL-6, neutrophil elastase, and lactate hydrogenase (LDH) levels have been suggested as markers of acute exacerbation of IPF (39).
Radiology
Early reports of acute exacerbation of IPF describe diffuse ground-glass opacities on plain chest radiograph (9, 10). High-resolution computed tomography (HRCT) generally demonstrates bilateral ground-glass abnormality with or without areas of consolidation, superimposed on the bibasilar subpleural reticular abnormality, traction bronchiectasis, and honeycomb change typical of usual interstitial pneumonia (UIP) pattern (Figure 1) (11, 12, 14).
Figure 1.
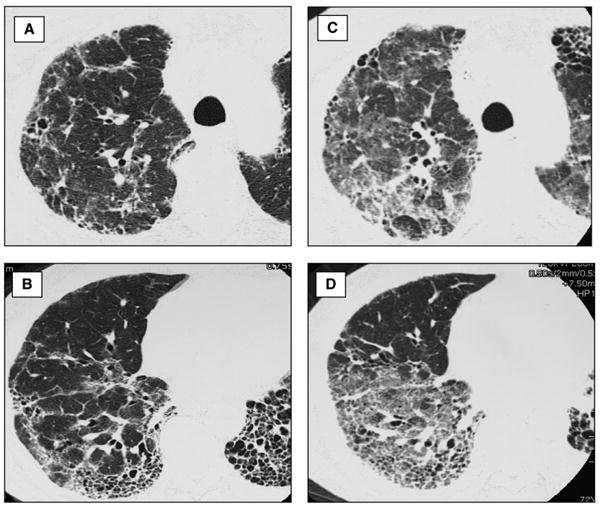
High-resolution computed tomography (HRCT) of acute exacerbation of idiopathic pulmonary fibrosis (IPF). (A, B) HRCT images through right upper and lower lung of a patient with stable IPF show typical features of usual interstitial pneumonia pattern: peripheral and basal predominant reticular abnormality with traction bronchiectasis and subpleural honeycombing. A small amount of ground-glass abnormality is present. (C, D) HRCT images through the right upper and lower lung of the same patient during an acute exacerbation of IPF show new, extensive ground-glass abnormality superimposed on the background of lung fibrosis.
Three HRCT patterns of abnormality have been suggested: peripheral, multifocal, and diffuse ground glass (11). The multifocal and diffuse patterns have been associated with diffuse alveolar damage superimposed on UIP on surgical lung biopsy. Survival may be related to the degree of CT involvement (11, 12, 14).
Histopathology
Diffuse alveolar damage superimposed on underlying UIP is the most commonly described finding when surgical lung biopsy is performed (Figure 2) (11, 10, 12, 14, 18, 24, 27). Diffuse alveolar damage has been described in nearly three-quarters of the lung specimens reported in the literature, and, in all but one case, the findings were associated with histologic evidence of underlying UIP. Organizing pneumonia without other evidence of organizing diffuse alveolar damage and extensive fibroblastic foci have also been described in a few cases (27).
Figure 2.
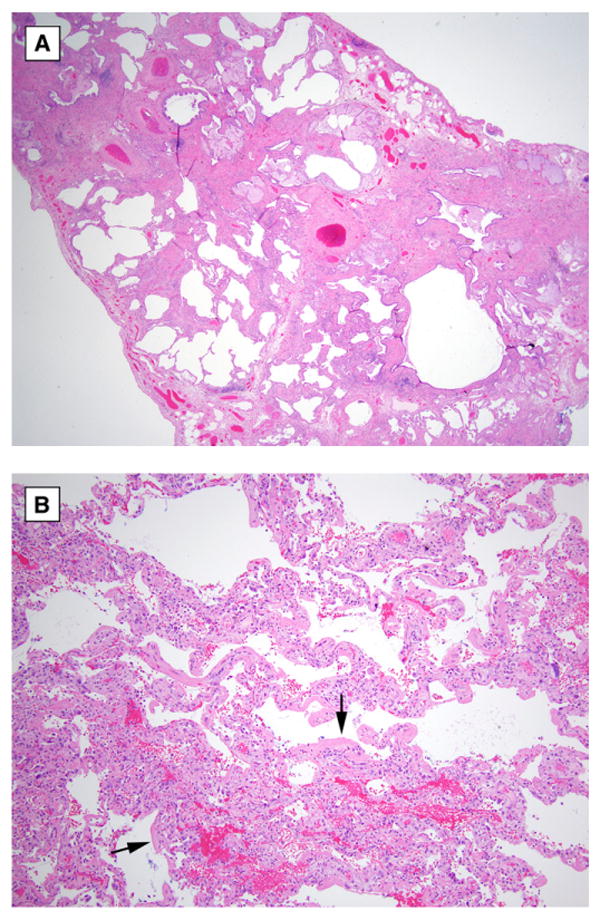
Surgical lung biopsy of acute exacerbation of idiopathic pulmonary fibrosis. (A) Low magnification photomicrograph shows chronic interstitial pneumonia with heterogeneous distribution and honeycomb change typical of usual interstitial pneumonia (original magnification, ×20; hematoxylin-and-eosin stain). (B) Higher magnification photomicrograph shows area of acute diffuse alveolar damage in same biopsy specimen characterized by well-developed eosinophilic hyaline membranes (arrows) (original magnification, ×100; hematoxylin-and-eosin stain).
Proposed Definition and Diagnostic Criteria
We propose the following definition for acute exacerbation of IPF: an acute, clinically significant deterioration of unidentifiable cause in a patient with underlying IPF. Proposed diagnostic criteria for acute exacerbation of IPF are listed in Table 2. Clinical deteriorations of IPF that are of unknown cause but do not fulfill the criteria listed due to missing data should be termed “suspected acute exacerbations of IPF.” This could include the following cases: those that are of more than 30 days' duration, those that have unilateral ground-glass abnormalities, or those that have not undergone endotracheal aspirate or BAL evaluation. In developing these criteria, we have tried to reconcile the desire for diagnostic specificity with our lack of understanding regarding the etiology and pathobiology of acute exacerbations. It is hoped that the diagnostic criteria presented here will serve to standardize inclusion criteria in future research studies, and that these criteria will be revised and modified as we gain additional insight and information.
