

Abstract
Furan-containing congeners of the histamine H2 receptor antagonist ranitidine were synthesized and tested for improgan-like antinociceptive activity. The most potent ligand of the series, VUF5498, is the most potent improgan-like agent described to date (ED50= 25 nmol, icv). This compound is approximately equal in potency with morphine. These non-imidazole, improgan-like pain relievers further define the structural requirements for analgesics of this class and are important tools for ongoing mechanism-based studies.
Keywords: ranitidine, improgan, pain, analgesia, H2 receptor
Cimetidine, burimamide, and ranitidine (Fig. 1) are histamine H2 receptor antagonists which do not significantly penetrate the blood brain barrier under physiological conditions1,2. When injected directly into the brain, however, these compounds are highly effective pain relievers3,4. Studies of cimetidine congeners led to the discovery of improgan (Fig. 1), an analgesic which lacks H2 receptor (H2R) antagonist properties and does not act on any of the four known histamine receptors3,5,6. Improgan acts in the brain stem to stimulate descending, non-opioid circuits7, and cannabinoid mechanisms may be involved8, but the molecular target(s) for improgan remain unknown5.
Figure 1.
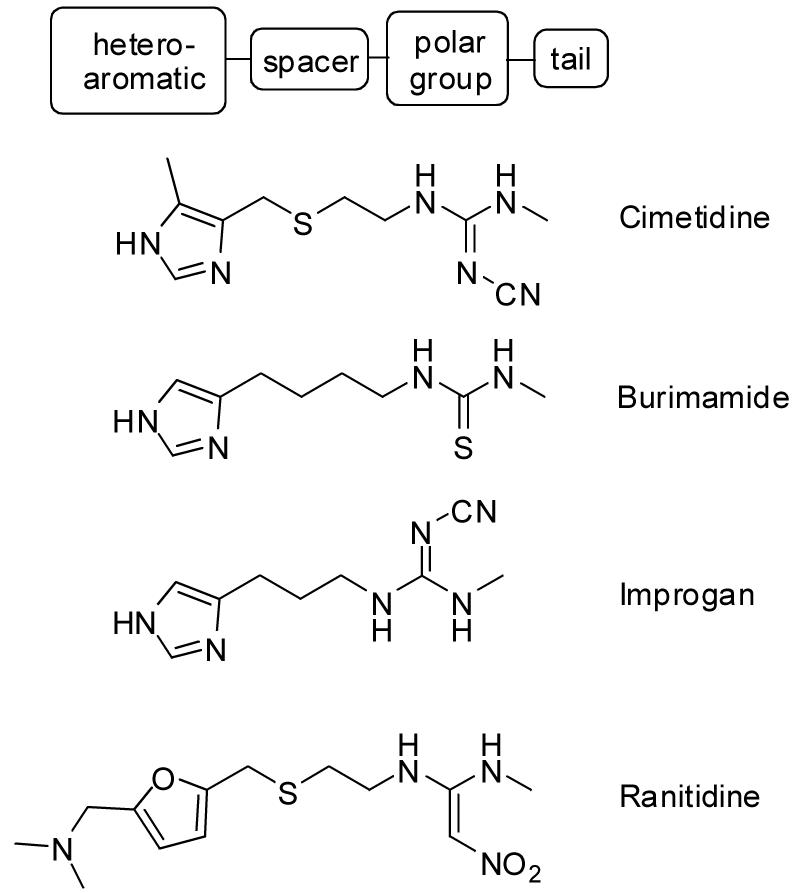
Structures of improgan and related congeners which have antinociceptive properties. The general schematic formula for improgan-like compounds is given at the top.
Even in the absence of a specific in vitro target for improgan, structure-activity relationship (SAR) studies can help to find the relevant analgesic mechanisms and lead to development of more potent derivatives of this class of analgesics. Among imidazole-containing compounds, it has been described that the analgesic potency differs from the H2R, H3R and H4R activity profile3,9. For example, burimamide (Fig. 1), an imidazole-containing histaminergic ligand with weak H2R and potent H3R and H4R activity10, is a potent analgesic, although other imidazole-containing H3R and H4R ligands (e.g. thioperamide) are not3,11. Studies of burimamide congeners in which the spacer length (Fig. 1) was varied confirmed activity in compounds containing spacers of two-, three, four- and six- methylene units3. Recently, systematic variation in the length of improgan's spacer led to new congeners with increased antinociceptive potency12.
