

Abstract
Leukocytes have been implicated in the pathogenesis of ischemic acute renal failure (ARF), but the roles of the individual cell types involved are largely unknown. Recent indirect evidence suggests that T cells may play an important role in a murine model of ARF. In the current study, we found that mice deficient in T cells (nu/nu mice) are both functionally and structurally protected from postischemic renal injury. Reconstitution of nu/nu mice with wild-type T cells restored postischemic injury. We then analyzed the contribution of the individual T cell subsets to postischemic injury and found that mice deficient in CD4+ T cells, but not mice deficient in CD8+ T cells, were significantly protected from ARF. Direct evidence for a pathophysiologic role of the CD4+ T cell was obtained when reconstitution of CD4-deficient mice with wild-type CD4+ T cells restored postischemic injury. In addition, adoptive transfers of CD4+ T cells lacking either the costimulatory molecule CD28 or the ability to produce IFN-γ were inadequate to restore injury phenotype. These results demonstrate that the CD4+ T cell is an important mediator of ischemic ARF, and targeting this cell may yield novel therapies.
Introduction
Ischemic acute renal failure (ARF) is the most common cause of intrinsic ARF in adults (1). In native kidneys, it is associated with an overall mortality rate of up to 50% (2). Despite developments in dialysis treatment, this mortality rate has not improved in the last 30 years (3). In transplant kidneys, ischemic injury increases length of hospitalization and leads to increased allograft loss (4).
Recently, leukocytes have been implicated in the pathogenesis of renal ischemia-reperfusion injury (IRI); most of the work has focused on the role of the neutrophil (5). Evidence from several studies suggests, however, that T cells could also be important leukocyte mediators of renal IRI. Lymphocytes have been found in postischemic human (6, 7) and rat (8) kidneys, particularly in the outer medulla. We recently found that genetically engineered mice deficient in both CD4+ and CD8+ lymphocytes had substantially less kidney dysfunction after renal ischemia than did wild-type control mice with renal IRI (9). T cells have also been found to be important in the pathogenesis of IRI in other organs (10). Importantly, the study of T cells in ARF is a novel approach in this area of investigation, and opens the potential of harnessing developments in T cell biology for novel ARF therapies.
We hypothesized that the T cell has an important role in the pathogenesis of renal IRI, and sought to directly identify which T cell subset might be more important. The hypothesis was investigated with a redundant approach using distinct strains of T cell–deficient mice and adoptive transfer techniques. T cell–deficient (nu/nu) mice were found to be protected from postischemic renal injury, while nu/nu mice reconstituted with wild-type T cells were found to have postischemic renal injury similar to that in wild-type mice. To elucidate the primary T cell subset that is important in renal IRI, mice deficient in CD4+ T cells alone or CD8+ T cells alone were subjected to renal IRI. The CD4-deficient mice had significantly improved recovery of renal function after ischemia compared with CD8-deficient mice and wild-type control mice. Furthermore, CD4-deficient mice reconstituted with CD4+ T cells had a restoration of the postischemic injury phenotype. To identify possible mechanisms involved in the protection from renal injury seen in the T cell–deficient mice, we studied nu/nu mice that had been adoptively transferred with an enriched CD4+ T cell population obtained from mice that were deficient in either CD28 (accessory molecule mediated pathway) or IFN-γ (to distinguish between a possible Th1 versus a Th2 response). We found that both CD28 and IFN-γ are important components of CD4+ T cell–mediated renal injury following IRI, as neither defective CD4+ population restored the postischemic injury phenotype.
Methods
Mice.
nu/nu mice (B6.Cg-Foxn1nu) and C57BL/6 wild-type littermates were purchased from The Jackson Laboratory (Bar Harbor, Maine, USA). The two main defects of mice homozygous for the nu/nu spontaneous mutation (Foxn1nu, formerly Hfh11nu) are abnormal hair growth and defective development of the thymic epithelium. nu/nu mice are therefore athymic due to a developmental failure of the thymus. Consequently, homozygous nu/nu mice lack T cells and cell-mediated immunity. CD4-deficient mice (B6.129S2-Cd4tm1Mak), CD8-deficient mice (B6.129S2-Cd8atm1Mak), CD28-deficient mice (B6.129S2-Cd28tm1Mak), IFN-γ–deficient mice (B6.129S7-Ifngtm1Ts), and wild-type littermates were also purchased from The Jackson Laboratory. Mice were housed under pathogen-free conditions, according to NIH guidelines. To confirm knockout and adoptive transfer status, spleens and lymph nodes from each group of mice were collected upon sacrifice and analyzed for CD4+ and CD8+ cells using flow cytometric analysis.
Renal ischemia reperfusion model.
An established model of renal IRI was used (9). Animals were anesthetized with 75 mg/kg intraperitoneal sodium pentobarbital. Abdominal incisions were made and the renal pedicles were bluntly dissected. A microvascular clamp was placed on both renal pedicles for 30 minutes. (An ischemia time of 30 minutes was chosen because it produces a sublethal kidney injury with many similarities to that seen in human IRI with acute tubular necrosis.) During the procedure, animals were kept well hydrated with saline, and were kept at a constant temperature (∼37°C). After the clamps were removed, the wounds were sutured and the animals were allowed to recover.
Assessment of postischemic renal function.