TABLE 2. DIAGNOSIS OF ACUTE EXACERBATION.
| Diagnostic Criteria |
|---|
| Previous or concurrent diagnosis of idiopathic pulmonary fibrosis* |
| Unexplained worsening or development of dyspnea within 30 days |
| High-resolution computed tomography with new bilateral ground-glass abnormality and/or consolidation superimposed on a background reticular or honeycomb pattern consistent with usual interstitial pneumonia pattern† |
| No evidence of pulmonary infection by endotracheal aspirate or bronchoalveolar lavage‡ |
| Exclusion of alternative causes, including the following: |
|
Patients with idiopathic clinical worsening who fail to meet all five criteria due to missing data should be termed “suspected acute exacerbations.”
If the diagnosis of idiopathic pulmonary fibrosis is not previously established according to American Thoracic Society/European Respiratory Society consensus criteria (2), this criterion can be met by the presence of radiologic and/or histopathologic changes consistent with usual interstitial pneumonia pattern on the current evaluation.
If no previous high-resolution computed tomography is available, the qualifier “new” can be dropped.
Evaluation of samples should include studies for routine bacterial organisms, opportunistic pathogens, and common viral pathogens.
Causes of acute lung injury include sepsis, aspiration, trauma, reperfusion pulmonary edema, pulmonary contusion, fat embolization, inhalational injury, cardiopulmonary bypass, drug toxicity, acute pancreatitis, transfusion of blood products, and stem cell transplantation (50).
Several items regarding the proposed diagnostic criteria deserve further emphasis and explanation. First, the choice of 30 days or less as the time course for acute exacerbation, while based on published experience, is recognized as arbitrary. Certainly, cases of idiopathic clinical deterioration over longer periods of time (e.g., 2 mo) may share a similar pathobiology. However, it was believed that a relatively short time period was important to separate acute worsening from the more gradual progression seen in many patients with IPF. Second, the requirement for documentation of abnormal gas exchange present in previously published diagnostic criteria has been removed. Any diagnostic specificity gained from including documentation of abnormal gas exchange as a requirement was believed to be outweighed by the decreased sensitivity (cases with clear exacerbation but without previous data points would be excluded, and there would be bias toward more severe disease; i.e., patients with comparison data would more likely be sicker and/or have more advanced disease) its inclusion would cause. Third, endotracheal aspirate or BAL is recommended to investigate a potentially infectious etiology. Evaluation of these samples should include routine studies for bacterial organisms, opportunistic pathogens (e.g., Pneumocystis jiroveci), and common respiratory viruses. Although the epidemiology of viral respiratory infections varies globally, relevant viruses in North America would include influenza A and B, parainfluenza 1–4, respiratory syncytial virus A and B, human metapneumovirus, adenoviruses, rhinovirus, and coronaviruses. In cases complicated by suspected nosocomial infection, the etiologic role of organisms identified on culture should be decided based on clinical judgment. Fourth, acute exacerbations of other forms of interstitial lung disease (e.g., nonspecific interstitial pneumonia) have been deliberately excluded, although they may turn out to have a similar etiology and/or pathobiology. As more is learned about acute exacerbations in IPF, it may be appropriate to broaden the definition to include acute exacerbations of other conditions. Fifth, the exclusion of alternative etiologies, such as heart failure and pulmonary embolism, is left intentionally vague, because the diagnostic approach to these conditions is complex and beyond the scope of this article to adequately describe.
Potential Etiology and Biology of Acute Exacerbations
Etiology
The etiology of acute exacerbations of IPF is unknown; we propose several hypotheses that should be tested in future research studies. First, perhaps acute exacerbations of IPF represent a distinct, pathobiological manifestation of the primary disease process, characterized by idiopathic lung injury. Second, acute exacerbations of IPF may represent clinically occult but biologically distinct conditions that go undiagnosed (e.g., viral infection, aspiration). Viral infections, especially herpesviruses, have been associated with IPF and may cause acute deterioration (40–46). Gastroesophageal reflux is common in patients with IPF and may lead to aspiration of gastric contents—a known cause of diffuse alveolar damage—and acute worsening (47–50). Third, acute exacerbations of IPF may be the sequelae of an acute direct stress to the lung, with a subsequent acceleration of the already abnormal fibroproliferative process intrinsic to IPF (51). No consensus opinion was reached regarding the relative likelihood of these competing hypotheses.
Biology
There is little known regarding the pathobiology of acute exacerbations of IPF. Disordered epithelial cell integrity, cellular inflammation, cytokines, matrix metalloproteinases (MMPs), and coagulation components are all likely involved in the pathogenesis of IPF (52), and rapid alteration in these processes may contribute to acute exacerbations. Below we expand on some of these biological processes and discuss how they might be involved in acute exacerbations of IPF.
Epithelial cell integrity
Loss of alveolar epithelial cell integrity and injury may play an important role in acute exacerbations, leading to the extrusion of fibrin onto the alveolar surface and remodeling (53). Acute exacerbation is morphologically characterized by BALF neutrophilia and histopathology showing diffuse alveolar damage. Both of these observations suggest loss of alveolar epithelial cell integrity and injury may play an important role (12, 18, 24). Environmental factors likely interact with a genetic variability in epithelial cell function, which may explain why only a subset of patients with IPF appears to develop acute exacerbations (54).
Fibrocyte function
Fibrocytes are circulating bone marrow–derived precursors (55–57) that migrate to the lung in both human disease (58–62) and animal models of fibrosis (58, 59). Fibrocytes can be recruited in response to chemokines generated by infection or injury and may potentiate fibrogenesis via extracellular matrix production and/or secretion of profibrotic factors (55, 58, 59, 63). Recent evidence suggests that the percentage of circulating fibrocytes is increased in IPF compared with normal control subjects (6–10 vs. 0.5–2.4%, respectively) (62, 64). In patients experiencing acute exacerbation, the level is further elevated. In one patient, the level of fibrocytes measured during acute exacerbation was 23.1% and this decreased to 3.7% upon recovery 6 weeks later (64). Whether fibrocyte recruitment and/or function is abnormal in acute exacerbation is unknown.
