

Abstract
The [4Fe-4S] cluster is ubiquitous to a class of base excision repair enzymes, in organisms ranging from bacteria to man, and was first considered as a structural element, owing to its redox stability under physiological conditions. When studied bound to DNA, two of these repair proteins (MutY and Endonuclease III from Escherichia coli) display DNA-dependent reversible electron transfer with characteristics typical of high potential iron proteins. These results have inspired a reexamination of the role of the [4Fe-4S] cluster in this class of enzymes. Might the [4Fe-4S] cluster be used as a redox cofactor to search for damaged sites using DNA-mediated charge transport, a process well known to be highly sensitive to lesions and mismatched bases? Described here are experiments demonstrating the utility of DNA-mediated charge transport in characterizing these DNA-binding metalloproteins, as well as efforts to elucidate this new function for DNA as an electronic signaling medium among the proteins.
1. Introduction
The double helix of DNA is a molecule rich with information. Not only does it contain, in its sequence of base pairs, the genetic code for the cell, but the complex topology of DNA provides a three-dimensional surface that a wide variety of proteins must parse to locate specific sequences and damaged sites for the purposes of replication, transcription and repair [1]. It is not well understood how these proteins accomplish this task in a timely fashion. In the case of DNA repair, where the entire genome must be constantly monitored for often very subtle modifications, it is even more difficult to imagine how proteins efficiently locate damage.
In base excision repair (BER), glycosylase enzymes are responsible for searching the genome for chemically modified bases and catalyzing their excision [2]. These enzymes must first locate their substrate in a vast excess of undamaged DNA, flip the substrate into the active site of the protein, and catalyze scission of the N-glycosidic bond between the errant base and the sugar-phosphate backbone. While much is known about the catalysis and substrate discrimination steps in this process, very little is known about the daunting initial search of the genome these enzymes must undertake. It has been demonstrated that many of these enzymes can move along the DNA helix in a processive manner [3–4], but the in vivo relevance of this search mechanism as the primary mode of damage detection by DNA-binding proteins is disputed [5–7].
Many BER glycosylases contain a [4Fe-4S] cluster [8–10], the function of which is unknown. Endonuclease III (Endo III) was the first glycosylase discovered to contain this metal cofactor [8]. Endo III removes a wide variety of oxidized pyrimidines from DNA and contains the helix-hairpin-helix (HhH) recognition motif [11–19]. MutY, structurally similar to Endo III [18–21], is another BER glycosylase that contains a [4Fe-4S] cluster [20]. However, MutY instead removes adenine from 8-oxo-guanine:adenine mispairs [22–34].
The role of the [4Fe-4S] cluster in these glycosylases is of great interest. Earlier, experiments were performed with Endo III to determine the properties and function of the [4Fe-4S] cluster [8,35]. Mossbauer and electron paramagnetic resonance (EPR) spectroscopy experiments confirmed that the protein contains the [4Fe-4S]2+ cluster when the protein is not bound to DNA. The cluster was unable to be oxidized by ferricyanide without degradation to the [3Fe-4S]1+ species as observed by EPR at 4K. Photoreduction of Endo III did give rise to the [4Fe-4S]1+ cluster but with an estimated reduction potential of less than −600 mV versus NHE. Since it appeared that stable oxidation of the [4Fe-4S] cluster was not possible, nor was reduction feasible in a biological environment, the cofactor was relegated to a structural role.
The [4Fe-4S] cluster was analogously assigned a structural role in MutY. However, the David laboratory has since performed several experiments to investigate the role of the cluster in this protein [36–38]. They have developed a method to remove reversibly the cluster from the protein and discovered that the cofactor is not necessary for protein folding nor does it contribute to the thermal stability of the protein. Nonetheless, the [4Fe-4S] cluster is necessary for DNA binding and enzyme activity. In addition, mutagenesis studies further highlight the necessity of the [4Fe-4S] cluster for MutY repair. In these experiments, the cysteines that ligate the cluster are mutated to both coordinating (histidine and serine) and non-coordinating (alanine) residues leading to, in some cases, quite dramatic effects on the repair capacity of MutY.
Crystal structures are available for MutY and Endo III both free and bound to DNA [18–20, 39–40]. These provide many clues about the environment of the cluster in both states. In each protein, the [4Fe-4S] cluster is ligated by a unique cysteine motif (C-X6-C-X2-C-X5-C). Some of these ligating residues are located in a loop termed the iron-sulfur cluster loop (FCL). This loop also contains many positively charged residues that interact with the DNA backbone. The overall structures of free and DNA-bound proteins are similar (backbone RMSD = 1.3 – 2.1 Å); large conformational changes do not occur in the protein upon binding to DNA. In both MutY and Endo III, the [4Fe-4S] cluster is located ~13 Å from the nearest DNA backbone atom, and ~20 Å from both the center of the DNA helix and the glycosylase active site.
Thus far we have discussed two major questions in the field of base excision repair. First we introduced the general question of how glycosylases quickly and efficiently locate damage in the genome. And second, what is the role of the [4Fe-4S] cluster in a select set of glycosylases? This review describes work in our laboratory that addresses both of these questions. Using electrochemical and spectroscopic techniques originally developed to study DNA-mediated charge transport (CT), we have now learned a great deal about the properties of the [4Fe-4S] cluster in these glycosylases when the protein is bound to DNA. Additionally, we have proposed a model describing how these proteins might be able to use DNA-mediated CT as a way to search for damage in the genome.
2. Electrochemical Investigation of MutY and Endo III
The double helical structure of DNA, with its π-stacked array of heterocyclic aromatic base pairs, allows DNA to serve as a medium for CT [41–44]. Long range DNA CT is efficient over distances of at least 200 Å [45] and displays exquisite sensitivity to mismatched or damaged base pairs [46–49]. While it has been demonstrated that DNA CT can occur in biologically relevant environments (in histone-wrapped DNA or in mamalian cell nuclei) [50–51], a biological role for DNA CT has not yet been established.
Many tools have been developed to study DNA CT, including DNA-modified electrode systems [52–53]. In these systems, modified DNA duplexes are self-assembled into a monolayer via a thiol terminated tether on gold or through a pyrene-terminated linker on highly oriented pyrolytic graphite (HOPG) electrode surfaces. CT through the duplex can then be monitored using redox-active intercalative probes. DNA-modified electrodes have been successfully used to evaluate DNA CT efficiency in duplexes containing a wide variety of mismatched [48] and damaged bases [48–49], or with proteins that bend DNA or flip a base out of the π-stack [54].