The H2R antagonist ranitidine (Fig. 1) also produces antinociception when given into the brain3. However, no antinociceptive structure-activity relationships of furan-containing compounds related to improgan or ranitidine have been previously described. Because the imidazole moiety can hamper clinical development of a compound13 and may also limit brain penetration14, the present studies describe the synthesis and antinociceptive activity of a series of furan-containing, improgan-like compounds.
Ligands containing a methyl ethyl thioether spacer (as in ranitidine, VUF8294, VUF5401, Table 1) were obtained as previously described15. Compounds containing simple alkyl spacers were synthesized as illustrated in Scheme 1. Furan (1) was reacted with nbutyllithium and 1,ω -dibromoalkanes to give intermediates 2 that were converted to the phthalimido-derivatives 3. A Mannich reaction via a modification of the method described by Price16, using paraformaldehyde and dimethylammonium chloride in ethanol, gave compounds 4 in moderate yield. Deprotection of the phthalimido moiety under basic conditions gave the 2-(ω-aminoalkyl)-5-(dimethylaminomethyl)furanes 5. Subsequent treatment of these key intermediates 5 with different electrophiles gave the desired compounds in moderate to good yields. Thus, nitroethenylguanidines 6 were prepared by addition of 1,1 bis-(methylthio)-2-nitroethene and methylamine. Cyanoguanidines 7 were prepared by addition of dimethyl cyanocarbonodithioimidate and subsequent treatment with methylamine. Ureas 8 were made by reaction with the corresponding isocyanate. Amide coupling by reaction of key intermediates 5 with the corresponding acid chlorides gave amides 9 in only moderate (isolated) yields. Reductive amination using intermediates 5 with the corresponding aldehydes and sodium borohydride gave secondary amines 10 in excellent yields. VUF 5500 (Table 1) was synthesized as described for 2, using furan and 1-bromo-4-phenylbutane. The product was isolated as an oxalate salt in low yield (17%).
Table 1.
Chemical structures and antinociceptive activity of congeners of improgan and ranitidine. Fourteen new furan derivatives (in shaded cells) were synthesized and tested presently. Two compounds (in bold) showed very high antinociceptive potency. Compounds are arranged in order of decreasing potency. Derivatives at the bottom of the table (below the solid line) were either inactive or had low potency.
| Ar-Y-P-Z | |||||
|---|---|---|---|---|---|
| Compound | Ar | Y | P | Z | ED50a |
| VUF 5498 | 2-DMAF | -(CH2)6- | NH-C(=CH-NO2)-NH- | methyl | 25 (15-36) |
| VUF 8294 | 2-DMAF | -CH2-S-(CH2)2- | NH-C(=N-CN)-NH- | methyl | 67 (18-117)b |
| VUF 5407 | 4(5)-IM | -(CH2)4- | NH-C(=O)-NH- | phenyl | 71 (49-93)c |
| VUF 4687 | 4(5)-IM | -(CH2)4- | NH-C(=S)-NH- | phenylethyl | 80 ± 9d |
| VUF 4685 | 4(5)-IM | -(CH2)4- | NH-C(=S)-NH- | phenyl | 81 ± 7d |
| VUF 5420 | 4(5)-IM | -(CH2)4- | NH-C(=N-CN)-NH- | methyl | 82 (74-90)e |
| VUF 6914 | 4(5)-IM | -(CH2)8- | NH-C(=N-CN)-NH- | methyl | 82 (70-94)e |
| VUF 4740 | 4(5)-IM | -(CH2)6- | NH-C(=S)-NH- | methyl | 87 ± 15d |