MMP-9 and transforming growth factor-β
MMPs regulate extracellular matrix turnover (65). Patients with rapidly progressive IPF show increases in active MMP-9 in BALF (66). Excessive MMP-9 may severely disrupt the structural and functional integrity of the alveolar–capillary basement membrane (67, 68) and may activate latent transforming growth factor (TGF)-β, a profibrotic cytokine (69, 70). TGF-β may be further activated by stretching of the lung through a mechanism involving integrins (71). For example, single-lung ventilation may lead to stretch-dependent TGF-β activation and acute exacerbation, a hypothesis supported by the observation that acute exacerbations after video-assisted thorascopic surgery (VATS) were more prominent in the intraoperatively ventilated lung (24). Thus, increases in MMP-9 and TGF-β activation may promote the development of acute exacerbation in IPF.
Disordered coagulation and fibrinolysis
Disordered coagulation and fibrinolysis may be important components of acute exacerbations of IPF (16). Studies of patients with stable IPF have demonstrated a procoagulant and antifibrinolytic alveolar environment (29, 72–74), and a similar environment has been described in the acute respiratory distress syndrome (ARDS) (74–76). The presence of disordered coagulation and fibrinolysis in stable IPF and the clinical and pathologic similarities of acute exacerbations with ARDS support a role for these mechanisms.
Genetic predisposition
A polymorphism in erythrocyte complement receptor 1 and mutations in surfactant protein genes are present in selected patients with IPF (77, 78). Recently, the presence of heterozygous mutations in the telomerase reverse transcriptase (hTERT) and/or RNA component (hTR) genes encoding telomerase components have been reported in several families with IPF and in one patient with sporadic IPF (79, 80). These mutations lead to shortened telomeres that may limit the regenerative capacity of alveolar epithelial cells and contribute to the pathobiology of acute exacerbation.
Future Directions
Our understanding of acute exacerbations of IPF is limited and key questions remain. There are several important areas for future research. Investigators should focus on characterizing the epidemiology of acute exacerbations. What is the true incidence of acute exacerbations? Are they a common complication of IPF? What is their prognostic significance? What are the risk factors (clinical, biological, and genetic) for their development? Are acute exacerbations of IPF different from acute exacerbations of other forms of interstitial lung disease? Large, prospective cohorts of well-defined patients are needed to answer these questions. The role of corticosteroids and other therapies, such as anticoagulation, for the treatment of acute exacerbation remains unclear. A better understanding of the epidemiology of this condition is essential to the design of future treatment trials aimed at clarifying treatment issues.
Studies of the underlying pathobiology of acute exacerbations of IPF are also critically important. Do acute exacerbations of IPF represent a distinct pathobiological manifestation of the primary disease process or are they caused by occult complications such as infection and aspiration? What are the roles of inflammation, coagulation, epithelial cell biology, and genetics? Future studies should prospectively collect biological samples from subjects experiencing acute exacerbations of IPF. A systematic search for evidence of infection, perhaps using pan-viral and bacterial microarrays to look for novel agents, is important. Additional studies could include gene expression arrays of lung tissues or of BALF cells, and proteomic analysis of plasma and BAL. Given the clinical, radiographic, and histopathologic similarities between acute exacerbation of IPF and ARDS, the role of factors associated with the pathobiology of ARDS should also be explored. It is possible that similar biological mechanisms are involved in these two conditions. If so, treatment approaches targeting patients with ARDS could be beneficial to patients with acute exacerbation of IPF.
To answer these and other questions, a systematic and concerted effort to identify patients undergoing acute exacerbations of IPF is an essential first step. We propose that the definition and diagnostic criteria presented above be used for these efforts. The development of multicenter, national, or international registries to prospectively collect clinical data and biological samples from patients with acute exacerbations of IPF would allow investigators sufficient numbers of subjects to successfully study this condition. It is only through the standardization of terminology and collaboration of effort that important progress will be made.
Acknowledgments
Supported by National Institutes of Health (NIH) HL86516 (H.R.C.), NHLBI SCOR HL6767 (K.K.B.), NIH HL080383 (R.J.K.), NIH HL083901 (J.A.L.), and NIH HL58655 and NIH 080685 (M.A.O.).