DNA-modified electrodes have also been shown to be useful in studying DNA-binding proteins that contain their own redox cofactor, such as MutY and Endo III [55–57]. In these experiments, modified DNA is self-assembled into a loosely packed monolayer and backfilled with an alkane or alkanethiol moiety to prevent any nonspecific interactions between the protein and the electrode surface (Figure 1). The protein is then introduced to the electrode and monitored with an electrochemical technique (typically cyclic or square wave voltammetry).
Figure 1.
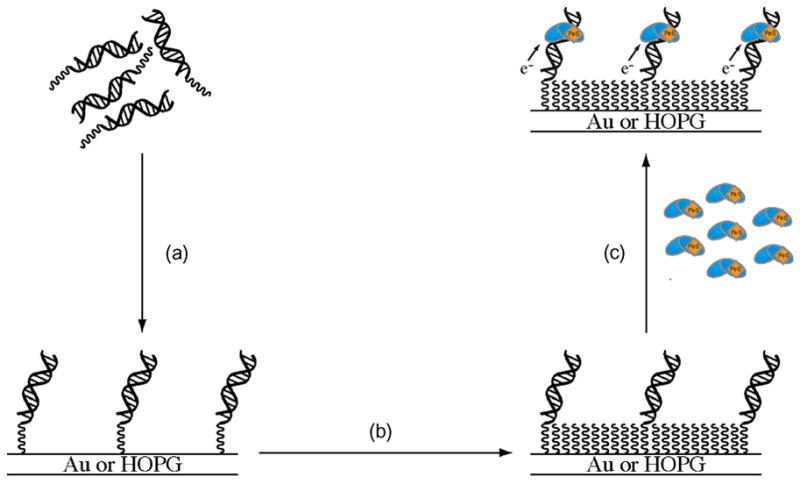
A schematic illustration showing the general strategy for protein electrochemistry experiments at DNA-modified electrodes. (a) Modified DNA self-assembles into a monolayer on an Au or HOPG electrode surface. (b) The DNA-modified surface is then passivated with an alkane or alkanethiol to prevent any interactions between the protein and the bare electrode surface. (c) A protein solution is introduced and monitored electrochemically with cyclic or square-wave voltammetry.
MutY and Endo III both exhibit a quasi-reversible signal when investigated with cyclic voltammetry at a DNA-modified Au electrode [55–56]. This signal is not observed at a mercaptohexanol-modified Au electrode. The signal grows in over ~10 minutes and remains stable up to 30 minutes at ambient temperature. The signal shape is typical of slow, diffusion-limited kinetic processes, and this is consistent with the linear relationship observed between the peak current and the square root of the scan rate. However, multiple buffer exchanges are required to eliminate the signal completely, indicating that the protein is tightly bound to the DNA film. The midpoint potentials measured from these signals are +59 mV and +90 mV versus NHE for Endo III and MutY, respectively.
These experiments raise several questions. Does this signal originate from the iron-sulfur cluster? Is an intact DNA π-stack required to mediate this redox process? Analysis of C199H MutY [37,38], where a cysteine that ligates the cluster is mutated to a histidine, revealed that this mutation leads to a shift in the midpoint potential of −30 mV [55]. C199H is a mutation that maintains near wild-type activity both in vivo and in vitro but the [4Fe-4S]2+ cluster appears to somewhat more likely to decompose to the [3Fe-4S]1+ state [37]. Nevertheless, this change in the coordination environment of the [4Fe-4S] cluster and attendant change in the measured potential is a clear indication that the signal observed is indeed associated with the [4Fe-4S] cluster.
To test the importance of DNA-mediated CT in the redox activity of the [4Fe-4S] cluster, we evaluated MutY and Endo III on an electrode containing a duplex with an abasic site incorporated only three bases away from the electrode surface [55–56]. Previous studies indicate that an abasic site drastically attenuates CT to a redox active intercalator bound above the lesion site [48]. If CT to the cluster is mediated by the DNA π-stack, then, the introduction of an abasic site into the duplex would be expected to attenuate the signal from the [4Fe-4S] cluster. Indeed, both MutY and Endo III exhibit a diminished signal at an electrode modified with DNA containing the intervening abasic site [55–56]. This result demonstrates that CT through the π-stack of DNA is required to observe [4Fe-4S] cluster oxidation electrochemically.
Recently we have monitored MutY and Endo III using DNA-modified HOPG as electrodes and similar potentials are observed (Figure 2) as well as comparable peak shapes and scan-rate dependence relationships [57]. Taking advantage of the wider potential window available with graphite electrodes, we were also able to obtain signals for Endo III on bare HOPG in the absence of DNA. An irreversible anodic peak at +250 mV versus NHE is assigned to the [4Fe-4S]2+/3+ couple in the absence of DNA. Additional scans lead to the appearance of new peaks at much lower potentials, consistent with previous observations that oxidation of the protein in the absence of DNA leads to decomposition of the [4Fe-4S] cluster. Reductive scans of Endo III at bare HOPG electrodes show evidence of a cathodic peak that is assigned to the [4Fe-4S]2+/1+ redox couple. Taken together, these results illustrate that binding of the protein to DNA shifts the redox potential of the [4Fe-4S]2+/3+ couple by ≥ 200 mV and stabilizes the [4Fe-4S]3+ oxidation state, such that reversible oxidation and reduction is possible at ambient temperature. DNA binding appears to convert the clusters in these proteins from ones that resemble low potential ferredoxins to those that resemble high potential iron proteins (HiPIPs).
Figure 2.
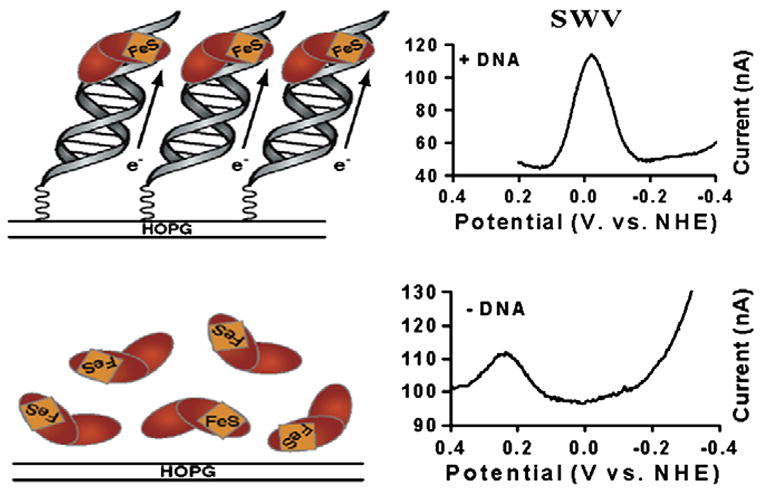
Endo III potentials are measured electrochemically with and without DNA. The top panel shows a square wave voltammogram (right) for Endo III at a DNA-modified HOPG electrode. A peak with a midpoint potential of +20 mV versus NHE is evident. At top left is a cartoon representation this electrode setup. The bottom panel (right) shows square wave voltammetry at a bare HOPG electrode where a peak at +250 mV is apparent. Note that a peak is not observed at +20 mV in the absence of DNA. At bottom left is a cartoon representation of Endo III analyzed at a bare HOPG electrode.