| VUF 4582 | 4(5)-IM | -(CH2)2- | NH-C(=S)-NH- | phenyl | 89 (64-114)c |
| VUF 5405 | 4(5)-IM | -(CH2)4- | NH-C(=CH-NO2)-NH- | methyl | 104 (78-130)c |
| VUF 4686 | 4(5)-IM | -(CH2)4- | NH-C(=S)-NH- | benzyl | 105 (est)d |
| VUF 5651 | 4-Me-5-IM | -(CH2)4- | NH-C(=N-CN)-NH- | methyl | 105 (92-119)e |
| VUF 6913 | 4(5)-IM | -(CH2)5- | NH-C(=N-CN)-NH- | methyl | 106 (90-121)e |
| CC10 | 4(5)-IM | Trans-cyclopropyl | NH-C(=N-CN)-NH- | methyl | 106 (87-125)e |
| Ranitidine | 2-DMAF | -CH2-S-(CH2)2- | NH-C(=CH-NO2)-NH- | methyl | 109 ± 16d |
| VUF 5497 | 2-DMAF | -(CH2)4- | NH-C(=CH-NO2)-NH- | methyl | 111 (54-168) |
| VUF 4577 | 4(5)-IM | -(CH2)2- | NH-C(=S)-NH- | methyl | 117 (90-144)c |
| VUF 5401 | 2-DMAF | -CH2-S-(CH2)2- | NH-C(=S)-NH- | methyl | 117 (41-194)b |
| VUF 5550 | 2-DMAF | -(CH2)6- | NH-C(=N-CN)-NH- | methyl | 120 (59-181) |
| VUF 5499 | 2-DMAF | -(CH2)4- | NH-C(=O)- | methyl | 126 (79-174) |
| VUF 5509 | 2-DMAF | -(CH2)4- | NH- | phenyl | 131 (31-232)h |
| VUF 5733 | 4(5)-IM | -(CH2)2- | NH-C(=N-CN)-NH- | methyl | 137 (121-153)e |
| VUF 5500 | 2-DMAF | -(CH2)3- | CH2-- | phenyl | 143 (103-255) |
| VUF 5496 | 2-DMAF | -(CH2)4- | NH-C(=N-CN)-NH- | methyl | 159 (36-283)h |
| Burimamide | 4(5)-IM | -(CH2)4- | NH-C(=S)-NH- | methyl | 184 ± 16d |
| VUF 5261 | 4(5)-IM | 1,4-piperidinyl | -C(=S)-NH- | methyl | 201 ± 8d |
| VUF 5520 | 2-DMAF | -(CH2)4- | NH-C(=O)- | phenyl | 203 (154-252) |
| Norburimamide | 4(5)-IM | -(CH2)3- | NH-C(=S)-NH- | methyl | 217 ± 29d |
| Improgan | 4(5)-IM | -(CH2)3- | NH-C(=N-CN)-NH- | methyl | 276 (193-359)f |
| VUF 5554 | 2-DMAF | -(CH2)6- | NH-C(=O)-NH- | phenyl | 284 (243-324) |
| VUF 4684 | 4(5)-IM | -(CH2)4- | NH-C(=S)-NH- | cyclohexyl | 285 ± 21d |
| Metiamide | 4-Me-5-IM | -CH2-S-(CH2)2- | NH-C(=S)-NH- | methyl | 370 ± 39d |
| Cimetidine | 4-Me-5-IM | -CH2-S-(CH2)2- | NH-C(=N-CN)-NH- | methyl | 464 ± 89d |
| VUF 5495 | 2-DMAF | -(CH2)4- | -NH2 | - | 689 (316-1,062)h |
| VUF 5547 | 2-DMAF | -(CH2)6- | NH-C(=O)-NH- | 4-iodophenyl | > 149 |
| VUF 5548 | 2-DMAF | -(CH2)4- | NH-C(=O)-NH- | 4-iodophenyl | > 226 |
| Thioperamide | 4(5)-IM | 1,4-piperidinyl | -C(=S)-NH- | cyclohexyl | > 342d |
| VUF 8299 | Phenyl | -CH2-S-(CH2)2- | NH-C(=N-CN)-NH- | methyl | > 403d |
| VUF 5394 | 1-IM | -(CH2)3- | NH-C(=S)-NH- | methyl | > 504c |
| VUF 8298 | 2-Pyridinyl | -CH2-S-(CH2)2- | NH-C(=N-CN)-NH- | methyl | > 602d |
| VUF 4741 | 4(5)-IM | -(CH2)6- | NH-C(=S)-NH- | phenyl | partial agonistd |
| VUF 5262 | 4(5)-IM | 1,4-piperidinyl | -C(=S)-NH- | phenyl | partial agonistd |
| VUF 6912 | 4(5)-IM | -(CH2)6- | NH-C(=N-CN)-NH- | methyl | partial agoniste |
| VUF 5393 | 1-IM | -(CH2)3- | NH-C(=CH-NO2)-NH- | methyl | toxicc,g |
Hot plate ED50 values (nmol, 10 min after drug) were estimated by non-linear regression following intracerebroventricular injection. Data from the present study (shaded cells) were derived from at least three doses of each drug (n=6-12). Error estimates are specified as either 95% confidence intervals (in parentheses) or as ± SEM. Results from the tail flick test (not shown) were virtually identical with hot plate data.