Footnotes
Conflict of Interest Statement: H.R.C. received $5,000 from InterMune for advisory board activities in 2006. B.B.M. does not have a financial relationship with a commercial entity that interest in the subject of this manuscript. K.R.F. received $4,100 in 2005 and $3,000 in 2007 from Boehringer Ingelheim; $2,500 in 2006 from InterMune; and $1,200 in 2005 from Fibrogen for serving on advisory boards related to pulmonary fibrosis. K.K.B. has served as a consultant or spoken to the following companies interested in IPF: Actelion, Amgen, Genzyme, Wyeth, Biogen, Boehringer Ingelheim, Novartis, and Lung Rx. He or his institution have received grants to support the performance of treatment trials in IPF from Actelion, InterMune, Biogen, and Genzyme. R.J.K. does not have a financial relationship with a commercial entity that interest in the subject of this manuscript. T.E.K. has served on advisory boards for InterMune and for GlaxoSmithKline (GSK); and served as a consultant for Nektar, Alexza, Astra-Zeneca, Biogen, Centocor, Fibrogen, Genzyme, Human Genome Sciences, Merck, and CoTherix. J.A.L. has been supported by Novartis for an investigator-initiated study, for service on an advisory board by InterMune, and by Actelion for service of advisory and data safety monitoring boards. J.E.L. does not have a financial relationship with a commercial entity that interest in the subject of this manuscript. I.N. has participated as a speaker for various pharmaceutical companies (GSK, Pfizer, Boehringer Ingelheim, in 2006 for various amounts). He has also received honoraria for participating in advisory board meetings for Novartis in 2005 and GSK in 2006. I.N.'s institution has received grants and contracts from Actelion, CoTherix, Centocor, Genzyme, InterMune, Novartis, and Roche, all ongoing in 2007. M.A.O. does not have a financial relationship with a commercial entity that interest in the subject of this manuscript. G.R. served as a steering committee member for BUILD-1 and serves as a steering committee member for BUILD-3 studies for IPF sponsored by Actelion. J.R. in 2006 received $10,000 for research conducted as part of a multicenter trial sponsored by InterMune. J.H.R. does not have a financial relationship with a commercial entity that interest in the subject of this manuscript. D.A.Z. received speaker honoraria and served on advisory board for InterMune ($7,000 in 2006, $6,000 in 2005; $6,000 in 2004). D.A.Z. has received research grants from InterMune, Actelion, and CoTherix for participating in multicenter clinical trials. G.W.H. does not have a financial relationship with a commercial entity that interest in the subject of this manuscript. T.V.C. does not have a financial relationship with a commercial entity that interest in the subject of this manuscript. J.J.E. does not have a financial relationship with a commercial entity that interest in the subject of this manuscript. D.M.H. received $6,000 for giving advice and grading HRCTs for clinical trial run by InterMune in 2005/2006. T.J. does not have a financial relationship with a commercial entity that interest in the subject of this manuscript. N.K. received $5,000 for serving on a Biogen Idec advisory board in November 2005. N.K. is also a recipient of an investigator-initiated grant from Biogen Idec in August 2006 ($674,800 for 2 yr). D.S.K. does not have a financial relationship with a commercial entity that interest in the subject of this manuscript. Y.K. does not have a financial relationship with a commercial entity that interest in the subject of this manuscript. D.A.L. has received less than $5,000 in 2004, 2005, and 2006 from InterMune, for interpretation of CT scans. He has received more than $5,000 from Encysive for consultation on clinical trials. He received $6,000 in 2006 for service on an advisory board for Actelion. J.M.-Q. received a research grant of $180,000 from Boehringer Ingelheim, Germany, in 2003 through 2006 for an in vitro study on fibrogenic mechanisms. J.M.-Q received a research grant of $80,000 from Altana, Germany, in 2003 through 2006 for an in vitro study on alveolar epithelia. J.L.M. does not have a financial relationship with a commercial entity that interest in the subject of this manuscript. A.G.N. received $4,000 for reviewing slides for InterMune Ltd in 2004–2006 and £8,600 for reviewing slides for Actelion Ltd in 2006 in relation to entry into drug trials for patients with IPF. M.S. does not have a financial relationship with a commercial entity that interest in the subject of this manuscript. G.B.T. does not have a financial relationship with a commercial entity that interest in the subject of this manuscript. A.U.W. does not have a financial relationship with a commercial entity that interest in the subject of this manuscript. F.J.M. is a consultant for Altana Pharma and has received compensation greater than $10K. F.J.M has been a member of several advisory boards, CME committees, and the speakers' bureau for Boehringer Ingelheim, Pfizer, and GSK. His total compensation per company was greater than $10K. In addition, F.J.M. is on an advisory board for Novartis and a speaker's bureau for Sepracor and Astra, receiving less than $10K per company. F.J.M. has been an investigator for industry-sponsored studies for GSK, Boehringer Ingelheim, and Actelion.