Blood samples were obtained from the tail vein at 0, 24, 48, and 72 hours after ischemia. Serum creatinine (SCr) (mg/dl) was measured on a Roche Cobas Fara automated system (Roche, Nutley, New Jersey, USA) using a Creatinine 557 kit (Sigma Diagnostics, St. Louis, Missouri, USA).
Tissue histological examination.
At 24 or 72 hours after ischemia, kidneys were dissected from mice, and coronal tissue slices were fixed in 10% formalin and processed for histologic examination using standard techniques. Formalin tissue was embedded in paraffin and 4-μm sections were cut. Basement membranes and other oxidizable structures containing 1,2-diol were visualized using periodic acid–Schiff reagent. These sections were examined in a blinded fashion by a nephrologist and a pathologist to evaluate the degree of tubular necrosis.
Immunohistochemistry.
Tissue sections were prepared as described above for routine histology. Sections (4 μm) were prepared on a cryostat and mounted on Fisher Superfrost Plus slides, fixed in ice-cold acetone for 1–2 minutes, and allowed to air dry. Sections were then blocked with 1:100 normal rabbit serum in PBS containing Vector Avidin DH (Vector Laboratories Inc., Burlingame, California, USA). The following primary antibodies were then added to the sections: GK 1.5 (rat anti-mouse CD4; American Type Culture Collection, Manassas, Virginia, USA), GD 2.43 (rat anti-mouse CD8; American Type Culture Collection), M1/70 (rat anti-mouse MAC-1; Caltag Laboratories Inc., Burlingame, California, USA), and 7/4 (rat anti-mouse neutrophil; Accurate Chemical & Scientific Corp., Westbury, New York, USA). Sections were then incubated for 1 hour at room temperature. An isotype control primary antibody was used as a background staining control. Sections were then rinsed in PBS and treated with 3% hydrogen peroxide in biotin (10 μg/l PBS) to block the biotin binding sites. After three washes in PBS, the slides were incubated with a biotin-conjugated rabbit anti-rat IgG secondary antibody (Vector Laboratories Inc.) for 35 minutes at room temperature. Sections were once again washed, and were then incubated for 45 minutes in Vector Elite ABC (Vector Laboratories Inc.). Sections were washed and developed with 3-amino-9-ethyl-carbazole, counterstained with hematoxylin, and mounted using Glycergel (DAKO Corp., Carpinteria, California, USA). After the entire kidney section was viewed, ten high-powered (×40) fields were counted in the area of the corticomedullary junction, and total numbers of cells were quantified in a blinded fashion.
T cell adoptive transfer.
Spleens and lymph nodes were collected from C57BL/6 wild-type littermates, CD28-deficient mice, and IFN-γ–deficient mice. Splenic cells were collected by centrifugation, and the red blood cells were removed by lysis in NH4Cl for 5 minutes. T cell enrichment was performed using nylon wool column chromatography. Briefly, sterile nylon wool–packed columns were equilibrated in RPMI with 5% FCS, and cells were incubated on the column for 1 hour at 37°C in 5% CO2. Nonadherent cells (predominantly T cells) were then eluted in 15 ml of RPMI and collected by centrifugation. The percentages of CD4+ and CD8+ T cells were assessed by flow cytometry before and after nylon wool T cell–enrichment. Enriched T cells were then washed three times in HBSS to remove serum from the preparation. Approximately 15 × 106 enriched T cells were injected intraperitoneally into each nu/nu mouse. IRI was induced in the mice 3 weeks after transfer. The 3-week timepoint was based on our preliminary studies in which earlier timepoints had less efficient reconstitution.
Purified populations of CD4+ T cells were obtained by negative selection of CD8+ T cells and B cells on an immunoaffinity column according to the manufacturer’s specifications (cellect-plus kits; Cytovax Biotechnologies Inc., Alberta, Canada). Enriched CD4+ T cells were eluted from each column, and approximately 5–10 × 106 cells were injected intraperitoneally. FACS analysis was performed before and after column purification to determine percentages of CD4+ and CD8+ cells in cell preparations. IRI was performed on the mice 3 weeks after transfer.
FACS analysis.
Postischemic spleens and/or lymph nodes were crushed in 5 ml HBSS-2 using five strokes in a glass homogenizer. Cells were collected by centrifugation at 250 g for 10 minutes. They were then resuspended in ammonium chloride/potassium buffer for 5 minutes to remove red blood cells. Cells were washed three times with ice-cold HBSS-2 and filtered through a 70-μm nylon screen to remove debris. For each analysis, 106 cells were treated with an Fc block for 5 minutes, and then stained with 0.5 μg R-PE rat anti-mouse CD4 (H129.19) antibody and FITC-conjugated anti-mouse CD8b.2 (53-5.8) antibody or isotype controls (PharMingen, San Diego, California, USA) for 1 hour on ice. Cells were then washed twice, fixed in 1% formalin, and analyzed on a Epics XL flow cytometer using System II software (Beckman Coulter, Fullerton, California, USA).
Statistical analysis.
Data are expressed as mean ± SE. Comparisons of group means were performed using 1-way ANOVA with the Student-Newman-Keuls multiple group comparison test. P < 0.05 was considered significant.
Results
Confirmation of T cell knockout and reconstitution status.