3. EPR Studies with Co(phen)33+ as an Oxidant in Solution
Previous investigations of Endo III with EPR spectroscopy at 10K after exposure to ferricyanide revealed a signal attributed to the [3Fe-4S]1+ cluster [8]. In HiPIPs, the [3Fe-4S]1+ species is a known oxidative and hydrolytic damage product following oxidation of [4Fe-4S]2+ to the [4Fe-4S]3+ cluster [58]. Given this relationship, the amount of [3Fe-4S]1+ could indirectly indicate how readily the [4Fe-4S]3+ species is formed.
A cationic oxidant, Co(phen)33+ (phen = 1,10-phenanthroline), that binds DNA, was employed to determine whether binding of the protein to DNA might promote oxidation of the [4Fe-4S]2+ cluster in solution [56]. When Endo III is incubated with three molar equivalents Co(phen)33+, a small signal is observed that is typical of the [3Fe-4S]1+ cluster. In the presence of DNA, the intensity of this signal increases significantly, perhaps indicating that when Endo III is DNA-bound, the Co(III) oxidant generates more of the [4Fe-4S]3+ species. A similar trend is observed with MutY where exposure of the protein to the Co(III) oxidant yields a four-fold greater signal in the presence of DNA. Local concentration effects may be ruled out as MutY incubated with ten equivalents of Co(III) in the absence of DNA exhibits only a small signal intensity. Thus, in these solution experiments as well, DNA binding appears to activate the proteins towards oxidation.
4. A Model for Damage Detection through DNA Charge Transport
Our laboratory has proposed a model describing how DNA-dependent oxidation of the [4Fe-4S]2+ cluster might allow these glycosylases to use DNA-mediated CT as a way to search for damaged sites in DNA [55–57, 59] (Figure 3). When the protein is not bound to DNA, as previously demonstrated, the [4Fe-4S] cluster is in the 2+ oxidation state and is not readily oxidized and without decomposition of the cluster. Upon binding non-specifically to DNA, however, our data indicate that the [4Fe-4S] cluster is more easily oxidized. Based on the measured ~280 mV shift in the redox potential observed when the protein binds DNA, it is estimated that the binding affinity to DNA of the protein bearing the [4Fe-4S]3+ cluster should be at least three orders of magnitude greater than the affinity of the protein with the [4Fe-4S]2+ cluster. The electron released in this oxidation process can then reduce a distally bound protein in the [4Fe-4S]3+ state in a DNA-mediated CT reaction. The newly reduced protein then dissociates from the DNA, owing to its lower affinity. This process can only occur, however, if the intervening DNA is undamaged and fully Watson-Crick base-paired. If a lesion is present between the two proteins, the CT reaction cannot occur and both proteins remain bound in the vicinity of the lesion. Thus DNA CT provides a means to monitor the integrity of DNA, and, in this model, DNA-mediated CT serves as an initial sorting mechanism, allowing glycosylases to eliminate rapidly undamaged regions of the genome from their search and redistribute themselves onto sites near damage. It is important to note that not only could this CT process occur between two proteins of the same type (between two MutY molecules, for example), but, due to the similarity in redox potentials measured for MutY and Endo III (within ~30 mV of each other), the CT reaction could also occur between a MutY and an Endo III. Given the low copy numbers of the proteins in the cell [60], this interprotein CT is beneficial, allowing a greater number of proteins to participate in the search process.
Figure 3.
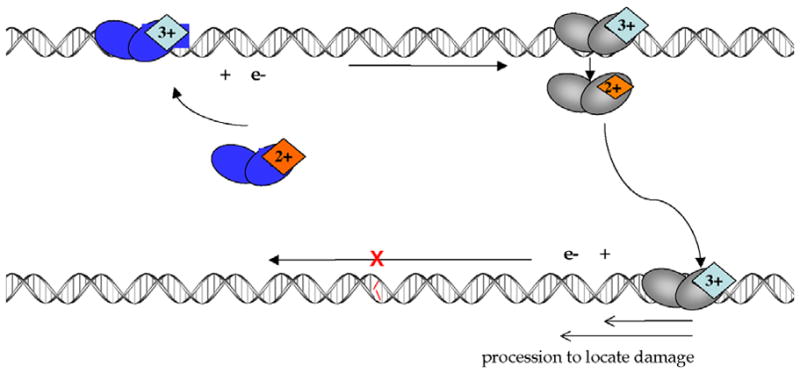
A model for DNA CT in DNA repair. Upon binding to DNA, a protein with an FeS cluster in the 2+ oxidation state can become oxidized to the 3+ state (top left). If the surrounding DNA is undamaged, the released electron can reduce another oxidized protein bound at a distal site (top right) causing the second protein to lose affinity for DNA. As described, DNA CT between two BER enzymes allows for a rapid search of the intervening region of the genome. If a damaged site is present between the two proteins, the DNA-mediated CT event does not occur among these two proteins; both enzymes remain oxidized and bound close to the aberrant site. This process allows proteins to be redistributed from undamaged sites to locations containing lesions.
It is interesting to note that other organisms also contain BER repair proteins with an iron-sulfur cofactor [9, 10]. Most notably, in a set of thermophilic organisms, there is a uracil DNA glycosylase (UDG) that contains a [4Fe-4S] cluster [9]. This is especially noteworthy given that the primary process that leads to uracil in DNA, cytosine deamination, has an enhanced rate at high temperatures [61]. Yet these organisms do not display a higher mutation rate [62]. Might the presence of this cofactor help fulfill this greater requirement for repair? One of these, Archaeoglobus fulgidus UDG, has been evaluated at DNA-modified electrodes and also exhibits DNA-mediated redox activity with a midpoint potential of +95 mV versus NHE [56].