The Idiopathic Pulmonary Fibrosis Clinical Research Network (IPFnet) consists of a central data coordinating center (Duke Clinical Research Institute) and 11 clinical sites, as follows: Emory University, Gulf South Research Consortium (Tulane University and University of Alabama, Birmingham); Mayo Clinic, Rochester; National Jewish Medical and Research Center; University of California, Los Angeles; University of California, San Francisco; University of Chicago; University of Michigan Health System; University of Washington; Vanderbilt University; and Weill Cornell Medical College.
References
- 1.American Thoracic Society/European Respiratory Society. American Thoracic Society/European Respiratory Society international multidisciplinary consensus classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2002;165:277–304. doi: 10.1164/ajrccm.165.2.ats01. [DOI] [PubMed] [Google Scholar]
- 2.American Thoracic Society/European Respiratory Society. Idiopathic pulmonary fibrosis: diagnosis and treatment. International consensus statement. Am J Respir Crit Care Med. 2000;161:646–664. doi: 10.1164/ajrccm.161.2.ats3-00. [DOI] [PubMed] [Google Scholar]
- 3.Bjoraker JA, Ryu JH, Edwin MK, Myers JL, Tazelaar HD, Schroeder DR, Offord KP. Prognostic significance of histopathologic subsets in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1998;157:199–203. doi: 10.1164/ajrccm.157.1.9704130. [DOI] [PubMed] [Google Scholar]
- 4.Flaherty KR, Travis WD, Colby TV, Toews GB, Kazerooni EA, Gross BH, Jain A, Strawderman RL, Flint A, Lynch JP, et al. Histopathologic variability in usual and nonspecific interstitial pneumonias. Am J Respir Crit Care Med. 2001;164:1722–1727. doi: 10.1164/ajrccm.164.9.2103074. [DOI] [PubMed] [Google Scholar]
- 5.Nicholson AG, Colby TV, Dubois RM, Hansell DM, Wells AU. The prognostic significance of the histologic pattern of interstitial pneumonia in patients presenting with the clinical entity of cryptogenic fibrosing alveolitis. Am J Respir Crit Care Med. 2000;162:2213–2217. doi: 10.1164/ajrccm.162.6.2003049. [DOI] [PubMed] [Google Scholar]
- 6.Carrington CB, Gaensler EA, Coutu RE, Fitzgerald MX, Gupta RG. Natural history and treated course of usual and desquamative interstitial pneumonia. N Engl J Med. 1978;298:801–809. doi: 10.1056/NEJM197804132981501. [DOI] [PubMed] [Google Scholar]
- 7.Stack BHR, Choo-Kang YEJ, Heard BE. The prognosis of cryptogenic fibrosing alveolitis. Thorax. 1972;27:535–542. doi: 10.1136/thx.27.5.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim DS, Collard HR, King TE., Jr Classification and natural history of the idiopathic interstitial pneumonias. Proc Am Thorac Soc. 2006;3:285–292. doi: 10.1513/pats.200601-005TK. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kondo A, Saiki S. Acute exacerbation in idiopathic interstitial pneumonia (IIP) In: Harasawa M, Fukuchi Y, Morinari H, editors. Interstitial pneumonia of unknown etiology. Tokyo, Japan: University of Tokyo Press; 1989. pp. 33–42. Intractable Diseases Research Foundation Publication No. 27. [Google Scholar]
- 10.Kondoh Y, Taniguchi H, Kawabata Y, Yokoi T, Suzuki K, Takagi K. Acute exacerbation in idiopathic pulmonary fibrosis: analysis of clinical and pathologic findings in three cases. Chest. 1993;103:1808–1812. doi: 10.1378/chest.103.6.1808. [DOI] [PubMed] [Google Scholar]
- 11.Akira M, Hamada H, Sakatani M, Kobayashi C, Nishioka M, Yamamoto S. CT findings during phase of accelerated deterioration in patients with idiopathic pulmonary fibrosis. AJR Am J Roentgenol. 1997;168:79–83. doi: 10.2214/ajr.168.1.8976924. [DOI] [PubMed] [Google Scholar]
- 12.Ambrosini V, Cancellieri A, Chilosi M, Zompatori M, Trisolini R, Saragoni L, Poletti V. Acute exacerbation of idiopathic pulmonary fibrosis: report of a series. Eur Respir J. 2003;22:821–826. doi: 10.1183/09031936.03.00022703. [DOI] [PubMed] [Google Scholar]
- 13.Azuma A, Nukiwa T, Tsuboi E, Suga M, Abe S, Nakata K, Taguchi Y, Nagai S, Itoh H, Ohi M, et al. Placebo-controlled trial of pirfenidone in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2005;171:1040–1047. doi: 10.1164/rccm.200404-571OC. [DOI] [PubMed] [Google Scholar]
- 14.Kim DS, Park JH, Park BK, Lee JS, Nicholson AG, Colby T. Acute exacerbation of idiopathic pulmonary fibrosis: frequency and clinical features. Eur Respir J. 2006;27:143–150. doi: 10.1183/09031936.06.00114004. [DOI] [PubMed] [Google Scholar]
- 15.Kondoh Y, Taniguchi H, Yokoi T, Nishiyama O, Ohishi T, Kato T, Suzuki K, Suzuki R. Cyclophosphamide and low-dose prednisolone in idiopathic pulmonary fibrosis and fibrosing nonspecific interstitial pneumonia. Eur Respir J. 2005;25:528–533. doi: 10.1183/09031936.05.00071004. [DOI] [PubMed] [Google Scholar]
- 16.Kubo H, Nakayama K, Yanai M, Suzuki T, Yamaya M, Watanabe M, Sasaki H. Anticoagulant therapy for idiopathic pulmonary fibrosis. Chest. 2005;128:1475–1482. doi: 10.1378/chest.128.3.1475. [DOI] [PubMed] [Google Scholar]
- 17.Okamoto T, Ichiyasu H, Ichikado K, Muranaka H, Sato K, Okamoto S, Iyonaga K, Suga M, Kohrogi H. Clinical analysis of the acute exacerbation in patients with idiopathic pulmonary fibrosis [in Japanese] Nihon Kokyuki Gakkai Zasshi. 2006;44:359–367. [PubMed] [Google Scholar]
- 18.Parambil JG, Myers JL, Ryu JH. Histopathologic features and outcome of patients with acute exacerbation of idiopathic pulmonary fibrosis undergoing surgical lung biopsy. Chest. 2005;128:3310–3315. doi: 10.1378/chest.128.5.3310. [DOI] [PubMed] [Google Scholar]