To confirm the absence or presence of CD4+ and/or CD8+ T cells, we used flow cytometry to analyze lymph nodes and spleen from each mouse upon sacrifice. Figure 1a shows the absence of both CD4+ and CD8+ cells in nu/nu mice, the presence of CD4+ and CD8+ cells in T cell–reconstituted nu/nu mice (Figure 1b), the absence of CD4+ cells in CD4-deficient mice (Figure 1c), and the presence of CD4+ T cells in CD4-deficient mice that were reconstituted with CD4+ cells (Figure 1d). CD4+ cells that were taken from mice deficient in either CD28 or IFN-γ were equivalent to wild-type cells in their ability to reconstitute after adoptive transfer. FACS analysis was also used to determine the enrichment of T cells in the adoptive transfer experiments. Nylon wool was used to obtain an enriched population of T cells from a lymphocyte cell preparation. FACS analysis performed before enrichment on nylon wool showed CD4+ staining of 11.4%, and CD8+ staining of 7.75%. Following nylon wool enrichment, these values were increased to 36.2% CD4+ stained cells and 39.7% CD8+ stained cells. For the CD4+ adoptive transfer experiment, the cell preparation before CD4+ column enrichment consisted of 19.1% CD4+ cells. Column FACS analysis after CD4+ enrichment showed an increase of CD4+ cells to 71.8%.
Figure 1.

Flow cytometry confirmation of the absence or presence of CD4+ and/or CD8+ T cells. (a) CD4+ and CD8+ T cells are absent from spleens obtained from nu/nu mice. (b) After nu/nu mice were adoptively transferred with a T cell population from a wild-type control mouse, both CD4+ and CD8+ T cells were detected in spleens. The reconstitution of these cells in nu/nu mice ranged between 2 and 6%. (c) CD4-deficient mice show an absence of CD4+ T cells, but a normal complement of CD8+ T cells when compared with a wild-type control (not shown). (d) CD4-reconstituted CD4-deficient mice show a CD4+ population of approximately 2%.
T cell–deficient mice show a return of the injury phenotype when adoptively transferred with an enriched T cell population.
To demonstrate the effect of T cell deficiency on early renal dysfunction following IRI, we first evaluated postischemic renal function in nu/nu mice. These mice underwent moderate renal ischemia (30 minutes of renal artery clamping) and were monitored for 72 hours afterward. Postischemic renal function in nu/nu mice and wild-type control mice is shown in Figure 2. SCr was significantly reduced in the nu/nu mice compared with wild-type control mice with IRI, both 24 hours (n = 8, 1.73 ± 0.19 vs. 2.86 ± 0.13, P < 0.05) and 48 hours (n = 8, 1.67 ± 0.15 vs. 2.7 ± 0.09, P < 0.05) after ischemia. These data demonstrate that T cell–deficient nu/nu mice had less renal dysfunction following IRI than did wild-type control mice.
Figure 2.

T cell–deficient mice are functionally protected from postischemic renal injury. SCr was significantly reduced at 24 and 48 hours (*P < 0.05) after IRI in nu/nu mice (circles) compared with SCr in wild-type control mice with IRI (squares). The injury phenotype was restored when nu/nu mice were adoptively transferred with wild-type T cells (triangles). Reconstituted nu/nu mice show a significantly higher SCr at 24 and 48 hours after ischemia (#P < 0.05) compared with nu/nu mice. In all experiments, SCr (mg/dl) was measured from tail blood samples obtained from animals at 0 (preischemia), 24, 48, and 72 hours after moderate ischemia (30 minutes of bilateral clamping). T cell–transferred mice received 15 × 106 purified T cells (obtained from wild-type control mice) intraperitoneally 3 weeks prior to ischemic injury.
nu/nu mice are genetically mutant mice, and thus may differ from wild-type control mice in ways other than lack of T cells. To confirm that reduced ischemic injury in nu/nu mice was due to the deficiency in T cells, we investigated whether reconstituting nu/nu mice with T cells from wild-type mice would return the renal IRI phenotype. An adoptive transfer technique was used to reconstitute the nu/nu mice with wild-type T cells. Postischemic renal function in the T cell–reconstituted nu/nu mice is also shown in Figure 2. A return of the renal IRI phenotype is evident: SCr was significantly elevated when compared with the nu/nu mice at 24 hours (n = 8, 1.73 ± 0.19 vs. 2.59 ± 0.24, P < 0.05) and 48 hours (n = 8, 1.67 ± 0.15 vs. 2.27 ± 0.25, P < 0.05) after ischemia. Moreover, SCr in T cell–reconstituted nu/nu mice with IRI was comparable to that in wild-type control mice with renal IRI.
To investigate the extent of tubular necrosis in nu/nu mice, we histologically examined kidney tissue at 24 and 72 hours after ischemia. nu/nu mice at 24 hours after ischemia showed minimal tubular injury (data not shown). We therefore compared 72-hour postischemic tissue. Representative photomicrographs are shown in Figure 3. Normal kidney (no IRI) is represented in Figure 3a. Kidneys from wild-type mice at 72 hours after ischemia (Figure 3b) exhibited significant tubular injury, characterized by extensive tubular epithelial necrosis and sloughing of epithelial cells into the tubular lumen. Many of the tubules were dilated. Tubules also contained proteinaceous casts. Kidneys from nu/nu mice (Figure 3c) had less tubular injury than did kidneys from wild-type control mice (Figure 3b) at 72 hours. In nu/nu mice, a few tubules showed dilation with proteinaceous material in the lumen. nu/nu mice that were reconstituted with T cells (Figure 3d) had more severe tubular injury at 72 hours, similar to that seen in wild-type mice, with many tubules exhibiting dilation and congestion with proteinaceous material, as well as a loss of epithelial cell structure.