5. Redox Activation of MutY by Guanine Radical
What provides the driving force to initiate this search process? Guanine radicals, with an oxidation potential of 1 V versus NHE [63], have sufficient driving force to oxidize both Endo III and MutY. Moreover, these radicals are being generated under conditions of oxidative stress in the cell [64], when the DNA is in need of repair. Experimentally, we can generate guanine radicals via flash-quench of a Ru(II)dipyridophenazine (dppz) complex intercalated into the DNA helix (Figure 4) [65]. Irradiation of the Ru(II) intercalator generates an excited Ru(II)* state that can be quenched by an electron-accepting molecule in solution (Q) to generate the powerful ground state oxidant, Ru(III). This species has sufficient driving force (1.2 V) to oxidize the guanine bases in a DNA-mediated CT reaction. Can the guanine radical, once generated, in turn promote the oxidation of DNA-bound repair protein, increasing the concentration of [4Fe-4S]3+ and driving the redistribution of repair enzymes? In these experiments EPR and transient absorption spectroscopy were employed to monitor the formation of the guanine radical upon flash-quench of [Ru(phen)2dppz]2+ and, with addition of MutY, formation of oxidized [4Fe-4S]3+ cluster [66].
Figure 4.
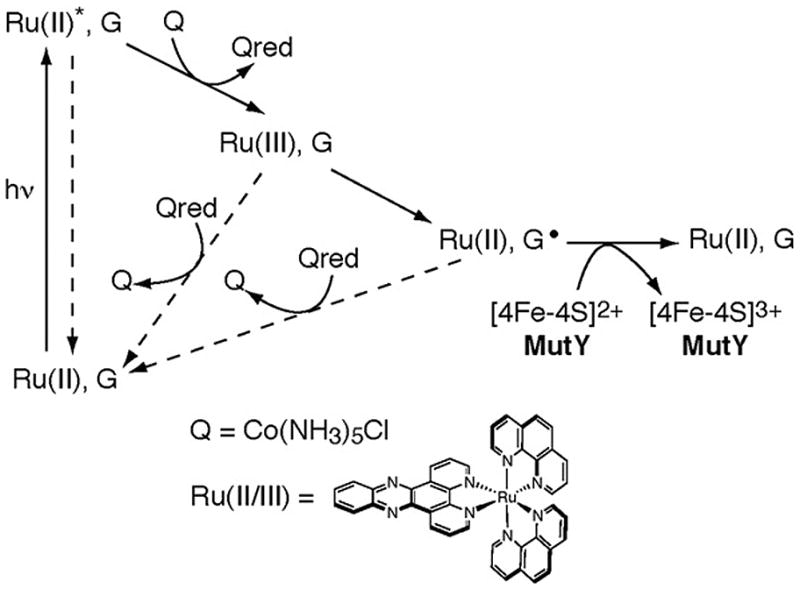
The flash-quench technique is used to generate Ru(III) and then guanine radicals by DNA-mediated CT. The dppz complex of Ru(II) intercalates into the DNA helix. Upon irradiation, the Ru(II) complex is excited to Ru(II)* which can then be quenched by Q in solution to generate Ru(III). This Ru(III) species has sufficient driving force to oxidize guanine in DNA to form the guanine radical cation and the original Ru(II) species. Guanine radical, a signal of oxidative stress, then can serve as an oxidant for the [4Fe-4S] cluster of the repair protein. Note that back electron transfer pathways are shown with dashed arrrows.
We first looked at a simplified system that included DNA (poly(dGC) or poly(dAT)), a sacrificial electron acceptor ([Co(NH3)5Cl]2+) and the photooxidant ([Ru(phen)2dppz]2+). Irradiation of these, in the presence or absence of MutY, was carried out while freezing to 77K and EPR spectra were recorded at 10K. Without MutY, irradiation of poly(dGC), [Ru(phen)2dppz]2+, and [Co(NH3)5Cl]2+ leads to an EPR signal with g = 2.004. This signal has been previously reported and is assigned to the guanine radical [67]. Irradiation of poly(dGC) in the presence of MutY, however, results in the appearance of new EPR signals with primary g values of 2.02 and 2.08 and a feature at 2.06. Interestingly, the guanine radical peak is not observed when MutY is introduced. The peak at g = 2.02 is instead typical of the [3Fe-4S]1+ cluster [58]. The signal at g = 2.08 and its accompanying secondary feature at g = 2.06 is assigned to the [4Fe-4S]3+ cluster [58, 68]. It is noteworthy that, with poly(dAT) and MutY, both signals are still observed, albeit at a much lower intensity. When bound to DNA, MutY can, in fact, be oxidized directly by the intercalated Ru(III) oxidant without an intermediate DNA base radical, but the initial formation of guanine radicals indeed facilitates efficient MutY oxidation.
We also performed, on a faster timescale, flash-quench reactions of [Ru(phen)2dppz]2+ bound non-specifically to poly(dGC) DNA at ambient temperatures. We monitored these assemblies with and without MutY by transient absorption spectroscopy to obtain the full absorption spectrum of the system after flash-quench of the Ru(II) intercalating complex. In the absence of MutY, the transient spectrum observed is characteristic of the spectrum of the guanine radical in duplex DNA, with broad maxima found at 390 and 510 nm [65]. In the presence of MutY, however, a more persistant signal shows the formation of a new species with an absorption maximum at ~405 nm. This spectrum corresponds well not to the guanine radical but to a [4Fe-4S]3+/2+ difference spectrum, which also has an absorption maximum near 405 nm [37, 69]. As observed in the EPR experiments, these transient absorption data then also support formation first of a guanine radical upon oxidative flash quench of [Ru(phen)2dppz]2+ bound to poly(dGC), but in the presence of MutY, formation of a second species, likely [4Fe-4S]3+, that is very long lived.
We further examined a more defined system where the photooxidant Ru(dppz)(phen)(bpy′)2+, (bpy′ = 4-butyric acid-4′-methyl bipyridine) is covalently linked to DNA at the 5′ end. Upon annealing to a complementary strand, the ruthenium complex specifically intercalates 3–4 bases from the 5′-end of the DNA duplex [70]; MutY is capable of binding non-specifically to this designed duplex. Using biochemical experiments to monitor sites of DNA damage, in the absence of MutY, oxidative damage localized on the 5′-G of a 5′-GG-3′ doublet is observed; this guanine damage pattern is a hallmark of oxidation from a distance via DNA-mediated CT from Ru(III) generated in situ. With 0.5–2 equivalents MutY, however, damage at this site is diminished, consistent with the idea that the guanine radical subsequently oxidizes the DNA-bound [4Fe-4S]2+ cluster, inhibiting the formation of irreversible oxidative damage at guanine sites.