- 19.Tiitto L, Bloigu R, Heiskanen U, Paakko P, Kinnula VL, Kaarteenaho-Wiik R. Relationship between histopathological features and the course of idiopathic pulmonary fibrosis/usual interstitial pneumonia. Thorax. 2006;61:1091–1095. doi: 10.1136/thx.2005.055814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Homma S, Sakamoto S, Kawabata M, Kishi K, Tsuboi E, Motoi N, Yoshimura K. Cyclosporin treatment in steroid-resistant and acutely exacerbated interstitial pneumonia. Intern Med. 2005;44:1144–1150. doi: 10.2169/internalmedicine.44.1144. [DOI] [PubMed] [Google Scholar]
- 21.Al-Hameed FM, Sharma S. Outcome of patients admitted to the intensive care unit for acute exacerbation of idiopathic pulmonary fibrosis. Can Respir J. 2004;11:117–122. doi: 10.1155/2004/379723. [DOI] [PubMed] [Google Scholar]
- 22.Blivet S, Philit F, Sab JM, Langevin B, Paret M, Guerin C, Robert D. Outcome of patients with idiopathic pulmonary fibrosis admitted to the ICU for respiratory failure. Chest. 2001;120:209–212. doi: 10.1378/chest.120.1.209. [DOI] [PubMed] [Google Scholar]
- 23.Kondoh Y, Taniguchi H, Kitaichi M, Yokoi T, Johkoh T, Oishi T, Kimura T, Nishiyama O, Kato K, du Bois RM. Acute exacerbation of interstitial pneumonia following surgical lung biopsy. Respir Med. 2006;100:1753–1759. doi: 10.1016/j.rmed.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 24.Rice AJ, Wells AU, Bouros D, du Bois RM, Hansell DM, Polychronopoulos V, Vassilakis D, Kerr JR, Evans TW, Nicholson AG. Terminal diffuse alveolar damage in relation to interstitial pneumonias: an autopsy study. Am J Clin Pathol. 2003;119:709–714. doi: 10.1309/UVAR-MDY8-FE9F-JDKU. [DOI] [PubMed] [Google Scholar]
- 25.Saydain G, Islam A, Afessa B, Ryu JH, Scott JP, Peters SG. Outcome of patients with idiopathic pulmonary fibrosis admitted to the intensive care unit. Am J Respir Crit Care Med. 2002;166:839–842. doi: 10.1164/rccm.2104038. [DOI] [PubMed] [Google Scholar]
- 26.Amin RS, Wert SE, Baughman RP, Tomashefski JF, Jr, Nogee LM, Brody AS, Hull WM, Whitsett JA. Surfactant protein deficiency in familial interstitial lung disease. J Pediatr. 2001;139:85–92. doi: 10.1067/mpd.2001.114545. [DOI] [PubMed] [Google Scholar]
- 27.Churg A, Muller NL, Silva CI, Wright JL. Acute exacerbation (acute lung injury of unknown cause) in UIP and other forms of fibrotic interstitial pneumonias. Am J Surg Pathol. 2007;31:277–284. doi: 10.1097/01.pas.0000213341.70852.9d. [DOI] [PubMed] [Google Scholar]
- 28.Kinder BW, Collard HR, King TE., Jr Anticoagulant therapy for idiopathic pulmonary fibrosis. Chest. 2006;130:302–303. doi: 10.1378/chest.130.1.302-a. [DOI] [PubMed] [Google Scholar]
- 29.Martinez FJ, Safrin S, Weycker D, Starko KM, Bradford WZ, King TE, Jr, Flaherty KR, Schwartz DA, Noble PW, Raghu G, et al. The clinical course of patients with idiopathic pulmonary fibrosis. Ann Intern Med. 2005;142:963–967. doi: 10.7326/0003-4819-142-12_part_1-200506210-00005. [DOI] [PubMed] [Google Scholar]
- 30.Hoshikawa Y, Kondo T. Perioperative lung injury: acute exacerbation of idiopathic pulmonary fibrosis and acute interstitial pneumonia after pulmonary resection [in Japanese] Nippon Geka Gakkai Zasshi. 2004;105:757–762. [PubMed] [Google Scholar]
- 31.Kumar P, Goldstraw P, Yamada K, Nicholson AG, Wells AU, Hansell DM, Dubois RM, Ladas G. Pulmonary fibrosis and lung cancer: risk and benefit analysis of pulmonary resection. J Thorac Cardiovasc Surg. 2003;125:1321–1327. doi: 10.1016/s0022-5223(03)00028-x. [DOI] [PubMed] [Google Scholar]
- 32.Sakamoto S, Homma S, Kawabata M, Kono T, Seki K, Nakata K, Yoshimura K. Fatal acute exacerbation of idiopathic pulmonary fibrosis/usual interstitial pneumonia initially in the right lung after surgery lobectomy for left lung cancer [in Japanese] Nihon Kokyuki Gakkai Zasshi. 2004;42:760–766. [PubMed] [Google Scholar]
- 33.Yuksel M, Ozyurtkan MO, Bostanci K, Ahiskali R, Kodalli N. Acute exacerbation of interstitial fibrosis after pulmonary resection. Ann Thorac Surg. 2006;82:336–338. doi: 10.1016/j.athoracsur.2005.09.036. [DOI] [PubMed] [Google Scholar]
- 34.Utz JP, Ryu JH, Douglas WW, Hartman TE, Tazelaar HD, Myers JL, Allen MS, Schroeder DR. High short-term mortality following lung biopsy for usual interstitial pneumonia. Eur Respir J. 2001;17:175–179. doi: 10.1183/09031936.01.17201750. [DOI] [PubMed] [Google Scholar]
- 35.Mukae H, Iiboshi H, Nakazato M, Hiratsuka T, Tokojima M, Abe K, Ashitani J, Kadota J, Matsukura S, Kohno S. Raised plasma concentrations of alpha-defensins in patients with idiopathic pulmonary fibrosis. Thorax. 2002;57:623–628. doi: 10.1136/thorax.57.7.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tajima S, Oshikawa K, Tominaga S, Sugiyama Y. The increase in serum soluble ST2 protein upon acute exacerbation of idiopathic pulmonary fibrosis. Chest. 2003;124:1206–1214. doi: 10.1378/chest.124.4.1206. [DOI] [PubMed] [Google Scholar]
- 37.Ziegenhagen MW, Zabel P, Zissel G, Schlaak M, Muller-Quernheim J. Serum level of interleukin 8 is elevated in idiopathic pulmonary fibrosis and indicates disease activity. Am J Respir Crit Care Med. 1998;157:762–768. doi: 10.1164/ajrccm.157.3.9705014. [DOI] [PubMed] [Google Scholar]
- 38.Akira M, Sakatani M, Hara H. Thin-section CT findings in rheumatoid arthritis-associated lung disease: CT patterns and their courses. J Comput Assist Tomogr. 1999;23:941–948. doi: 10.1097/00004728-199911000-00021. [DOI] [PubMed] [Google Scholar]