Figure 3.

Histological assessment of tubular injury at 72 hours after ischemia in T cell–deficient and T cell–reconstituted mice. (a) Normal wild-type mouse kidney that has not undergone IRI. (b) Wild-type kidney 72 hours after ischemia demonstrates extensive tubular damage. (c) nu/nu postischemic kidney shows a significant reduction in renal structural injury. (d) In contrast, T cell–reconstituted nu/nu mice show a return of postischemic renal injury with structural damage similar to that seen in the wild-type kidney. ×50.
Leukocyte infiltration following ischemia injury in T cell–deficient mice and reconstituted T cell–deficient mice.
Many studies have proposed that neutrophils play an important role in the postischemic stage of IRI, while others have argued otherwise (5). We quantified the infiltrating neutrophils with a specific antibody at 24 and 72 hours after ischemia in wild-type control, nu/nu, and T cell–reconstituted nu/nu mice; the results are represented in Figure 4. Our results show significant neutrophil infiltration in both wild-type, nu/nu, and T cell–reconstituted nu/nu mice. Thus in the current study, it does not appear that neutrophil infiltration into the kidney is correlated with T cell–mediated renal injury. We also analyzed the infiltration of macrophages into the postischemic kidney. Macrophage infiltration, prominent after ischemia, was also not significantly different between the T cell–deficient nu/nu mice and wild-type controls (Figure 5).
Figure 4.

Neutrophil infiltration into the postischemic kidney at 24 and 72 hours after ischemia in T cell–deficient and T cell–reconstituted mice compared with wild-type control mice. At 24 and 72 hours after ischemia there was no significant difference in the number of infiltrating neutrophils between wild-type mice (white bars), nu/nu mice (black bars), and T cell–reconstituted nu/nu mice (gray bars). n = 6 per group. HPF, high-powered fields.
Figure 5.

Macrophages infiltrating into the postischemic kidney at 72 hours after ischemia in T cell–deficient and T cell–reconstituted mice compared with wild-type control mice. At 72 hours after ischemia there was no significant difference in the number of infiltrating macrophages between wild-type mice (white bars), nu/nu mice (black bars), and T cell–reconstituted nu/nu mice (gray bars). n = 6 per group.
CD4-deficient mice are functionally and structurally protected from IRI.
After demonstrating a direct role for T cells in our model of renal IRI, we focused on identifying the important T cell subset involved in the pathogenesis of renal IRI. Mice deficient in either CD4+ T cells alone or CD8+ T cells alone were subjected to renal IRI, and postischemic structure and function were analyzed. SCr values at 24, 48, and 72 hours after ischemia are shown in Figure 6. CD4-deficient mice had significantly decreased SCr compared with wild-type control mice at each timepoint after ischemia (n = 8, P < 0.05). CD4-deficient mice also had significantly decreased SCr compared with CD8-deficient mice at 24 hours (n = 8, 2.67 ± 0.12 vs. 3.48 ± 0.14, P < 0.05) and 48 hours (2.3 ± 0.1 vs. 2.89 ± 0.19, P < 0.05) after ischemia. In contrast, CD8-deficient mice did not show significant improvement in renal function after ischemia compared with wild-type control mice. CD4-deficient mice were also significantly protected from mortality following renal IRI. CD4-deficient mice showed 88% survival compared with 20% in wild-type controls at 72 hours after ischemia (P < 0.05) (data not shown). Taken together, these results demonstrate that it is the CD4+ T cell that is an important T cell subset involved in the pathogenesis of renal injury following IRI.
Figure 6.

CD4-deficient mice are functionally protected from renal injury compared with CD8-deficient mice. CD4-deficient mice (diamonds) show significantly reduced SCr at all timepoints compared with wild-type mice (squares) (*P < 0.05) and with CD8-deficient mice (triangles) (#P < 0.05) at 24 and 48 hours after renal IRI.
To verify that the lack of CD4+ T cells was responsible for protection from renal IRI, we adoptively transferred CD4+ T cells from wild-type control littermates into CD4-deficient mice. SCr values for these mice are shown in Figure 7. CD4-deficient mice were significantly protected compared with wild-type mice at each timepoint (n = 8, P < 0.05). The transfer of CD4+ T cells into CD4-deficient mice restored the IRI phenotype, with a significant increase in SCr at 72 hours after ischemia compared with that in CD4-deficient mice (n = 8, 1.06 ± 0.07 vs. 1.95 ± 0.25, P < 0.05).
Figure 7.

Adoptive transfer of CD4+ T cells into CD4-deficient mice restores injury phenotype. CD4-deficient mice that have been reconstituted with wild-type CD4+ T cells (triangles, dashed line) have a return of the injury phenotype, with SCr values similar to wild-type control mice (squares). A significant increase in SCr in CD4-reconstituted mice was seen at 72 hours after ischemia compared with CD4-deficient mice (diamonds) (#P < 0.05). CD4-reconstituted mice received 5–10 × 106 purified T cells (obtained from wild-type control mice) intraperitoneally 3 weeks prior to ischemic injury.