The flash-quench reaction was monitored by EPR spectroscopy for the ruthenium-tethered oligonucleotide in the presence of MutY. As with poly(dGC), signals with g = 2.08, g = 2.06, and g = 2.02 are found at 10K, consistent with formation of the oxidized [4Fe-4S]3+ cluster as well as its decomposition product, [3Fe-4S]1+.
These results indicate that guanine radical formation, one of the first DNA damage products generated under oxidizing conditions [64], facilitates the oxidation of DNA-bound MutY. By oxidizing the [4Fe-4S] cluster in MutY, guanine radical may initiate the searching process for BER enzymes in accordance with our proposed model (Figure 5). Guanine radical formation promotes the oxidation of the [4Fe4S]-containing BER enzyme, and in so doing, repairs the guanine radical. This event could encourage non-specific binding of a MutY molecule near a DNA site undergoing oxidative stress. As illustrated in Figure 5, after this initial enzyme is oxidized, another enzyme then binds to the DNA helix at a distal site and reduces the first in a DNA-mediated reaction, thus initiating the search process for damaged bases. Hence, guanine radicals could provide the trigger for scanning of the genome via DNA CT by repair enzymes.
Figure 5.
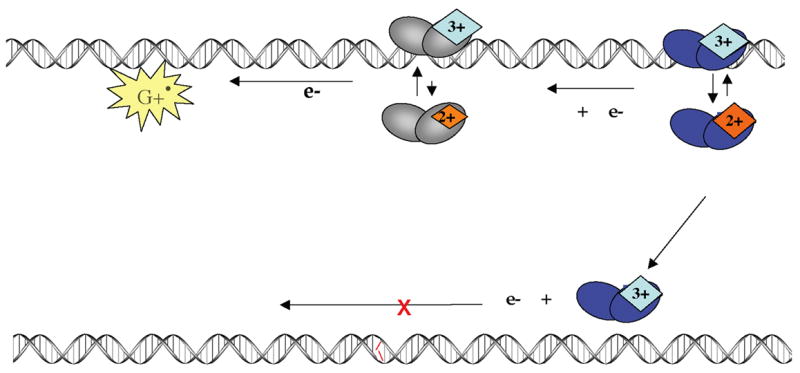
A model for guanine radical initiated searching via DNA CT. Guanine radical, one of the first signs of oxidative stress, can initiate scanning by oxidizing the [4Fe-4S]2+ cluster in MutY. The first oxidized protein can then be reduced by a second protein via the DNA base-pair stack. Again, if a lesion is present, the proteins stay oxidized and bound in the vicinity of the damaged site. Oxidative stress thus provides the driving force for the DNA CT search.
6. Modified nitroxyl radical as an electron trap in DNA-mediated CT
We have also investigated a stable nitroxyl radical tethered to a uridine base as an electron trap for this DNA-mediated CT reaction between repair proteins [71]. Here we have the advantage of monitoring electron trapping by EPR spectroscopy in a physiologically buffered solution at ambient temperature (without the need for freezing to 10K).
The strategy employed to introduce an electron trap into the DNA duplex is illustrated in Figure 6. These duplexes were synthesized containing a uracil base modified with an alkyne linked to a nitroxyl radical spin label [71]. The spin-label moiety is EPR-active at ambient temperature. Oxidation of the nitroxyl radical to the EPR-silent diamagnetic N-oxo-ammonium ion (R-N=O+) is achieved with a mild oxidant (IrCl62−) that cannot damage any of other bases [72]. Upon oxidation, this probe can serve as an electron trap located within the DNA helix. Since the reduction potential of the R-N=O+ modified uridine base is ~ 1 V versus NHE and the [4Fe-4S]3+/2+ midpoint potential is 0.1 V versus NHE for the protein bound to DNA [56], it should be favorable for an electron to transfer from the [4Fe-4S] cluster of the protein to the N-oxo-ammonium ion. Thus, we expect that the repair protein will reduce the EPR-silent N-oxo-ammonium species back to the EPR-active nitroxyl radical. This reaction would provide a positive signal for electron trapping from the DNA-bound protein. Significantly, the midpoint potentials of the BER enzymes (~0.1 V versus NHE) render them incapable of reducing the intervening bases [73].
Figure 6.
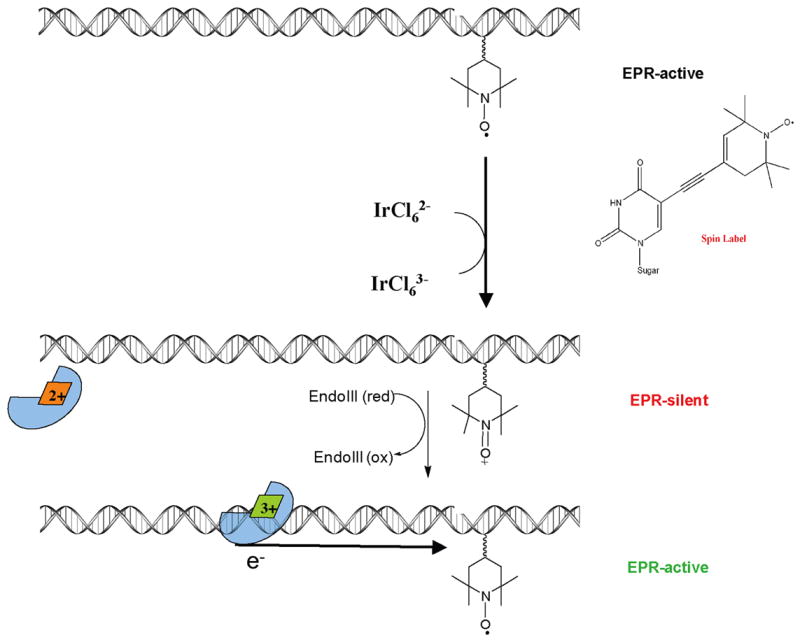
Strategy for electron trapping from the DNA-bound repair protein. (Top) A stable, EPR-active, nitroxyl radical is incorporated as a modified base in the DNA duplex. (Middle) A mild Ir(IV) oxidant is used to generate the EPR-silent N-oxo-ammonium ion. (Bottom) Protein binding and cluster oxidation releases the electron, which reduces the modified base back to the EPR-active nitroxide radical.
EPR spectra of a fully matched 36-mer DNA duplex containing the spin-label modified base were obtained at ambient temperature before and after addition of IrCl62−. A large decrease in the EPR signal is observed upon addition of Ir(IV), indicating that the spin label is readily reduced from the nitroxyl radical to the EPR-silent N-oxo ammonium ion. However, a rapid regeneration of the EPR signal is evident upon addition of MutY or Endo III. Importantly, addition of a DNA-binding protein that lacks the cluster (e.g. EcoRI) does not regenerate the signal (E.Y., unpublished results).