- 39.Yokoyama A, Kohno N, Hamada H, Sakatani M, Ueda E, Kondo K, Hirasawa Y, Hiwada K. Circulating KL-6 predicts the outcome of rapidly progressive idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1998;158:1680–1684. doi: 10.1164/ajrccm.158.5.9803115. [DOI] [PubMed] [Google Scholar]
- 40.Egan JJ, Stewart JP, Hasleton PS, Arrand JR, Carroll KB, Woodcock AA. Epstein-Barr virus replication within pulmonary epithelial cells in cryptogenic fibrosing alveolitis. Thorax. 1995;50:1234–1239. doi: 10.1136/thx.50.12.1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hogg JC. Epstein-Barr virus and cryptogenic fibrosing alveolitis. Thorax. 1995;50:1232. doi: 10.1136/thx.50.12.1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tsukamoto K, Hayakawa H, Sato A, Chida K, Nakamura H, Miura K. Involvement of Epstein-Barr virus latent membrane protein 1 in disease progression in patients with idiopathic pulmonary fibrosis. Thorax. 2000;55:958–961. doi: 10.1136/thorax.55.11.958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yonemaru M, Kasuga I, Kusumoto H, Kunisawa A, Kiyokawa H, Kuwabara S, Ichinose Y, Toyama K. Elevation of antibodies to cytomegalovirus and other herpes viruses in pulmonary fibrosis. Eur Respir J. 1997;10:2040–2045. doi: 10.1183/09031936.97.10092040. [DOI] [PubMed] [Google Scholar]
- 44.Semenzato G, de Rossi A, Agostini C. Human retroviruses and their aetiological link to pulmonary diseases. Eur Respir J. 1993;6:925–929. [PubMed] [Google Scholar]
- 45.Stewart JP, Egan JJ, Ross AJ, Kelly BG, Lok SS, Hasleton PS, Woodcock AA. The detection of Epstein-Barr virus DNA in lung tissue from patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1999;159:1336–1341. doi: 10.1164/ajrccm.159.4.9807077. [DOI] [PubMed] [Google Scholar]
- 46.Tang YW, Johnson JE, Browning PJ, Cruz-Gervis RA, Davis A, Graham BS, Brigham KL, Oates JA, Jr, Loyd JE, Stecenko AA. Herpesvirus DNA is consistently detected in lungs of patients with idiopathic pulmonary fibrosis. J Clin Microbiol. 2003;41:2633–2640. doi: 10.1128/JCM.41.6.2633-2640.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Raghu G, Freudenberger TD, Yang S, Curtis JR, Spada C, Hayes J, Sillery JK, Pope CE, II, Pellegrini CA. High prevalence of abnormal acid gastro-oesophageal reflux in idiopathic pulmonary fibrosis. Eur Respir J. 2006;27:136–142. doi: 10.1183/09031936.06.00037005. [DOI] [PubMed] [Google Scholar]
- 48.Raghu G, Yang ST, Spada C, Hayes J, Pellegrini CA. Sole treatment of acid gastroesophageal reflux in idiopathic pulmonary fibrosis: a case series. Chest. 2006;129:794–800. doi: 10.1378/chest.129.3.794. [DOI] [PubMed] [Google Scholar]
- 49.Sweet MP, Patti MG, Leard LE, Golden JA, Hays SR, Hoopes C, Theodore PR. Gastroesophageal reflux in patients with idiopathic pulmonary fibrosis referred for lung transplantation. J Thorac Cardiovasc Surg. 2007;133:1078–1084. doi: 10.1016/j.jtcvs.2006.09.085. [DOI] [PubMed] [Google Scholar]
- 50.Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med. 2000;342:1334–1349. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- 51.Selman M, King TE, Jr, Pardo A. Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann Intern Med. 2001;134:136–151. doi: 10.7326/0003-4819-134-2-200101160-00015. [DOI] [PubMed] [Google Scholar]
- 52.Selman M, Thannickal VJ, Pardo A, Zisman DA, Martinez FJ, Lynch JP. Idiopathic pulmonary fibrosis: pathogenesis and therapeutic approaches. Drugs. 2004;64:405–430. doi: 10.2165/00003495-200464040-00005. [DOI] [PubMed] [Google Scholar]
- 53.Kuhn C, III, Boldt J, King TE, Jr, Crouch E, Vartio T, McDonald JA. An immunohistochemical study of architectural remodeling and connective tissue synthesis in pulmonary fibrosis. Am Rev Respir Dis. 1989;140:1693–1703. doi: 10.1164/ajrccm/140.6.1693. [DOI] [PubMed] [Google Scholar]
- 54.Schwartz DA. The importance of gene-environment interactions and exposure assessment in understanding human diseases. J Expo Sci Environ Epidemiol. 2006;16:474–476. doi: 10.1038/sj.jes.7500531. [DOI] [PubMed] [Google Scholar]
- 55.Bucala R, Spiegel LA, Chesney J, Hogan M, Cerami A. Circulating fibrocytes define a new leukocyte subpopulation that mediates tissue repair. Molecular Med. 1994;1:71–81. [PMC free article] [PubMed] [Google Scholar]
- 56.Chesney J, Bacher M, Bender A, Bucala R. The peripheral blood fibrocyte is a potent antigen-presenting cell capable of priming naive T cells in situ. Proc Natl Acad Sci USA. 1997;94:6307–6312. doi: 10.1073/pnas.94.12.6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chesney J, Metz C, Stavitsky AB, Bacher M, Bucala R. Regulated production of type I collagen and inflammatory cytokines by peripheral blood fibrocytes. J Immunol. 1998;160:419–425. [PubMed] [Google Scholar]
- 58.Moore BB, Kolodsick JE, Thannickal VJ, Cooke K, Moore TA, Hogaboam C, Wilke CA, Toews GB. CCR2-mediated recruitment of fibrocytes to the alveolar space after fibrotic injury. Am J Pathol. 2005;166:675–684. doi: 10.1016/S0002-9440(10)62289-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Phillips RJ, Burdick MD, Hong K, Lutz MA, Murray LA, Xue YY, Belperio JA, Keane MP, Strieter RM. Circulating fibrocytes traffic to the lungs in response to CXCL12 and mediate fibrosis. J Clin Invest. 2004;114:438–446. doi: 10.1172/JCI20997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schmidt M, Sun G, Stacey M, Mori L, Mattoli S. Identification of circulating fibrocytes as precursors of bronchial myofibroblasts in asthma. J Immunol. 2003;170:380–389. doi: 10.4049/jimmunol.171.1.380. [DOI] [PubMed] [Google Scholar]