Histological assessment of tubular necrosis demonstrated that the CD4-deficient mice had reduced renal structural injury. Figure 8a shows a wild-type (no IRI) kidney. At 24 hours after ischemia there was already distinct protection from structural injury seen in CD4-deficient mouse kidneys (Figure 8c) compared with wild-type control mouse kidneys (Figure 8b). In contrast, the CD8-deficient mouse kidneys at 24 hours after ischemia (Figure 8d) showed injury similar to that in the wild-type control mice.
Figure 8.

Histological assessment of tubular injury at 24 hours after ischemia in CD4-deficient and CD8-deficient mice. (a) Normal wild-type mouse kidney that has not undergone IRI. (b) At 24 hours after ischemia, there is severe tubular damage in the wild-type control kidney. However, the CD4-deficient mouse kidney shows a significant reduction in tubular injury (c) compared with the wild-type control. In contrast, the CD8-deficient mouse kidney (d) shows tubular injury similar to that seen in the wild-type control kidney. ×50.
Leukocyte infiltration into the postischemic kidney of CD4-deficient and wild-type mice.
Neutrophil and macrophage infiltration into postischemic kidneys was quantified at 24 and 72 hours after ischemia in CD4-deficient and wild-type control mice (Figures 9 and 10). These results were similar to those obtained for the T cell–deficient nu/nu mouse experiments, demonstrating no significant difference in the amount of infiltrating neutrophils and macrophages between these groups.
Figure 9.

Neutrophils infiltrating into the postischemic kidney at 24 and 72 hours after ischemia in CD4-deficient and wild-type control mice. At 24 and 72 hours after ischemia, there is no significant difference seen in the number of infiltrating neutrophils between wild-type mice (open bars) and CD4-deficient mice (filled bars). n = 6 per group.
Figure 10.

Macrophages infiltrating into the postischemic kidney at 24 and 72 hours after ischemia in CD4-deficient and wild-type control mice. At 24 and 72 hours after ischemia, there was no significant difference seen in the amount of infiltrating macrophages between wild-type mice (open bars) and CD4-deficient mice (filled bars). n = 6 per group.
CD4+ T cell infiltration into the postischemic kidney.
Because our results demonstrated the importance of the CD4+ T cell in influencing the course of renal IRI, we stained postischemic kidney tissue to evaluate a potential infiltration of CD4+ T cells. CD4+ T cell staining is abundant in spleens (Figure 11a), and is represented by a brown stained ring around positive cells. Figure 11b shows a normal (no IRI) wild-type kidney showing no positive staining for CD4+ T cells. Neither the CD4-deficient nor nu/nu mice showed any positive staining for CD4+ T cells in postischemic kidney. However, the wild-type kidney 24 hours after ischemia (Figure 11c) did show a small number of CD4+ T cells in the outer medulla.
Figure 11.
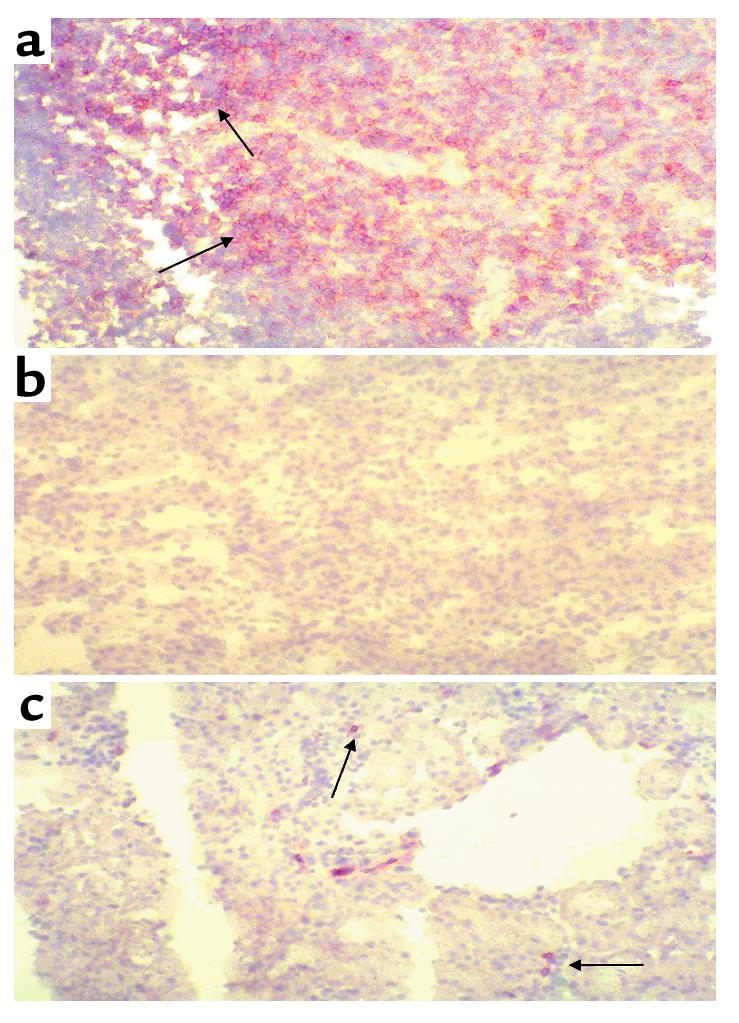
CD4+ T cells infiltrating into the postischemic kidney. (a) Positively stained wild-type spleen showing the presence of CD4+ T cells. This is represented by brown rings surrounding positive cells. (b) Wild-type (no IRI) kidney stained with an antibody for CD4. (c) A 24-hour postischemic kidney obtained from a wild-type control mouse, showing a small infiltration of CD4+ T cells. ×50.