Preliminary report of these results15.
Literature value11.
Literature value3.
Literature value12.
Toxicity observed below antinociceptive doses.
Dose-response curve had a steep slope, yielding large confidence intervals for these drugs. Structures for the Ar substituent abbreviations (left) and spacer Y abbreviations (right) are given below.
![]()
Scheme 1.

Reagents and conditions: (a) (i) BuLi, THF, 45°C. (ii) Br(CH2)4Br, THF, H2O, −30 °C → RT, 67% (b) KNPht, DMF, RT, 98% (c) HCHO, (CH3)2NH2Cl, EtOH, reflux, 16 h, 48%. (d) N2H4, NaOH, EtOH, reflux, 16h, 98%. (e) (i) 1,1-bis(methylthio)-2-nitroethylene, MeCN, Δ, 16h, 81%. (ii) MeNH2, EtOH, reflux, 16 h, 83% (f) (i) dimethyl cyanocarbonodithioimidate, EtOH, reflux, 16h, 76% (ii) MeNH2, EtOH, reflux, 16h, 88%. (g) RNCO, CH3CN, RT, 16h, 43%. (h) RCOCl, TEA, DCM, RT, 16h, 33%.(i) RCHO, MeOH, NaBH4, 5h, 95%.
Compounds in Table 1 were assessed for antinociceptive activity in male Sprague–Dawley rats (200-320g, Taconic Farms, Inc., Germantown, NY). Subjects were maintained on a reverse 12 L:12 D cycle (lights on 19:00, lights off 07:00). All experiments were reviewed and approved by the Albany Medical College Institutional Animal Care and Use Committee. Salts were dissolved in isotonic saline. Bases were dissolved in HCl (1.0-1.2 N), titrated to a pH between 5.5-6.5 and diluted with saline. Vehicle injections consisted of either saline, or neutralized, diluted HCl. Subjects were surgically fitted with a chronically-implanted guide cannula along with a stylet under general anesthesia exactly as previously described11. Coordinates (in mm from bregma17) for the guide cannulas were: AP −0.8, ML +1.5, DV −3.3, 0° angle. Injection cannulas extended 1 mm ventrally beyond the tip of the guides such that injections were made into the left lateral ventricle. Two nociceptive tests were used, the radiant heat tail flick test18 and the hot plate test19. For the tail flick test, the heat source was set to produce baseline latencies between 3 and 4 s, with a 15 s cutoff, exactly as recently described12. For the hot plate test, animals were placed on a 52 °C surface and the latency to a hind lift or lick was recorded, with a maximal exposure of 60 s. Baseline latencies were 8-14 sec. At least one week after surgery, animals were baseline tested, injected with drug (5 μl over 5 min), and then re-tested with single hot plate and tail flick tests at 5, 10 and 30 min after the replacement of the stylet exactly as described previously 11. Cannula placement was verified after each experiment. As described previously12, data were fitted by use of iterative nonlinear regression methods (Graphpad Prism, San Diego, CA) to the following equation:
where E is latency (sec), D is the dose of drug injected (nmol), BL is the baseline latency (sec), Top is the cutoff latency (sec), n is the slope function, and ED50 is the dose of drug inducing a 50% of maximum effect (nmol). Robust fits were obtained to estimate ED50 by constraint of the following variables as indicated: Top: 15 and 60 sec for tail flick and hot plate results, respectively; BL: 3.5 and 11.0 sec, respectively. All fits converged with statistically significant (P < 0.01) regression parameters, and ED50 values and 95% confidence intervals were obtained.
Fourteen newly-synthesized compounds which retain the heteroaromatic nucleus of ranitidine were tested for antinociceptive activity (shaded cells in Table 1). Except where indicated, all compounds produced maximal, dose-dependent, antinociceptive activity with no observable motor or behavioral side effects. Many of these compounds retained the antinociceptive properties of improgan. The drugs can be divided into three potency groups, high (ED50 <70 nmol, bold in Table 1), intermediate (ED50 = 111-284 nmol) and low (below line in Table 1).
All improgan-like analgesic drugs described to date consist of a hetero-aromatic nucleus, a spacer, a polar group and tail3,5 (Fig. 1). Previous studies have shown that congeners of improgan and cimetidine can retain antinociceptive activity with several kinds of variations in the spacer, the polar group, or the tail, as long as an imidazole moiety was retained as the hetero-aromatic nucleus (Table 1). However, replacement of the 4-methyl-5-imidazolyl group in cimetidine with either phenyl (VUF8299) or 2-pyridinyl (VUF8298) nuclei led to loss of activity.