Is this rapid signal regeneration associated with a DNA-mediated CT reaction or is does it occur as a consequence of direct binding of the protein to the spin-label modified base? This was explored in two separate experiments. MutY is known to bind preferentially to a 7-deaza-adenine:guanine (ZG) base pair without excising the modified adenine [74]. We observe that addition of MutY to the assembly containing the ZG base pair located 19 base pairs from the electron trap leads to a significantly greater regeneration of the EPR signal when compared to an identical duplex containing a CG base pair at the analogous site. Since MutY has a greater affinity for the ZG site when compared to a CG site, this increase in regeneration likely reflects that a greater fraction of MutY molecules are bound to this duplex. Furthermore, this indicates that protein reduction of the modified base largely occurs from a distance, as the ZG base pair is separated from the N-oxo-ammonium ion by 19 base pairs.
To examine further the DNA-mediated nature of CT from the protein, we also tested a different electron trap that is not well coupled into the base pair stack. Using a modified base where the nitroxyl radical is located in a saturated ring, addition of Ir(IV) also causes a loss of signal, consistent with complete oxidation to form the N-oxo-ammonium ion. Addition of MutY, however, now causes only a very small regeneration of the signal. This small regeneration could be due to very limited binding of the protein directly at the probe.
These data demonstrate that a uridine modified with a nitroxyl radical can serve as an electron trap within the DNA helix. Effective reduction of the well-coupled trap in the DNA duplex by MutY and Endo III further highlights the necessity for an intact π-stack to facilitate [4Fe-4S] cluster oxidation in these proteins.
7. Conclusions and Implications
While few diseases are associated with deficiencies in BER, a lower level of overall DNA repair capacity is often observed in tumor cells [1]. Quite recently, though, it has been established that inherited mutations in the human MutY gene (MutYH) can lead to a severe predisposition to colorectal cancer in a condition termed MutYH-associated polyposis (MAP) [75, 76]. Two of the most common mutations implicated in MAP, Y165C and G382D, involve highly conserved positions in the protein. In E. coli MutY, the corresponding mutations (Y82C and G253D) lead to modest decreases in substrate binding affinity and rate of excision [77]. In addition, structural studies show that Y82 and G253 interact with the DNA near the 8-oxo-guanine lesion site [39–40]. It is likely that Y82 and G253 are involved in substrate recognition, but it is still not completely understood how all of the mutations implicated in MAP give rise to cancer. However, given this connection between deficiencies in damage recognition in MutY and human disease, it is clearly important to understand how these enzymes search for their substrates.
Is the redox activity of the [4Fe-4S] cluster relevant in vivo? MutY and Endo III have similar redox potentials and could cooperatively search for damage using DNA CT inside the cell [56]. In this instance, if Endo III were inactivated, a decrease in the in vivo activity of MutY should be observed. The CC104 E. coli strain, which uses a mutation in lacZ to report the frequency of G:C to T:A transversion mutations, is often used as an indicator of MutY activity in vivo [22,78]. When the Endo III gene (nth) is knocked out in the CC104 genetic background, a small increase in the G:C to T:A mutation rate is observed [79]. While this observed effect appears at first to be attributed to overlapping substrate specificity with MutY, in vitro evidence to support this idea is lacking [17]. Could this relationship instead have something to do with the iron-sulfur cofactor harbored by each protein?
The experiments reviewed herein provide evidence for a DNA-dependent electron transfer role for the [4Fe-4S] cluster in a set of BER glycosylases. This apparent redox activity might allow these proteins quickly and efficiently to seek out damaged sites in the genome using DNA-mediated CT. In our proposed model, the DNA π-stack serves as a signaling medium whereby two proteins can communicate with each other and gain information about the integrity of the surrounding genome.
However, many questions remain. Do DNA repair proteins actually use DNA-mediated CT inside the cell? Is a metal cofactor necessary or can redox-active amino acids also serve as electron donors and acceptors? Can other DNA-binding proteins (i.e. transcription factors, chromatin remodeling factors) use DNA CT as a signaling mechanism? These questions form the basis of our ongoing exploration of biological DNA-mediated CT.
Acknowledgments
We are grateful to the NIH (GM49216) for their financial support. We also thank our coworkers and collaborators for their efforts, and we are grateful to Ed Stiefel for getting one of us (J.K.B.) excited about Fe-S clusters at the beginning.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T, editors. DNA Repair and Mutagenesis. ASM Press; Washington DC: 2006. [Google Scholar]
- 2.David SS, Williams SD. Chem Rev. 1998;98:1221–1262. doi: 10.1021/cr980321h. [DOI] [PubMed] [Google Scholar]