The role of CD28 and IFN-γ in T cell–mediated renal IRI.
To begin to explore the mechanism of how CD4+ T cells mediate renal IRI, we studied nu/nu mice that had been adoptively transferred with an enriched CD4+ T cell population obtained from mice that were deficient in either CD28 (accessory molecule mediated pathway) or IFN-γ (to distinguish between a possible Th1 versus a Th2 response). Figure 12 demonstrates the successful adoptive transfer of nu/nu mice with CD4+ T cells from CD28-deficient mice (Figure 12b) and IFN-γ–deficient mice (Figure 12c). Numbers of CD4+ cells and CD8+ cells in wild-type mice are shown in Figure 12a. Postischemic renal function in nu/nu mice adoptively transferred with CD4+ T cells from CD28- and IFN-γ–deficient mice is represented in Figure 13. Wild-type mice show an increase in SCr, as previously demonstrated. Also as previously found, nu/nu mice had a significant reduction in SCr compared with wild-type mice. Adoptive transfer of nu/nu mice with CD4+ T cells from either CD28-deficient mice or IFN-γ–deficient mice caused no increase in SCr compared with nu/nu mice. Thus, adoptive transfer of T cells from either CD28-deficient or IFN-γ–deficient mice did not produce a return of the injury phenotype.
Figure 12.

Flow cytometry confirmation of adoptive transfer of CD28-deficient and IFN-γ–deficient CD4+ T cells into T cell–deficient mice. (a) Wild-type spleen showing normal numbers of cells stained positively for CD4 (15.8%) and CD8 (13.1%). (b) nu/nu mice were reconstituted with CD4+ T cells (5.5% positive stained) from mice deficient in CD28. (c) nu/nu mice were reconstituted with CD4+ T cells (4% positive stained) from mice deficient in IFN-γ.
Figure 13.

Adoptive transfer with CD4+ T cells from mice deficient in either CD28 or IFN-γ does not return the injury phenotype. nu/nu mice (diamonds) show significantly reduced SCr at all timepoints when compared with wild-type mice (circles). nu/nu mice reconstituted with CD4+ T cells from mice deficient in either CD28 (squares) or IFN-γ (triangles) also show significantly reduced SCr at all timepoints compared with wild-type control mice.
Discussion
The current study provides direct evidence that T cells are mediators of renal IRI in mice, and that the CD4+ T cell is the major T cell subset responsible for renal injury following ischemia. Furthermore, the pathophysiologic role of the CD4+ T cell is dependent on CD28 and IFN-γ. These results, though novel in the kidney, are consistent with emerging studies in other organ systems. CD4+ T cells have been shown to mediate murine liver ischemic injury in studies of T cell–deficient mice (10). A recent study in a model of gut ischemia reperfusion showed that reconstitution of T cell–deficient mice with T cell–enriched splenocytes allowed mice to respond to gut ischemia reperfusion in a manner similar to that seen in wild-type mice (11). Using techniques similar to those used in our study, a recent study in a liver model of cold ischemia reperfusion showed that tissue injury was reduced by 51% in nu/nu mice, and that injury was restored by T cell transfer into nu/nu mice (12).
A potential limitation in the use of mutant mice is that physiologic and structural changes observed in IRI could be due to abnormalities independent of the one being studied. To confirm that T cell deficiency was responsible for reduced renal IRI, we used an adoptive transfer technique to reconstitute both the T cell–deficient mice and the CD4-deficient mice with their respective T cell populations. An important and intriguing finding of the present study was that a return of the injury phenotype in T cell–deficient mice was accomplished with a small degree of T cell reconstitution. Indeed, a splenic reconstitution of T cells of just 2–6% was sufficient to restore renal dysfunction and damage. All T cell–transferred populations were verified by FACS analysis both prior to cell transfer and at the conclusion of the experiment to ensure a minimal effect of contaminating cells. We cannot completely exclude the possibility that contaminating cells may have been involved in the restoration of the injury phenotype; however, the inability of CD4+ T cell transfers from CD28- and IFN-γ–deficient mice to restore the injury phenotype make it unlikely that contaminating cells are playing a significant role. While our results indicate that CD4+ T cells are important to the pathogenesis of ischemic ARF, they do not exclude the possible contribution of other T cells (e.g., CD8+ cells) to the development of IRI. The present results, however, indicate that these cells are probably not the main mediators of early IRI.
The mechanism by which CD4+ T cells might induce postischemic renal injury is not known; two potential pathways were studied. CD4+ T cells from CD28-deficient mice were adoptively transferred into nu/nu mice to determine whether accessory molecule expression was important in T cell–mediated renal IRI. Our results suggest that CD28 is important in the pathogenesis of renal IRI: our experiments showed that adoptive transfer of nu/nu mice with CD4+ T cells deficient in CD28 was unable to restore injury. A potential role for CD28 was hypothesized by Takada et al., who demonstrated that use of CTLA4Ig, which can block CD28 binding to CD80, is protective in a rat model of cold renal IRI (13).