Interestingly, ranitidine, the first non-imidazole H2 antagonist, was reported to produce antinociception after intracerebroventricular administration.3 Here we describe the first SAR studies of furan-containing antinociceptive compounds and compare the results with the SAR that was previously described for the imidazole-containing improgan analogues. Presently, many of the furan-containing compounds in Table 1 are more potent than improgan, which might suggest that replacement of imidazole by dimethylaminomethyl-furan increases antinociceptive potency. However, a more careful comparison of the activities of some of these compounds with previous results shows that this is not uniformly the case. For example, furan VUF5497 (ED50=111 nmol, Table 1) has the same potency as the imidazole-containing equivalent compound (VUF5405, ED50=104 nmol). The furan-containing VUF5550 showed full antinociceptive agonist activity (ED50=120 nmol), whereas the imidazole equivalent of this drug (VUF 6912) behaved as a partial agonist. Most notably, the furan-containing VUF5496 (ED50=159 nmol) has only one-half the potency of its imidazole equivalent VUF5420 (ED50=86 nmol). These findings illustrate subtle differences in the SARs of the two series of heteroaromatic compounds.
Because compounds combining an imidazole heterocycle with a urea polar group and an aromatic tail have been previously reported to have antinociceptive activity (VUF5407), furan-containing congeners with these characteristics were synthesized and tested presently. Table 1 shows that when 4 or 6 carbon spacers were combined with phenyl or iodophenyl tails, little or no activity was found (VUF5547, VUF5548, and VUF5554). Although a urea polar group does not seem allowed for activity in the furan series, a thiourea polar group is tolerated (compare ranitidine and VUF5401). The intermediate-to-low potency of congeners containing carbamate (VUF5520, VUF5499) and amine (VUF5509, VUF5495) polar groups was somewhat unexpected, and requires further study.
VUF5498 and VUF8294 are the two most potent improgan-like antinociceptive agents described to date. VUF5498, (ED50=25 nmol, Table 1) is four-fold more potent than ranitidine, and more than ten-fold more potent than improgan. When administered by the icv route, VUF5498 has the approximate analgesic potency of morphine12. Both the nitroethenylguanidine polar group and the extended alkyl spacer in VUF5498 seem to contribute to this high potency. Both the furan-containing VUF5497 and its imidazole equivalent (VUF5405) contain the nitroethenylguani-dine polar group and both are about three times as potent as improgan. Extension of the spacer from four carbons (VUF5497) to six carbons (VUF5498) seems to result in a further four-fold increase in potency. Consistent with this finding, we recently reported that extension of the spacer length in improgan from three to four carbons (i.e., VUF5420) more than doubles its potency12, although further lengthening of the spacer did not further increase potency (see VUF6913 and VUF6914). VUF8294 (ED50=67 nmol), the other high potency compound discovered presently, bears a strong resemblance to the structure of cimetidine, except that the latter's 4-methylimidazole ring has been replaced by the aminofuran nucleus. Cimetidine (ED50 =464 nmol, Table 1) has a very low antinociceptive potency, and the reasons for the high potency of VUF8294 are not clear.