- 61.Nihlberg K, Larsen K, Hultgardh-Nilsson A, Malmstrom A, Bjermer L, Westergren-Thorsson G. Tissue fibrocytes in patients with mild asthma: a possible link to thickness of reticular basement membrane? Respir Res. 2006;7:50. doi: 10.1186/1465-9921-7-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mehrad B, Burdick MD, Zisman DA, Keane MP, Belperio JA, Strieter RM. Circulating peripheral blood fibrocytes in human fibrotic interstitial lung disease. Biochem Biophys Res Commun. 2007;353:104–108. doi: 10.1016/j.bbrc.2006.11.149. [DOI] [PubMed] [Google Scholar]
- 63.Moore BB, Murray L, Das A, Wilke CA, Herrygers AB, Toews GB. The role of CCL12 in the recruitment of fibrocytes and lung fibrosis. Am J Respir Cell Mol Biol. 2006;35:175–181. doi: 10.1165/rcmb.2005-0239OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gilpin S, Moeller A, Baatjes A, Martin G, Ask K, O'Bryne P, Cox G, Gauldie J, Kolb M. Circulating human matrix-producing progenitor cells in pulmonary fibroproliferative disorders; Presented at the International Colloquium on Lung Fibrosis; 2006 September 7–10; Eltville, Germany. [Google Scholar]
- 65.Nagase H, Visse R, Murphy G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc Res. 2006;69:562–573. doi: 10.1016/j.cardiores.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 66.Suga M, Iyonaga K, Okamoto T, Gushima Y, Miyakawa H, Akaike T, Ando M. Characteristic elevation of matrix metalloproteinase activity in idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2000;162:1949–1956. doi: 10.1164/ajrccm.162.5.9906096. [DOI] [PubMed] [Google Scholar]
- 67.Atkinson JJ, Senior RM. Matrix metalloproteinase-9 in lung remodeling. Am J Respir Cell Mol Biol. 2003;28:12–24. doi: 10.1165/rcmb.2002-0166TR. [DOI] [PubMed] [Google Scholar]
- 68.Pardo A, Ruiz V, Arreola JL, Ramirez R, Cisneros-Lira J, Gaxiola M, Barrios R, Kala SV, Lieberman MW, Selman M. Bleomycin-induced pulmonary fibrosis is attenuated in gamma-glutamyl transpeptidase-deficient mice. Am J Respir Crit Care Med. 2003;167:925–932. doi: 10.1164/rccm.200209-1007OC. [DOI] [PubMed] [Google Scholar]
- 69.Wang H, Kochevar IE. Involvement of UVB-induced reactive oxygen species in TGF-beta biosynthesis and activation in keratinocytes. Free Radic Biol Med. 2005;38:890–897. doi: 10.1016/j.freeradbiomed.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 70.Yu Q, Stamenkovic I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 2000;14:163–176. [PMC free article] [PubMed] [Google Scholar]
- 71.Munger JS, Huang X, Kawakatsu H, Griffiths MJ, Dalton SL, Wu J, Pittet JF, Kaminski N, Garat C, Matthay MA, et al. The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell. 1999;96:319–328. doi: 10.1016/s0092-8674(00)80545-0. [DOI] [PubMed] [Google Scholar]
- 72.Gunther A, Mosavi P, Ruppert C, Heinemann S, Temmesfeld B, Velcovsky HG, Morr H, Grimminger F, Walmrath D, Seeger W. Enhanced tissue factor pathway activity and fibrin turnover in the alveolar compartment of patients with interstitial lung disease. Thromb Haemost. 2000;83:853–860. [PubMed] [Google Scholar]
- 73.Perez RL, Duncan A, Hunter RL, Staton GW., Jr Elevated D dimer in the lungs and blood of patients with sarcoidosis. Chest. 1993;103:1100–1106. doi: 10.1378/chest.103.4.1100. [DOI] [PubMed] [Google Scholar]
- 74.Olman MA. Mechanisms of fibroproliferation in acute lung injury. In: Matthay MA, Lenfant C, editors. Acute respiratory distress syndrome. New York: Marcel Dekker; 2003. pp. 313–354. [Google Scholar]
- 75.Gaggar A, Olman MA. Biologic markers of mortality in acute lung injury. Clin Chim Acta. 2006;372:24–32. doi: 10.1016/j.cca.2006.04.019. [DOI] [PubMed] [Google Scholar]
- 76.Prabhakaran P, Ware LB, White KE, Cross MT, Matthay MA, Olman MA. Elevated levels of plasminogen activator inhibitor-1 in pulmonary edema fluid are associated with mortality in acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2003;285:L20–L28. doi: 10.1152/ajplung.00312.2002. [DOI] [PubMed] [Google Scholar]
- 77.Zorzetto M, Ferrarotti I, Trisolini R, Agli LL, Scabini R, Novo M, De Silvestri A, Patelli M, Martinetti M, Cuccia M, et al. Complement receptor 1 gene polymorphisms are associated with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2003;168:330–334. doi: 10.1164/rccm.200302-221OC. [DOI] [PubMed] [Google Scholar]
- 78.Lawson WE, Loyd JE. The genetic approach in pulmonary fibrosis: can it provide clues to this complex disease? Proc Am Thorac Soc. 2006;3:345–349. doi: 10.1513/pats.200512-137TK. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Armanios MY, Chen JJ, Cogan JD, Alder JK, Ingersoll RG, Markin C, Lawson WE, Xie M, Vulto I, Phillips JA, III, et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med. 2007;356:1317–1326. doi: 10.1056/NEJMoa066157. [DOI] [PubMed] [Google Scholar]
- 80.Tsakiri K, Cronkhite J, Kuan P, Xing C, Raghu G, Weissler J, Rosenblatt R, Shay J, Garcia C. Adult-onset pulmonary fibrosis caused by mutations in telemorase. Proc Natl Acad Sci USA. 2007;104:7552–7557. doi: 10.1073/pnas.0701009104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Stern JB, Mal H, Groussard O, Brugiere O, Marceau A, Jebrak G, Fournier M. Prognosis of patients with advanced idiopathic pulmonary fibrosis requiring mechanical ventilation for acute respiratory failure. Chest. 2001;120:213–219. doi: 10.1378/chest.120.1.213. [DOI] [PubMed] [Google Scholar]