We also examined the effects of reconstituting nu/nu mice with CD4+ T cells from IFN-γ–deficient mice. This experiment was designed to determine whether CD4+ T cell–mediated renal IRI was associated primarily with a Th1 or a Th2 response. We found that IFN-γ–deficient CD4+ T cells did not restore injury in nu/nu mice. This suggests that the Th1 polarization of CD4+ cells might be more important in mediating renal IRI. It also suggests that IFN-γ may play an important role in the regulation of the early injury response to IRI.
T cell activation in renal IRI would not be conventionally expected. Classically, T cell activation is thought to require foreign antigens bound to self-MHC molecules, together with costimulatory signals by antigen-presenting cells. Nonetheless, T cell activation and the production of associated cytokines have been described in experimental renal IRI (5, 8, 14). The absence of foreign antigens in renal IRI leads us to speculate that alloantigen-independent T cell activation may be involved in renal IRI. In this regard, hypoxia itself might be sufficient for T cell activation. CD4+ T cells have been shown to increase adhesion to endothelial monolayers following anoxia/reoxygenation (15). Recently, it has also been suggested that increased adhesion of infiltrating T cells to renal tubular epithelial cells under hypoxic conditions may provide a potential mechanism underlying postischemic tubular dysfunction (9). The danger signals activated in renal IRI could thus activate the CD4+ T cell and lead it to function in more of an innate manner, rather than in its classical adaptive immunologic role. (16, 17).
Localization of T cells in the kidney would help elucidate the mechanism of T cell–induced renal dysfunction. However, immunohistochemistry with specific antibodies to CD4+ T cells demonstrated a paucity of T cell infiltration into the postischemic kidney. It may be possible that only a small number of infiltrating T cells, relative to the amount of neutrophil or macrophage infiltration, is sufficient to exert an effect. Alternatively, the T cells could be orchestrating an effect on renal injury from a distant site, possibly via soluble cytokines.
In view of previous work suggesting an important role of neutrophils in renal IRI (18, 19), we investigated in this study the effect of T cell manipulation on neutrophil infiltration. We have previously used methods such as Leder staining and myeloperoxidase assays to identify neutrophils (20, 21). A recent study, however, has shown that these techniques also detect macrophages, and are nonspecific (22). Therefore, in the current study, we used a specific monoclonal antibody to neutrophils to detect their infiltration into postischemic kidney tissue. Brisk neutrophil infiltration to the corticomedullary junction after ischemia was observed in both wild-type mice with full expression of renal injury, and in T cell–manipulated mice that were protected. Similarly, we found significant macrophage infiltration after ischemia, with little difference between wild-type and T cell–deficient groups. Therefore we cannot clearly identify regulation of neutrophil or macrophage infiltration as the mechanism by which T cells participate in this model. This finding, however, does not exclude a possible effect of neutrophils or macrophages, independent of mere tissue infiltration, in renal IRI. In a previous study that evaluated leukocyte migration in experimental asthma, we found that leukocyte blockade can protect tissue structure and function despite not attenuating leukocyte migration and infiltration (23, 24).
This study demonstrates the important role for CD4+ T cells in renal IRI, using redundant approaches including the use of spontaneous T cell–deficient mice, targeted T cell–deficient mice, mice with targeted T cell dysfunction, and adoptive transfer techniques. Although many underlying mechanisms still need to be elucidated, our current data support studies in humans targeting CD4+ T cells in renal IRI, particularly in the transplant patient.
Acknowledgments
This study was supported by a National Kidney Foundation Postdoctoral Research Fellowship grant (M.J. Burne), a National Kidney Foundation Clinical Scientist award (H. Rabb), NIH grant RO1 DK-54770 (H. Rabb), and by the Hermundslie Trust. Publication costs were supported by an unrestricted educational grant from Sangstat Medical Corporation, Fremont, California, USA. We thank Alexander Khoruts for critical review of the manuscript.
References
- 1.Molitoris BA. Ischemic acute renal failure: exciting times at our fingertips. Curr Opin Nephrol Hypertens. 1998;7:405–406. doi: 10.1097/00041552-199807000-00009. [DOI] [PubMed] [Google Scholar]
- 2.Humes HD. Acute renal failure: prevailing challenges and prospects for the future. Kidney Int. 1995;50(Suppl.):S26–S32. [PubMed] [Google Scholar]
- 3.Star RA. Treatment of acute renal failure. Kidney Int. 1998;54:1817–1831. doi: 10.1046/j.1523-1755.1998.00210.x. [DOI] [PubMed] [Google Scholar]
- 4.Terasaki PI, Cecka JM, Gjertson DW, Takemoto S. High survival rates of kidney transplants from spousal and living unrelated donors. N Engl J Med. 1995;333:333–336. doi: 10.1056/NEJM199508103330601. [DOI] [PubMed] [Google Scholar]
- 5.Rabb H, O’Meara YM, Maderna P, Coleman P, Brady HR. Leukocytes, cell adhesion molecules and ischemic acute renal failure. Kidney Int. 1997;51:1463–1468. doi: 10.1038/ki.1997.200. [DOI] [PubMed] [Google Scholar]
- 6.Solez K, Morel-Maroger L, Sraer JD. The morphology of “acute tubular necrosis” in man: analysis of 57 renal biopsies and a comparison with the glycerol model. Medicine. 1979;58:362–376. [PubMed] [Google Scholar]
- 7.Olsen, S., and Solez, K. 1979. Acute tubular necrosis and toxic renal injury. In Renal pathology. C.C. Tisher and B.M. Brenner, editors. J.B. Lippincott Co. Philadelphia, Pennsylvania, USA. 769–809.