- 3.Blainey PC, van Oijen AM, Banerjee A, Verdine GL, Xie XS. Proc Natl Acad Sci USA. 2006;103:5752–5757. doi: 10.1073/pnas.0509723103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Francis AW, David SS. Biochemistry. 2003;42:801–810. doi: 10.1021/bi026375+. [DOI] [PubMed] [Google Scholar]
- 5.Halford SE, Szczelkun MD. Eur Biophys J. 2002;31:257–267. doi: 10.1007/s00249-002-0224-4. [DOI] [PubMed] [Google Scholar]
- 6.Gowers DM, Halford SE. EMBO J. 2003;22:1410–1418. doi: 10.1093/emboj/cdg125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gerland U, Moroz JD, Hwa T. Proc Natl Acad Sci USA. 2002;99:12015–12020. doi: 10.1073/pnas.192693599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cunningham RP, Asahara H, Bank JF, Scholes CP, Salerno JC, Surerus K, Munck E, McCracken J, Peisach J, Emptage MH. Biochemistry. 1989;28:4450–4455. doi: 10.1021/bi00436a049. [DOI] [PubMed] [Google Scholar]
- 9.Hinks JA, Evans MCW, de Miguel Y, Sartori AA, Jiricny J, Pearl LH. J Biol Chem. 2002;277:16936–16940. doi: 10.1074/jbc.M200668200. [DOI] [PubMed] [Google Scholar]
- 10.Rebeil R, Sun Y, Chooback L, Pedraza-Reyes M, Kinsland C, Begley TP, Nicholson WL. J Bacteriol. 1998;180:4879–4885. doi: 10.1128/jb.180.18.4879-4885.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Demple B, Linn S. Nature. 1980;287:203–208. doi: 10.1038/287203a0. [DOI] [PubMed] [Google Scholar]
- 12.Katcher HL, Wallace SS. Biochemistry. 1983;22:4071–4081. doi: 10.1021/bi00286a013. [DOI] [PubMed] [Google Scholar]
- 13.Breimer LH, Lindahl T. J Biol Chem. 1984;259:5543–5548. [PubMed] [Google Scholar]
- 14.Weiss RB, Duker NJ. Nucleic Acids Res. 1986;14:6621–6631. doi: 10.1093/nar/14.16.6621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boorstein RJ, Hilbert TP, Cadet J, Cunningham RP, Teebor GW. Biochemistry. 1989;28:6164–6170. doi: 10.1021/bi00441a007. [DOI] [PubMed] [Google Scholar]
- 16.Wagner JR, Blount BC, Weinfeld M. Anal Biochem. 1996;233:76–86. doi: 10.1006/abio.1996.0010. [DOI] [PubMed] [Google Scholar]
- 17.Dizdaroglu M, Laval J, Boiteux S. Biochemistry. 1993;32:12105–12111. doi: 10.1021/bi00096a022. [DOI] [PubMed] [Google Scholar]
- 18.Kuo CF, McRee DE, Fisher CL, O’Handley SF, Cunningham RP, Tainer JA. Science. 1992;258:434–440. doi: 10.1126/science.1411536. [DOI] [PubMed] [Google Scholar]
- 19.Thayer MM, Ahern H, Xing D, Cunningham RP, Tainer JA. EMBO J. 1995;14:4108–4120. doi: 10.1002/j.1460-2075.1995.tb00083.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guan Y, Manuel RC, Arvai AS, Parikh SS, Mol CD, Miller JH, Lloyd S, Tainer JA. Nat Struct Biol. 1998;5:1058–1064. doi: 10.1038/4168. [DOI] [PubMed] [Google Scholar]
- 21.Michaels ML, Pham L, Nghiem Y, Cruz C, Miller JH. Nucleic Acids Res. 1990;18:3841–3845. doi: 10.1093/nar/18.13.3841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nghiem Y, Cabrera M, Cupples CG, Miller JH. Proc Natl Acad Sci USA. 1988;85:2709–2713. doi: 10.1073/pnas.85.8.2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Michaels ML, Cruz C, Grollman AP, Miller JH. Proc Natl Acad Sci USA. 1992;89:7022–7025. doi: 10.1073/pnas.89.15.7022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Michaels ML, Miller JH. J Bacteriol. 1992;174:6321–6325. doi: 10.1128/jb.174.20.6321-6325.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Michaels ML, Tchou J, Grollman AP, Miller JH. Biochemistry. 1992;31:10964–10968. doi: 10.1021/bi00160a004. [DOI] [PubMed] [Google Scholar]
- 26.Gogos A, Cillo J, Clarke ND, Lu AL. Biochemistry. 1996;35:16665–16671. doi: 10.1021/bi960843w. [DOI] [PubMed] [Google Scholar]
- 27.Lu AL, Tsai-Wu JJ, Cillo J. J Biol Chem. 1995;270:23582–23588. doi: 10.1074/jbc.270.40.23582. [DOI] [PubMed] [Google Scholar]
- 28.Manuel RC, Czerwinski EW, Lloyd RS. J Biol Chem. 1996;271:16218–16226. doi: 10.1074/jbc.271.27.16218. [DOI] [PubMed] [Google Scholar]
- 29.Francis AW, Helquist SA, Kool ET, David SS. J Am Chem Soc. 2003;125:16235–16242. doi: 10.1021/ja0374426. [DOI] [PubMed] [Google Scholar]
- 30.Chepanoske CL, Porello SL, Fujiwara T, Sugiyama H, David SS. Nucleic Acids Res. 1999;27:3197–3204. doi: 10.1093/nar/27.15.3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vidmar JJ, Cupples CG. Can J Microbiol. 1993;39:892–894. doi: 10.1139/m93-133. [DOI] [PubMed] [Google Scholar]
- 32.Bulychev NV, Varaprasad CV, Dorman G, Miller JH, Eisenberg M, Grollman AP, Johnson F. Biochemistry. 1996;35:13147–13156. doi: 10.1021/bi960694h. [DOI] [PubMed] [Google Scholar]
- 33.Porello SL, Leyes AE, David SS. Biochemistry. 1998;37:14756–14764. doi: 10.1021/bi981594+. [DOI] [PubMed] [Google Scholar]
- 34.Radicella JP, Clark EA, Fox MS. Proc Natl Acad Sci USA. 1988;85:9674–9678. doi: 10.1073/pnas.85.24.9674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fu W, O’Handley S, Cunningham RP, Johnson MK. J Biol Chem. 1992;267:16135–16137. [PubMed] [Google Scholar]
- 36.Porello SL, Cannon MJ, David SS. Biochemistry. 1998;37:6465–6475. doi: 10.1021/bi972433t. [DOI] [PubMed] [Google Scholar]
- 37.Messick TE, Chmiel NH, Golinelli MP, Langer MR, Joshua-Tor L, David SS. Biochemistry. 2002;41:3931–3942. doi: 10.1021/bi012035x. [DOI] [PubMed] [Google Scholar]
- 38.Golinelli MP, Chmiel NH, David SS. Biochemistry. 1999;38:6997–7007. doi: 10.1021/bi982300n. [DOI] [PubMed] [Google Scholar]
- 39.Fromme JC, Verdine GL. EMBO J. 2003;22:3461–3471. doi: 10.1093/emboj/cdg311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fromme JC, Banerjee A, Huang SJ, Verdine GL. Nature. 2004;427:652–656. doi: 10.1038/nature02306. [DOI] [PubMed] [Google Scholar]