In conclusion, the present results demonstrate the pain-relieving properties of several compounds possessing the dimethylaminomethylfuran nucleus of ranitidine. Antinociceptive activity was retained in furan-containing compounds possessing alkyl spacers, polar groups, and tails similar to those of previously found in improgan-like drugs. Replacement of the methyl ethyl thioether chain in ranitidine with an extended hexyl chain resulted in VUF5498, a drug ten times more potent than improgan, and the most potent compound in this series described to date. The mechanism of action of neither improgan nor VUF5498 is presently known, but similarities in the SARs suggest that they may act by similar means. The discovery of high potency, imidazole-free improgan-like analgesics further defines the improgan receptor pharmacophore, will facilitate the search for the relevant pain-relieving mechanism(s), and may lead to the development of clinically useful non-opioid analgesics
Acknowledgments
This work was supported by a grant (DA-015915) from the National Institute on Drug Abuse. We thank Dr. M. VanAlstine (Albany Medical College) and Prof. M. Wentland (Rensselaer Polytechnic Institute, Troy, NY) for valuable discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Ganellin CR, Ganellin CR, Parsons ME. Vol. 10. John Wright & Sons, Ltd.; Bristol: 1982. [Google Scholar]
- 2.Hough LB, Glick SD, Su KL. Pharmacol. Biochem. Behav. 1986;24:1257. doi: 10.1016/0091-3057(86)90181-4. [DOI] [PubMed] [Google Scholar]
- 3.Hough LB, Nalwalk JW, Li BY, Leurs R, Menge WMPB, Timmerman H, Cioffi C, Wentland M. J. Pharmacol. Exp. Ther. 1997;283:1534. [PubMed] [Google Scholar]
- 4.Netti C, Bossa R, Galatulas I, Sibilia V, Pecile A. Pharmacology. 1984;28:262. doi: 10.1159/000137972. [DOI] [PubMed] [Google Scholar]
- 5.Hough LB, Nalwalk JW, Barnes WG, Leurs R, Menge WM, Timmerman H, Watanabe T, Timmerman H, Yanai K. Histamine Research in the New Millenium. Vol. 23. Elsevier; Amsterdam: 2001. (Excerpta Medica International Congress Series 1224). [Google Scholar]
- 6.Li BY, Nalwalk JW, Barker LA, Cumming P, Parsons ME, Hough LB. J. Pharmacol. Exp. Ther. 1996;276:500. [PubMed] [Google Scholar]
- 7.Nalwalk JW, Svokos K, Taraschenko O, Leurs R, Timmerman H, Hough LB. Brain Res. 2004;1021:248. doi: 10.1016/j.brainres.2004.06.066. [DOI] [PubMed] [Google Scholar]
- 8.Nalwalk JW, Svokos K, Hough LB. Eur.J.Pharmacol. 2006;549:79. doi: 10.1016/j.ejphar.2006.08.035. [DOI] [PubMed] [Google Scholar]
- 9.Zhu Y, Michalovich D, Wu H-L, Tan KB, Dytko GM, Mannan IJ, Boyce R, Alston J, Tierney LA, Li X, Herrity NC, Vawter L, Sarau HM, Ames RS, Davenport CM, Hieble JP, Wilson S, Bergsma DJ, Fitzgerald LW. Mol. Pharmacol. 2001;59:434. doi: 10.1124/mol.59.3.434. [DOI] [PubMed] [Google Scholar]
- 10.Lim HD, van Rijn RM, Ling P, Bakker RA, Thurmond RL, Leurs R. J. Pharmacol. Exp. Ther. 2005;314:1310. doi: 10.1124/jpet.105.087965. [DOI] [PubMed] [Google Scholar]
- 11.Hough LB, Nalwalk JW, Leurs R, Menge WMPB, Timmerman H. Pharmacol. Biochem. Beha.v. 2000;65:61. doi: 10.1016/s0091-3057(99)00187-2. [DOI] [PubMed] [Google Scholar]
- 12.Hough LB, De Esch IJ, Janssen E, Phillips J, Svokos K, Kern B, Trachler J, Abood ME, Leurs R, Nalwalk JW. Neuropharmacol. 2006;51:447. doi: 10.1016/j.neuropharm.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 13.Esbenshade TA, Krueger KM, Miller TR, Kang CH, Denny LI, Witte DG, Yao BB, Fox GB, Faghih R, Bennani YL, Williams M, Hancock AA. J. Pharmacol. Exp. Ther. 2003;305:887. doi: 10.1124/jpet.102.047183. [DOI] [PubMed] [Google Scholar]
- 14.Sakurai E, Gunji E, IIzuka Y, Hikichi N, Maeyama K, Watanabe T. J.Pharm.Pharmacol. 1994;46:209. doi: 10.1111/j.2042-7158.1994.tb03780.x. [DOI] [PubMed] [Google Scholar]
- 15.Hough LB. Med. Chem.Res. 2004;13:78. [Google Scholar]
- 16.Price BJ, Clitherow JW, Bradshaw J. Ger. Offen. 1978 [2734070] [Google Scholar]
- 17.Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. Vol. 2. Academic Press; Sydney: 1986. [DOI] [PubMed] [Google Scholar]
- 18.D'Amour FE, Smith DL. J. Pharmacol. Exp. Ther. 1941;72:74. [Google Scholar]
- 19.Eddy NB, Leimbach D. J. Pharmacol. Exp. Ther. 1953;107:385. [PubMed] [Google Scholar]