- 8.Takada M, Nadeau KC, Shaw GD, Marquette KA, Tilney NL. The cytokine-adhesion molecule cascade in ischemia/reperfusion injury of the rat kidney. Inhibition by a soluble P-selectin ligand. J Clin Invest. 1997;99:2682–2690. doi: 10.1172/JCI119457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rabb H, et al. Pathophysiological role of T lymphocytes in renal ischemia reperfusion injury in mice. Am J Physiol. 2000;279:F525–F531. doi: 10.1152/ajprenal.2000.279.3.F525. [DOI] [PubMed] [Google Scholar]
- 10.Zwacka RM, et al. CD4(+) T-lymphocytes mediate ischemia/reperfusion-induced inflammatory responses in mouse liver. J Clin Invest. 1997;100:279–289. doi: 10.1172/JCI119533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Horie Y, Wolf R, Chervenak RP, Jennings SR, Granger DN. T-lymphocytes contribute to hepatic leukostasis and hypoxic stress induced by gut ischemia-reperfusion. Microcirculation. 1999;6:267–280. [PubMed] [Google Scholar]
- 12.Le Moine O, et al. Cold liver ischemia-reperfusion injury critically depends on liver T cells and is improved by donor pretreatment with interleukin 10 in mice. Hepatology. 2000;31:1266–1274. doi: 10.1053/jhep.2000.7881. [DOI] [PubMed] [Google Scholar]
- 13.Takada M, Chandraker A, Nadeau KC, Sayegh MH, Tilney NL. The role of the B7 co-stimulatory pathway in experimental cold ischemia/reperfusion injury. J Clin Invest. 1997;100:1199–1203. doi: 10.1172/JCI119632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goes N, Urmson J, Ramassar V, Halloran PF. Ischemic acute tubular necrosis induces an extensive local cytokine response. Evidence for induction of interferon-gamma, transforming growth factor-beta 1, granulocyte-macrophage colony-stimulating factor, interleukin-2, and interleukin-10. Transplantation. 1995;59:565–572. [PubMed] [Google Scholar]
- 15.Kokura S, et al. Endothelial cells exposed to anoxia/reoxygenation are hyperadhesive to T-lymphocytes: kinetics and molecular mechanisms. Microcirculation. 2000;7:13–23. [PubMed] [Google Scholar]
- 16.Matzinger P. An innate sense of danger. Semin Immunol. 1998;10:399–415. doi: 10.1006/smim.1998.0143. [DOI] [PubMed] [Google Scholar]
- 17.Lu CY, Penfield JG, Kielar ML, Vazquez MA, Jeyarajah DR. Hypothesis: is renal allograft rejection initiated by the response to injury sustained during the transplant process? Kidney Int. 1999;55:2157–2168. doi: 10.1046/j.1523-1755.1999.00491.x. [DOI] [PubMed] [Google Scholar]
- 18.Kelly KJ, et al. Intracellular adhesion molecule-1 deficient mice are protected against ischemic renal injury. J Clin Invest. 1996;97:1056–1063. doi: 10.1172/JCI118498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Okusa MD, et al. A(2A) adenosine receptor-mediated inhibition of renal injury and neutrophil adhesion. Am J Physiol Renal Physiol. 2000;279:F809–F818. doi: 10.1152/ajprenal.2000.279.5.F809. [DOI] [PubMed] [Google Scholar]
- 20.Rabb H, et al. Role of CD11a and CD11b in ischemic acute renal failure in rats. Am J Physiol. 1994;267:F1052–F1058. doi: 10.1152/ajprenal.1994.267.6.F1052. [DOI] [PubMed] [Google Scholar]
- 21.Rabb H, et al. Renal ischemia reperfusion injury in L-selectin mice. Am J Physiol. 1996;271:F408–F413. doi: 10.1152/ajprenal.1996.271.2.F408. [DOI] [PubMed] [Google Scholar]
- 22.Ysebaert DK, et al. Identification and kinetics of leukocytes after severe ischemia/reperfusion renal injury. Nephrol Dial Transplant. 2000;15:1562–1574. doi: 10.1093/ndt/15.10.1562. [DOI] [PubMed] [Google Scholar]
- 23.Rabb H, Oliverstein R, Issekutz TB, Renzi PM, Martin JG. The role of the leukocyte adhesion molecules VLA-4, LFA-1, and Mac-1 in allergic airway responses in the rat. Am J Respir Crit Care Med. 1994;5:1186–1191. doi: 10.1164/ajrccm.149.5.8173758. [DOI] [PubMed] [Google Scholar]
- 24.Rabb H, Martin JG. An emerging paradigm shift on the role of leukocyte adhesion molecules. J Clin Invest. 1997;100:2937–2938. doi: 10.1172/JCI119844. [DOI] [PMC free article] [PubMed] [Google Scholar]