- 41.Kelley SO, Barton JK. Science. 1999;283:375–381. doi: 10.1126/science.283.5400.375. [DOI] [PubMed] [Google Scholar]
- 42.O’Neill MA, Barton JK. In: Topics in Current Chemistry: Electron Transfer in DNA: I. Schuster GB, editor. Springer-Verlag; Heidelberg: 2004. pp. 67–115. [Google Scholar]
- 43.Schuster GB. Acc Chem Res. 2000;33:253–260. doi: 10.1021/ar980059z. [DOI] [PubMed] [Google Scholar]
- 44.Giese B. Annu Rev Biochem. 2002;71:51–70. doi: 10.1146/annurev.biochem.71.083101.134037. [DOI] [PubMed] [Google Scholar]
- 45.Nunez ME, Hall DB, Barton JK. Chem Biol. 1999;6:85–97. doi: 10.1016/S1074-5521(99)80005-2. [DOI] [PubMed] [Google Scholar]
- 46.Bhattacharya PK, Barton JK. J Am Chem Soc. 2001;123:8649–8656. doi: 10.1021/ja010996t. [DOI] [PubMed] [Google Scholar]
- 47.Hall DB, Barton JK. J Am Chem Soc. 1997;119:5045–5046. [Google Scholar]
- 48.Boon EM, Ceres DM, Drummond TG, Hill MG, Barton JK. Nat Biotechnol. 2000;18:1096–1100. doi: 10.1038/80301. [DOI] [PubMed] [Google Scholar]
- 49.Boal AK, Barton JK. Bioconjug Chem. 2005;16:312–321. doi: 10.1021/bc0497362. [DOI] [PubMed] [Google Scholar]
- 50.Nunez ME, Holmquist GP, Barton JK. Biochemistry. 2001;40:12465–12471. doi: 10.1021/bi011560t. [DOI] [PubMed] [Google Scholar]
- 51.Nunez ME, Noyes KT, Barton JK. Chem Biol. 2002;9:403–415. doi: 10.1016/s1074-5521(02)00121-7. [DOI] [PubMed] [Google Scholar]
- 52.Kelley SO, Boon EM, Barton JK, Jackson NM, Hill MG. Nucleic Acids Res. 1999;27:4830–4837. doi: 10.1093/nar/27.24.4830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gorodetsky AA, Barton JK. Langmuir. 2006;22:7917–7922. doi: 10.1021/la0611054. [DOI] [PubMed] [Google Scholar]
- 54.Boon EM, Salas JE, Barton JK. Nat Biotechnol. 2002;20:282–286. doi: 10.1038/nbt0302-282. [DOI] [PubMed] [Google Scholar]
- 55.Boon EM, Livingston AL, Chmiel NH, David SS, Barton JK. Proc Natl Acad Sci USA. 2003;100:12543–12547. doi: 10.1073/pnas.2035257100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Boal AK, Yavin E, Lukianova OA, O’Shea VL, David SS, Barton JK. Biochemistry. 2005;44:8397–8407. doi: 10.1021/bi047494n. [DOI] [PubMed] [Google Scholar]
- 57.Gorodetsky AA, Boal AK, Barton JK. J Am Chem Soc. 2006;128:12082–12083. doi: 10.1021/ja064784d. [DOI] [PubMed] [Google Scholar]
- 58.Cowan JA, Lui SM. Adv Inorg Chem. 1998;45:313–350. [Google Scholar]
- 59.Rajski SR, Jackson BA, Barton JK. Mutat Res. 2000;447:49–72. doi: 10.1016/s0027-5107(99)00195-5. [DOI] [PubMed] [Google Scholar]
- 60.Demple B, Harrison L. Annu Rev Biochem. 1994;63:915–948. doi: 10.1146/annurev.bi.63.070194.004411. [DOI] [PubMed] [Google Scholar]
- 61.Lindahl T, Nyberg B. Biochemistry. 1974;13:3405–3410. doi: 10.1021/bi00713a035. [DOI] [PubMed] [Google Scholar]
- 62.Jacobs KL, Grogan DW. J Bacteriol. 1997;179:3298–3303. doi: 10.1128/jb.179.10.3298-3303.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Steenken S, Jovanovic SV. J Am Chem Soc. 1997;119:617–618. [Google Scholar]
- 64.Cadet J, Bellon S, Berger M, Bourdat AG, Douki T, Duarte V, Frelon S, Gasparutto D, Muller E, Ravanat JL, Sauvaigo S. Biol Chem. 2002;383:933–943. doi: 10.1515/BC.2002.100. [DOI] [PubMed] [Google Scholar]
- 65.Stemp EDA, Arkin MR, Barton JK. J Am Chem Soc. 1997;119:2921–2925. [Google Scholar]
- 66.Yavin E, Boal AK, Stemp EDA, Boon EM, Livingston AL, O’Shea VL, David SS, Barton JK. Proc Natl Acad Sci USA. 2005;102:3546–3551. doi: 10.1073/pnas.0409410102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cullis PM, Malone ME, Merson Davies LA. J Am Chem Soc. 1996;118:2775–2781. [Google Scholar]
- 68.Dilg AWE, Mincione G, Achterhold K, Iakovleva O, Mentler M, Luchinat C, Bertini I, Parak FG. J Biol Inorg Chem. 1999;4:727–741. doi: 10.1007/s007750050345. [DOI] [PubMed] [Google Scholar]
- 69.Johnson MK, Duderstadt RE, Duin EC. Adv Inorg Chem. 1999;47:1–82. [Google Scholar]
- 70.Arkin MR, Stemp EDA, Coates Pulver S, Barton JK. Chem Biol. 1997;4:389–400. doi: 10.1016/s1074-5521(97)90129-0. [DOI] [PubMed] [Google Scholar]
- 71.Yavin E, Stemp EDA, O’Shea VL, David SS, Barton JK. Proc Natl Acad Sci USA. 2006;103:3610–3614. doi: 10.1073/pnas.0600239103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hickerson RP, Prat F, Miller JG, Foote CS, Burrows CJ. J Am Chem Soc. 1999;121:9423–9428. [Google Scholar]
- 73.Seidel CAM, Schulz A, Sauer MHM. J Phys Chem. 1996;100:5541–5553. [Google Scholar]
- 74.Porello SL, Williams SD, Kuhn H, Michaels ML, David SS. J Am Chem Soc. 1996;118:10684–10692. [Google Scholar]
- 75.Al-Tassan N, Chmiel NH, Maynard J, Fleming N, Livingston AL, Williams GT, Hodges AK, Davies DR, David SS, Sampson JR, Cheadle JP. Nat Genet. 2002;30:227–232. doi: 10.1038/ng828. [DOI] [PubMed] [Google Scholar]
- 76.Sampson JR, Jones S, Dolwani S, Cheadle JP. Biochem Soc Trans. 2005;33:679–683. doi: 10.1042/BST0330679. [DOI] [PubMed] [Google Scholar]
- 77.Livingston AL, Kundu S, Henderson Pozzi M, Anderson DW, David SS. Biochemistry. 2005;44:14179–14190. doi: 10.1021/bi050976u. [DOI] [PubMed] [Google Scholar]
- 78.Cupples CG, Miller JH. Proc Natl Acad Sci USA. 1989;86:5345–5349. doi: 10.1073/pnas.86.14.5345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tano K, Iwamatsu Y, Yasuhira S, Utsumi H, Takimoto K. J Radiat Res. 2001;42:409–413. doi: 10.1269/jrr.42.409. [DOI] [PubMed] [Google Scholar]